3.2. Syntheses
((4-Azido-2-fluorocyclopent-2-en-1-yl)oxy)(t-butyl)diphenylsilane (±)-4. To a stirred mixture of (±)-3 (1.15 g, 3.22 mmol) and PPh3 (2.52 g, 9.61 mmol) in anhydrous THF/CH2Cl2 (1/1 v/v, 60 mL) at 0 °C were added, under an argon atmosphere, diphenylphosphoryl azide (DPPA, 2 mL, 9.31 mmol) and diisopropyl azodicarboxylate (DIAD, 1.8 mL, 9.14 mmol). The reaction mixture was stirred at 0 °C for 30 min then concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (petroleum ether/CH2Cl2 100/0 to 85/15), affording (±)-4 (825 mg, 67% yield) as a yellowish oil. Due to its instability (±)-4 was quickly used for the next step. Rf = 0.75 (petroleum ether/EtOAc, 7/3). 1H-NMR (CDCl3, 300 MHz): δ 7.69–7.65 (m, 4H, ArH), 7.48–7.37 (m, 6H, ArH), 5.29 (dd, J = 2.5, 0.5 Hz, 1H, H3), 4.91–4.86 (m, 1H, H1), 4.49–4.43 (m, 1H, H4), 2.20 (dddd, J = 14.4, 7.6, 3.9, 0.9 Hz, 1H, H5a), 1.97 (dddd, J = 14.4, 7.2, 2.5, 1.1 Hz, 1H, H5b), 1.08 (s, 9H, 3 × CH3).
4-((tert-Butyldimethylsilyl)oxy)-3-fluorocyclopent-2-enamine (±)-5. To a stirred solution of (±)-4 (825 mg, 2.16 mmol) in anhydrous THF (70 mL) at 0 °C was added dropwise, under an argon atmosphere, LiAlH4 (1 M in THF, 3.3 mL, 3.3 mmol). The reaction mixture was allowed to warm to room temperature and stirred for additional 30 min. THF/H2O (15 mL, 9/1 v/v) were added dropwise then the mixture was filtrated through a short pad of celite® and silica gel which was washed with MeOH. The filtrate was concentrated under reduced pressure to afford (±)-5 (646 mg, 84% yield) as a colorless oil. Rf = 0.3 (CH2Cl2/MeOH, 9/1). 1H-NMR (CDCl3, 400 MHz): δ 7.69–7.65 (m, 4H, ArH), 7.45–7.35 (m, 6H, ArH), 5.21 (d, J = 2.1 Hz, 1H, H2), 4.87–4.83 (m, 1H, H4), 4.07–4.02 (m, 1H, H1), 2.25–2.19 (m, 1H, H5a), 1.69–1.61 (ddd, J = 14.0, 7.3, 3.4 Hz, 1H, H5b), 1.40 (br s, 1H, NH2), 1.07 (s, 9H, 3 × CH3). 13C-NMR (CDCl3, 100 MHz): δ 163.5 (d, J = 287 Hz, C3), 136.0 (CAr), 135.9 (CAr), 134.0 (Cq), 133.5 (Cq), 130.0 (CAr), 129.9 (CAr), 127.9 (CAr), 127.7 (CAr), 111.3 (d, J = 6.8 Hz, C2), 72.7 (d, J = 21.5 Hz, C4), 50.7 (d, J = 9.8 Hz, C1), 43.7 (d, J = 5.6 Hz, C5), 27.0 (3 × CH3), 19.3 (Cq). 19F-NMR (CDCl3, 376 MHz): δ −127.2.
