
Boosting Electrochemical Nitrogen Reduction Performance over Binuclear Mo Atoms on N-Doped Nanoporous Graphene: A Theoretical Investigation


{kind=link}
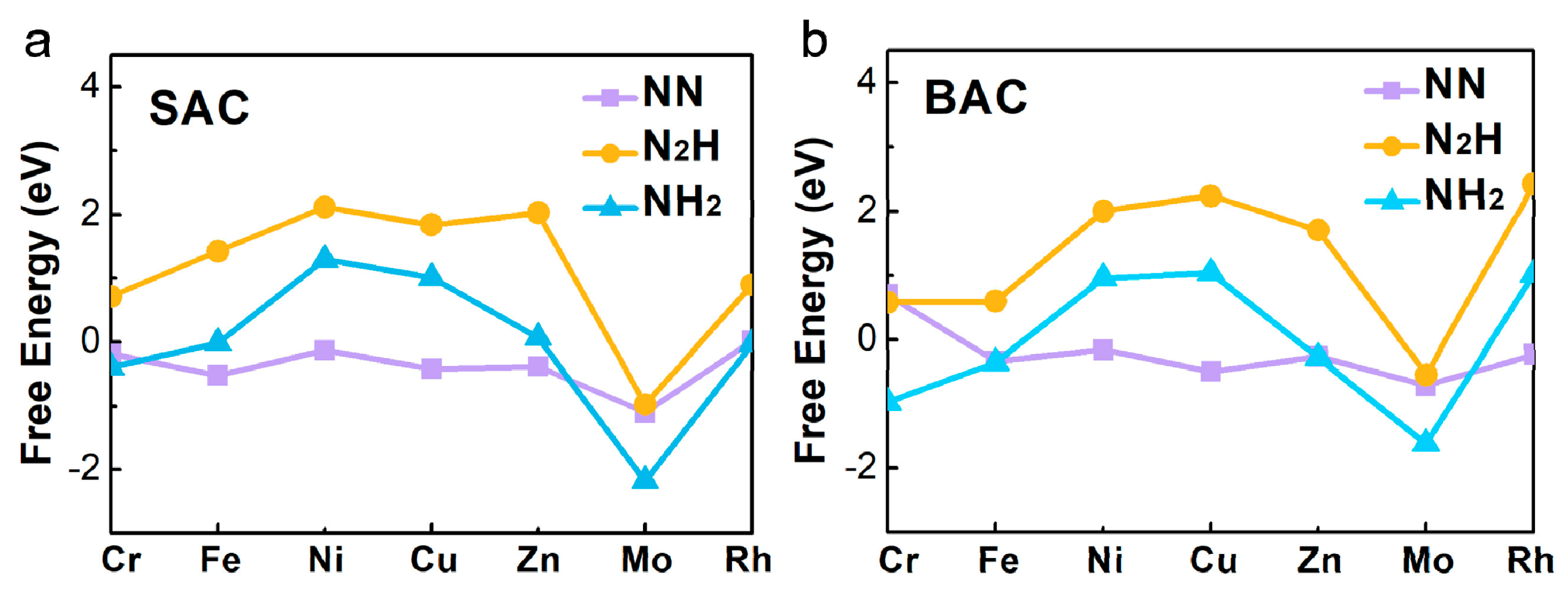{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Stability for Various Mono- and Binuclear N-C Catalysts
2.2. Screen of Potential NRR Electrocatalyst Combining Different Descriptors
2.3. Mo- and Mo2-N-C Monolayer for NRR
2.4. Hydrogen Evolution Reaction
3. Models and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Amar, I.A.; Lan, R.; Petit, C.T.G.; Tao, S. Solid-state electrochemical synthesis of ammonia: A review. J. Solid State Electrochem. 2011, 15, 1845–1860. [Google Scholar] [CrossRef]
- Honkala, K. Ammonia synthesis from first-principles calculations. Science 2005, 307, 555–558. [Google Scholar] [CrossRef]
- Erisman, J.W.; Sutton, M.A.; Galloway, J.; Klimont, Z.; Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008, 1, 636–639. [Google Scholar] [CrossRef]
- Giddey, S.; Badwal, S.P.S.; Kulkarni, A. Review of electrochemical ammonia production technologies and materials. Int. J. Hydrogen Energy 2013, 38, 14576–14594. [Google Scholar] [CrossRef]
- Licht, S.; Cui, B.; Wang, B.; Li, F.F.; Lau, J.; Liu, S. Ammonia synthesis by N2 and steam electrolysis in molten hydroxide suspensions of nanoscale Fe2O3. Science 2014, 345, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Service, R.F. New recipe produces ammonia emissions from the air, water, and sunlight. Science 2014, 345, 610. [Google Scholar] [CrossRef]
- Schlögl, R. Catalytic synthesis of ammonia—A “never-ending story”? Angew. Chem. Int. Ed. 2003, 42, 2004–2008. [Google Scholar] [CrossRef]
- Lv, C.; Yan, C.S.; Chen, G.; Ding, Y.; Sun, J.X.; Zhou, Y.S.; Yu, G. An amorphous noble-metal-free electrocatalyst that enables nitrogen fixation under ambient conditions. Angew. Chem. Int. Ed. 2018, 57, 6073–6076. [Google Scholar] [CrossRef]
- Kuriyama, S.; Arashiba, K.; Nakajima, K.; Matsuo, Y.; Tanaka, H.; Ishii, K.; Yoshizawa, K.; Nishibayashi, Y. Catalytic transformation of dinitrogen into ammonia and hydrazine by iron-dinitrogen complexes bearing pincer ligand. Nat. Commun. 2016, 7, 12181. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Shang, J.; Ai, Z.; Zhang, L. Efficient visible light nitrogen Fixation with BiOBr nanosheets of oxygen vacancies on the exposed {001} facets. J. Am. Chem. Soc. 2015, 137, 6393–6399. [Google Scholar] [CrossRef]
- Li, X.; Wang, W.; Jiang, D.; Sun, S.; Zhang, L.; Sun, X. Efficient solar-driven nitrogen fixation over carbon-tungstic-acid hybrids. Chem. Eur. J. 2016, 22, 13819–13822. [Google Scholar] [CrossRef]
- Liu, J.C.; Ma, X.L.; Li, Y.; Wang, Y.G.; Xiao, H.; Li, J. Heterogeneous Fe3 single-cluster catalyst for ammonia synthesis via an associative mechanism. Nat. Commun. 2018, 9, 1610. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, L.; Hu, L.; Chen, G.; Xin, H.; Feng, X. Ambient ammonia synthesis via palladium-catalyzed electrohydrogenation of dinitrogen at low overpotential. Nat. Commun. 2018, 9, 1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Ma, C.; Wu, X.; Deng, D.; Wei, M.; et al. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef]
