
Inhibition of Tyrosyl-DNA Phosphodiesterase 1 by Lipophilic Pyrimidine Nucleosides

 , ,
, ,  and
and
Abstract
:
1. Introduction
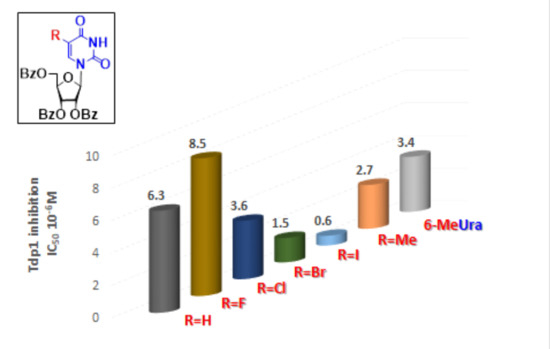
2. Results
2.1. DNA Repair Enzyme Inhibition
2.2. Cytotoxicity and Sensitization of Tumor Cells to the Effect of Topotecan
2.2.1. Cytotoxicity
2.2.2. Tumor Cell Sensitization to Topotecan
3. Discussion
4. Materials and Methods
4.1. General
4.2. Real-Time Detection of Tdp1 Activity
4.3. Cell Culture Assays
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
References
- Hosoya, N.; Miyagawa, K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014, 105, 370–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laev, S.S.; Salakhutdinov, N.F.; Lavrik, O.I. Tyrosyl-DNA phosphodiesterase inhibitors: Progress and potential. Bioorg. Med. Chem. 2016, 24, 5017–5027. [Google Scholar] [CrossRef] [PubMed]
- Brettrager, E.J.; van Waardenburg, R.C.A.M. Targeting tyrosyl-DNA phosphodiesterase I to enhance toxicity of phosphodiester linked DNA adducts. Cancer Drug Resist 2019, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharenko, A.; Dyrkheeva, N.; Lavrik, O. Dual DNA topoisomerase 1 and tyrosyl-DNA phosphodiesterase 1 inhibition for improved anticancer activity. Med. Res. Rev. 2019, 39, 1427–1441. [Google Scholar] [CrossRef]
- Kelley, M.R.; Logsdon, D.; Fishel, M.L. Targeting DNA repair pathways for cancer treatment: What’s new? Future Oncol. 2014, 10, 1215–1237. [Google Scholar] [CrossRef] [Green Version]
- Francica, P.; Rottenberg, S. Mechanisms of PARP inhibitor resistance in cancer and insights into the DNA damage response. Genome Med. 2018, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Matsuno, Y.; Hyodo, M.; Fujimori, H.; Shimizu, A.; Yoshioka, K.I. Sensitization of cancer cells to radiation and topoisomerase I inhibitor camptothecin using inhibitors of PARP and other signaling molecules. Cancers 2018, 10, 364. [Google Scholar] [CrossRef]
- Minchom, A.; Aversa, C.; Lopez, J. Dancing with the DNA damage response: Next-generation anti-cancer therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP-1 and beyond. Nat. Rev. 2010, 10, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Drenichev, M.S.; Mikhailov, S.N. Poly(ADP-ribose)–a unique natural polymer. Structural features, biological role and approaches to the chemical synthesis. Nucleosides Nucleot. Nucl. Acids 2015, 34, 258–276. [Google Scholar] [CrossRef]
- Drenichev, M.S.; Mikhailov, S.N. Poly(ADP-ribose): From chemical synthesis to drug design. Digest paper. Bioorg. Med. Chem. Lett. 2016, 26, 3395–3403. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. Inhibiting the DNA damage response as a therapeutic manoeuvre in cancer. Br. J. Pharmacol. 2013, 169, 1745–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moor, N.A.; Lavrik, O.I. Protein–protein interactions in DNA base excision repair. Biochemistry (Moscow) 2018, 83, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy. Cancers 2018, 10, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, S.; Nandi, S. DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun. Signal. 2017. [Google Scholar] [CrossRef] [Green Version]