- Jones, J.; Xiong, H.F.; Delariva, A.T.; Peterson, E.J.; Pham, H.; Challa, S.R.; Qi, G.S.; Oh, S.; Wiebenga, M.H.; Hernandez, X.I.P.; et al. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 2016, 353, 150–154. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Wang, A.; Qiao, B.; Liu, X.; Yang, X.; Wang, X.; Liang, J.; Li, J.; Liu, J.; Zhang, T. Remarkable performance of Ir1/FeO(x) single-atom catalyst in water gas shift reaction. J. Am. Chem. Soc. 2013, 135, 15314–15317. [Google Scholar] [CrossRef] [PubMed]
- Moses-DeBusk, M.; Yoon, M.; Allard, L.F.; Mullins, D.R.; Wu, Z.; Yang, X.; Veith, G.; Stocks, G.M.; Narula, C.K. CO oxidation on supported single Pt atoms: Experimental and ab initio density functional studies of CO interaction with Pt atom on Theta-Al2O3(010) surface. J. Am. Chem. Soc. 2013, 135, 12634–12645. [Google Scholar] [CrossRef]
- Narula, C.K.; Allard, L.F.; Stocks, G.M.; Moses-DeBusk, M. Remarkable NO oxidation on single supported platinum atoms. Sci. Rep. 2014, 4, 7238. [Google Scholar] [CrossRef] [Green Version]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Alam, M.Z.; De Leon, I.; Boyd, R.W. Large optical nonlinearity of indium tin oxide in its epsilon-near-zero region. Science 2016, 352, 795–797. [Google Scholar] [CrossRef]
- Zhao, J.X.; Chen, Z.F. Single Mo atom supported on defective boron nitride monolayer as an efficient electrocatalyst for nitrogen fixation: A computational study. J. Am. Chem. Soc. 2017, 139, 12480–12487. [Google Scholar] [CrossRef]
- Han, L.L.; Liu, X.J.; Chen, J.P.; Lin, R.Q.; Liu, H.X.; Lu, F.; Bak, S.; Liang, Z.X.; Zhao, S.Z.; Stavitski, E.; et al. Atomically dispersed molybdenum catalysts for high-efficiency ambient N2 fixation. Angew. Chem. Int. Ed. 2018, 58, 2321–2325. [Google Scholar] [CrossRef]
- Li, X.F.; Li, Q.K.; Cheng, J.; Liu, L.; Yan, Q.; Wu, Y.; Zhang, X.H.; Wang, Z.Y.; Qiu, Q.; Luo, Y. Conversion of dinitrogen to ammonia by FeN3-embedded graphene. J. Am. Chem. Soc. 2016, 138, 8706–8709. [Google Scholar] [CrossRef]
- Skulason, E.; Bligaard, T.; Gudmundsdottir, S.; Studt, F.; Rossmeisl, J.; Abild-Pedersen, F.; Vegge, T.; Jonsson, H.; Norskov, J.K. A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction. Phys. Chem. Chem. Phys. 2012, 14, 1235–1245. [Google Scholar] [CrossRef]
- Montoya, J.H.; Tsai, C.; Vojvodic, A.; Norskov, J.K. The challenge of electrochemical ammonia synthesis: A new perspective on the role of nitrogen scaling relations. ChemSusChem 2015, 8, 2180–2186. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, K.R.; Wang, Z.; Yan, X.; Cao, S.; Ye, Y.; Dong, Q.; Zhang, X.; Thorne, J.E.; Jin, L.; et al. Stable iridium dinuclear heterogeneous catalysts supported on metal-oxide substrate for solar water oxidation. Proc. Natl. Acad. Sci. USA 2018, 115, 2902–2907. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, H.; Chen, Y.; Zhu, J.; Gao, L.; Jin, Z.; Ge, J.; Jiang, Z.; Chen, S.; Liu, C.; et al. Identification of binuclear Co2N5 active sites for oxygen reduction reaction with more than one magnitude higher activity than single atom CoN4 Site. Nano Energy 2018, 46, 396–403. [Google Scholar] [CrossRef]