- Ferraris, D.V. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J. Med. Chem. 2010, 53, 4561–4584. [Google Scholar] [CrossRef]
- Comeaux, E.Q.; Waardenburg, R.C. Tyrosyl-DNA phosphodiesterase I resolves both naturally and chemically induced DNA adducts and its potential as a therapeutic target. Drug Metab Rev. 2014, 46, 494–507. [Google Scholar] [CrossRef]
- Rechkunova, N.I.; Lebedeva, N.A.; Lavrik, O.I. Tyrosyl-DNA phosphodiesterase Tdp1 is a new player in repair of apurinic/apirimidinic sites in DNA. Rus. J. Bioorg. Chem. 2015, 41, 474–480. [Google Scholar] [CrossRef]
- Komarova, A.O.; Drenichev, M.S.; Dyrkheeva, N.S.; Kulikova, I.V.; Oslovsky, V.E.; Zakharova, O.D.; Zakharenko, A.L.; Mikhailov, S.N.; Lavrik, O.I. Novel group of tyrosyl-DNA-phosphodiesterase 1 inhibitors based on disaccharide nucleosides as drug prototypes for anti-cancer therapy. J. Enz. Inh. Med. Chem. 2018, 33, 1415–1429. [Google Scholar] [CrossRef] [Green Version]
- Efremova, A.S.; Zakharenko, A.L.; Shram, S.I.; Kulikova, I.V.; Drenichev, M.S.; Sukhanova, M.V.; Khodyreva, S.N.; Myasoedov, N.F.; Lavrik, O.I.; Mikhailov, S.N. Disaccharide pyrimidine nucleosides and their derivatives: A novel group of cell-penetrating inhibitors of poly(ADP-ribose) polymerase-1. Nucleosides Nucleot. Nucl. Acids 2013, 32, 510–528. [Google Scholar] [CrossRef]
- Mikhailov, S.N.; Efimtseva, E.V.; Rodionov, A.A.; Bobkov, G.V.; Kulikova, I.V.; Herdewijn, P. Synthesis of 2-O-β-d-ribofuranosylnucleosides. Curr. Prot. Nucl. Acids Chem. 2006, 27, 1.14.1–1.14.18. [Google Scholar] [CrossRef] [PubMed]
- Efremova, A.S.; Shram, S.I.; Drenichev, M.S.; Posypanova, G.A.; Myasoedov, N.F.; Mikhailov, S.N. The selective toxic effect of dialdehyde derivatives of pyrimidine nucleosides on human ovarian cancer cells. Biochem. (Moscow) Suppl. Ser. B Biomed. Chem. 2014, 8, 318–322. [Google Scholar] [CrossRef]
- Mikhailov, S.N.; Kulikova, I.V.; Nauwelaerts, K.; Herdewijn, P. Synthesis of 2′-O-α-d-ribofuranosyladenosine, monomeric unit of poly(ADP-ribose). Tetrahedron 2008, 64, 2871–2876. [Google Scholar] [CrossRef]
- Mikhailov, S.N.; Drenichev, M.S.; Oslovsky, V.E.; Kulikova, I.V.; Herdewijn, P. Synthesis of poly(ADP-ribose) monomer containing 2′-O-α-d-ribofuranosyl adenosine. Curr. Prot. Nucl. Acids Chem. 2019, e92. [Google Scholar] [CrossRef] [PubMed]
- Vorbrüggen, H. Adventures in silicon-organic chemistry. Acc. Chem. Res. 1995, 28, 509–520. [Google Scholar] [CrossRef]
- Prasad, A.K.; Kumar, V.; Malhotra, S.; Ravikumar, V.T.; Sanghvi, Y.S.; Parmar, V.S. ‘Green’ methodology for efficient and selective benzoylation of nucleosides using benzoyl cyanide in an ionic liquid. Bioorg. Med. Chem. 2005, 13, 4467–4472. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, A.; Khomenko, T.; Zhukova, S.; Koval, O.; Zakharova, O.; Anarbaev, R.; Lebedeva, N.; Korchagina, D.; Komarova, N.; Vasiliev, V.; et al. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg. Med. Chem. 2015, 23, 2044–2052. [Google Scholar] [CrossRef]
- Shimokawa, S.; Kimura, J.; Mitsunobu, O. Studies on nucleosides and nucleotides. V. Selective aroylation of 5ʹ-hydroxyl group of uridine and adenosine. Bull. Chem. Soc. Jpn. 1976, 49, 3357–3358. [Google Scholar] [CrossRef] [Green Version]
- Preiss, J.; Schlaeger, R.; Hilz, H. Specific inhibition of poly ADP-ribose polymerase by thymidine and nicotinamide in HeLa cells. FEBS Lett. 1971, 19, 244–246. [Google Scholar] [CrossRef] [Green Version]
- Pivazyan, A.D.; Birks, E.M.; Wood, T.G.; Lin, T.S.; Prusoff, W.H. Inhibition of poly(ADP-ribose)polymerase activity by nucleoside analogs of thymidine. Biochem. Pharmacol. 1992, 44, 947–953. [Google Scholar] [CrossRef]
- Pouliot, J.J.; Robertson, C.A.; Nash, H.A. Pathways for repair of topoisomerase I covalent complexes in Saccharomyces cerevisiae. Genes Cells 2001, 6, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, J.J.; Yao, K.C.; Robertson, C.A.; Nash, H.A. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science 1999, 286, 552–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.W.; Burgin, A.B., Jr.; Huizenga, B.N.; Robertson, C.A.; Yao, K.C.; Nash, H.A. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. USA 1996, 93, 11534–11539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Interthal, H.; Chen, H.J.; Champoux, J.J. Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages. J. Biol. Chem. 2005, 280, 36518–36528. [Google Scholar] [CrossRef] [Green Version]
- Beretta, G.L.; Cossa, G.; Gatti, L.; Zunino, F.; Perego, P. Tyrosyl-DNA phosphodiesterase 1 targeting for modulation of camptothecin-based treatment. Curr. Med. Chem. 2010, 17, 1500–1508. [Google Scholar] [CrossRef]
- Interthal, H.; Chen, H.J.; Kehl-Fie, T.; Zotzmann, J.; Leppard, J.; Champoux, J. SCAN1 mutant Tdp1 accumulates the enzyme–DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005, 24, 2224–2233. [Google Scholar] [CrossRef] [Green Version]
- El-Khamisy, S.; Saifi, G.; Weinfeld, M.; Johansson, F.; Helleday, T.; Lupski, J.; Caldecott, K. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 2005, 434, 108–113. [Google Scholar] [CrossRef]
- Miao, Z.-H.; Agama, K.; Sordet, O.; Povirk, L.; Kohn, K.; Pommier, Y. Hereditary ataxia SCAN1 cells are defective for the repair of transcription-dependent topoisomerase I cleavage complexes. DNA Repair 2006, 5, 1489–1494. [Google Scholar] [CrossRef]
- Huang, H.-C.; Liu, J.; Baglo, Y.; Rizvi, I.; Anbil, S.; Pigula, M.; Hasan, T. Mechanism-informed Repurposing of Minocycline Overcomes Resistance to Topoisomerase Inhibition for Peritoneal Carcinomatosis. Mol. Cancer Ther. 2018, 17, 508–520. [Google Scholar] [CrossRef] [Green Version]
- Nivens, M.C.; Felder, T.; Galloway, A.H.; Pena, M.M.O.; Pouliot, J.J.; Spencer, H.T. Engineered Resistance to Camptothecin and Antifolates by Retroviral Coexpression of Tyrosyl DNA phosphodiesterase-I and Thymidylate Synthase. Cancer Chemother. Pharmacol. 2004, 53, 107–115. [Google Scholar] [CrossRef]