- Yan, H.; Lin, Y.; Wu, H.; Zhang, W.; Sun, Z.; Cheng, H.; Liu, W.; Wang, C.; Li, J.; Huang, X.; et al. Bottom-up precise synthesis of stable platinum dimers on graphene. Nat. Commun. 2017, 8, 1070. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Huang, Z.; Liu, W.; Chang, C.; Tang, H.; Li, Z.; Chen, W.; Jia, C.; Yao, T.; Wei, S.; et al. Design of N-coordinated dual-metal sites: A stable and active Pt-free catalyst for acidic oxygen reduction reaction. J. Am. Chem. Soc. 2017, 139, 17281–17284. [Google Scholar] [CrossRef]
- Ma, X.L.; Liu, J.C.; Xiao, H.; Li, J. Surface single-cluster catalyst for N2-to-NH3 thermal conversion. J. Am. Chem. Soc. 2018, 140, 46–49. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, Z.; Zhao, J. Computational screening of a single transition metal atom supported on the C2N monolayer for electrochemical ammonia synthesis. Phys. Chem. Chem. Phys. 2018, 20, 12835–12844. [Google Scholar] [CrossRef]
- Pignedoli, C.A.; Laino, T.; Treier, M.; Fasel, R.; Passerone, D. A simple approach for describing metal-supported cyclohexaphenylene dehydrogenation. Eur. Phys. J. B 2010, 75, 65–70. [Google Scholar] [CrossRef]
- Hinnemann, B.; Moses, P.G.; Bonde, J.; Jørgensen, K.P.; Nielsen, J.H.; Horch, S.; Chorkendorff, I.; Nørskov, J.K. Biomimetic hydrogen evolution: MoS2 nanoparticles as catalyst for hydrogen evolution. J. Am. Chem. Soc. 2005, 127, 5308–5309. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, 23–26. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A. Catalysts for hydrogen evolution from the [NiFe] hydrogenase to the Ni2P (001) surface: The importance of ensemble effect. J. Am. Chem. Soc. 2005, 127, 14871–14878. [Google Scholar] [CrossRef]
- Pasti, I.A.; Jovanovic, A.; Dobrota, A.S.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Atomic adsorption on graphene with a single vacancy: Systematic DFT study through the periodic table of elements. Phys. Chem. Chem. Phys. 2018, 20, 858–865. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [Green Version]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. CP2K: Atomistic simulations of condensed matter systems. WIREs Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: not available. |






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, R.; Hu, M.; Zhang, W.; He, J. Boosting Electrochemical Nitrogen Reduction Performance over Binuclear Mo Atoms on N-Doped Nanoporous Graphene: A Theoretical Investigation. Molecules 2019, 24, 1777. https://doi.org/10.3390/molecules24091777
Guo R, Hu M, Zhang W, He J. Boosting Electrochemical Nitrogen Reduction Performance over Binuclear Mo Atoms on N-Doped Nanoporous Graphene: A Theoretical Investigation. Molecules. 2019; 24(9):1777. https://doi.org/10.3390/molecules24091777
Chicago/Turabian StyleGuo, Ruijie, Min Hu, Weiqing Zhang, and Jia He. 2019. "Boosting Electrochemical Nitrogen Reduction Performance over Binuclear Mo Atoms on N-Doped Nanoporous Graphene: A Theoretical Investigation" Molecules 24, no. 9: 1777. https://doi.org/10.3390/molecules24091777