- Barthelmes, H.U.; Habermeyer, M.; Christensen, M.O.; Mielke, C.; Interthal, H.; Pouliot, J.J.; Boege, F.; Marko, D. TDP1 Overexpression in Human Cells Counteracts DNA Damage Mediated by Topoisomerases I and II. J. Biol. Chem. 2004, 279, 55618–55625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisenberg, C.; Gilbert, D.; Chalmers, A.; Haley, V.; Gollins, S.; Ward, S.; El-Khamisy, S. Clinical and cellular roles for TDP1 and TOP1 in modulating colorectal cancer response to irinotecan. Mol. Cancer Ther. 2015, 14, 575–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Aerschot, A.; Everaert, D.; Balzarini, J.; Augustyns, K.; Jie, L.; Janssen, G.; Peeters, O.; Blaton, N.; De Ranter, C.; De Clercq, E.; et al. Synthesis and anti-HIV evaluation of 2′,3′-dideoxyribo-5-chloropyrimidine analogues: Reduced toxicity of 5-chlorinated 2′,3′-dideoxynucleosides. J. Med. Chem. 1990, 33, 1833–1839. [Google Scholar] [CrossRef] [PubMed]
- Niedballa, U.; Vorbruggen, H. A general synthesis of N-glycosides. IV. Synthesis of nucleosides of hydroxyl and mercapto N-heterocycles. J. Org. Chem. 1977, 39, 3668–3671. [Google Scholar] [CrossRef]
- Vorbrüggen, H.; Bennua, B. A new simplified nucleoside synthesis. Chem. Ber. 1981, 114, 1279–1286. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–9 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | Structure | LogP 1 | IC50 µM | HeLa CC50 µM |
|---|---|---|---|---|
| 1a |  | −2.28 | >50 | ND 2 |
| 1b |  | −2.64 | >50 | ND |
| 1f |  | −2.24 | >50 | ND |
| 1c |  | −1.97 | >50 | ND |
| 1d |  | −1.44 | >50 | ND |
| 1e |  | −1.39 | >50 | ND |
| 2a |  | 5.07 | 6.3 ± 0.4 | ND |
| 2b |  | 5.27 | 8.5 ± 1.4 | >100 |
| 2f |  | 5.73 | 3.6 ± 1.1 | >100 |
| 2c |  | 5.90 | 1.5 ± 0.9 | >100 |
| 2d |  | 6.06 | 0.6 ± 0.9 | >100 |
| 2e |  | 5.47 | 2.7 ± 0.6 | >100 |
| 2g |  | 5.27 | 3.4 ± 0.2 | >100 |
| 3a |  | 0.08 | >100 | ND |
| 4a |  | 2.58 | 23 ± 6 | >100 |
| 5 |  | 5.07 | 18 ± 1 | ND |
| 6 |  | 5.28 | 6.0 ± 0.7 | >100 |
| 7 |  | 5.55 | 2.91 ± 0.01 | >100 |
| 8 |  | 4.69 | 4.3 ± 0.7 | >100 |
| 9 |  | 5.62 | 4.2 ± 0.3 | ND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakharenko, A.L.; Drenichev, M.S.; Dyrkheeva, N.S.; Ivanov, G.A.; Oslovsky, V.E.; Ilina, E.S.; Chernyshova, I.A.; Lavrik, O.I.; Mikhailov, S.N. Inhibition of Tyrosyl-DNA Phosphodiesterase 1 by Lipophilic Pyrimidine Nucleosides. Molecules 2020, 25, 3694. https://doi.org/10.3390/molecules25163694
Zakharenko AL, Drenichev MS, Dyrkheeva NS, Ivanov GA, Oslovsky VE, Ilina ES, Chernyshova IA, Lavrik OI, Mikhailov SN. Inhibition of Tyrosyl-DNA Phosphodiesterase 1 by Lipophilic Pyrimidine Nucleosides. Molecules. 2020; 25(16):3694. https://doi.org/10.3390/molecules25163694
Chicago/Turabian StyleZakharenko, Alexandra L., Mikhail S. Drenichev, Nadezhda S. Dyrkheeva, Georgy A. Ivanov, Vladimir E. Oslovsky, Ekaterina S. Ilina, Irina A. Chernyshova, Olga I. Lavrik, and Sergey N. Mikhailov. 2020. "Inhibition of Tyrosyl-DNA Phosphodiesterase 1 by Lipophilic Pyrimidine Nucleosides" Molecules 25, no. 16: 3694. https://doi.org/10.3390/molecules25163694