(E)-N-((4-((tert-Butyldiphenylsilyl)oxy)-3-fluorocyclopent-2-en-1-yl)carbamoyl)-3-ethoxyacrylamide (±)-6. (E)-3-ethoxyacryloyl chloride (280 mg, 2.08 mmol) was added to a suspension of AgOCN (465 mg, 3.12 mmol) in anhydrous toluene (4 mL) under an argon atmosphere. The mixture was refluxed for 30 min then cooled to room temperature and filtrated through a pad of celite®. The filtrate was added dropwise at −20 °C to a solution of amine (±)-5 (184 mg, 0.52 mmol) in anhydrous DMF (7 mL) under an argon atmosphere. The reaction mixture was allowed to reach room temperature and stirred for 5 h. The solvents were evaporated under reduced pressure and the residue was purified by flash chromatography on silica gel (petroleum ether/EtOAc, 100/0 to 60/40), affording (±)-6 (252 mg, 98% yield) as a colorless oil. Rf = 0.24 (petroleum ether/EtOAc, 7/3). 1H-NMR (CDCl3, 400 MHz): δ 9.24 (s, 1H, NH), 8.53 (d, J = 7.7 Hz, 1H, NH), 7.68–7.64 (m, 4H, ArH), 7.58 (d, J = 12.2 Hz, 1H, H3), 7.45–7.35 (m, 6H, ArH), 5.28–5.25 (m, 2H, H2 and H2′), 4.98–4.93 (m, 1H, H1′), 4.87–4.85 (m, 1H, H4′), 3.93 (q, J = 7.1 Hz, 2H, CH2-CH3), 2.30 (ddd, J = 14, 7.9, 2.9 Hz, 1H, H5′a), 1.88 (ddd, J = 14, 7.3, 3.3 Hz, 1H, H5′b), 1.33 (t, J = 7.1 Hz, 3H, CH2-CH3), 1.07 (s, 9H, 3 × CH3). 13C-NMR (CDCl3, 100 MHz): δ 168.0 (CO), 164.6 (d, J = 287 Hz, C3′), 163.0 (C3), 154.6 (CO), 135.9 (CAr), 135.7 (CAr), 133.6 (Cq), 133.1 (Cq), 129.8 (2 × CAr), 127.7 (CAr), 127.6 (CAr), 107.1 (d, J = 10.2 Hz, C2′), 97.8 (C2), 71.9 (d, J = 21.1 Hz, C4′), 67.5 (CH2-CH3), 49.0 (d, J = 11.5 Hz, C1′), 40.8 (d, J = 5.1 Hz, H5′), 26.8 (3 × CH3), 19.2 (Cq), 14.4 (CH2-CH3). 19F-NMR (CDCl3, 376 MHz): δ −124.4. HRMS (ESI−): calculated for C27H32FN2O4Si [M − H]− 495.2115; found: 495.2116.
Trans-1-(3-fluoro-4-hydroxycyclopent-2-en-1-yl)pyrimidine-2,4(1H,3H)-dione (±)-7. Hydrochloric acid (2N aqueous solution, 3.9 mL, 7.33 mmol) was added to a stirred solution of (±)-6 (1.2 g, 2.43 mmol) in EtOH (35 mL). The reaction mixture was refluxed overnight, then cooled to room temperature and concentrated under reduced pressure. The crude material was purified by flash chromatography on silica gel (CH2Cl2/MeOH, 100/0 to 90/10) to give (±)-7 (401 mg, 78% yield) as a white foam. Rf = 0.34 (CH2Cl2/MeOH, 9/1). 1H-NMR (CD3OD, 400 MHz): δ 7.48 (d, J = 8.0 Hz, 1H, H6), 5.67 (d, J = 8.0 Hz, 1H, H5), 5.68–5.62 (m, 1H, H1′), 5.29 (d, J = 2.2 Hz, 1H, H2′), 4.90–4.86 (m, 1H, H4′), 2.34 (dddd, J = 14.8, 8.2, 3.2, 1.3 Hz, 1H, H5′a), 2.19 (ddd, J = 14.8, 7.5, 3.6 Hz, 1H, H5′b). 13C-NMR (CD3OD, 100 MHz): δ 168.5 (d, J = 288 Hz, C3′), 166.4 (CO), 152.8 (CO), 143.2 (C6), 105.8 (d, J = 13.0 Hz, C2′), 102.8 (C5), 71.1 (d, J = 21.4 Hz, C4′), 57.5 (d, J = 12.0 Hz, C1′), 40.1 (d, J = 5.7 Hz, C5′). 19F-NMR (CD3OD, 376 MHz): δ −123.4. UV (EtOH 95) λmax = 265 nm (εmax = 9800). HRMS (ESI−): calculated for C9H8FN2O3 [M − H]− 211.0519; found: 211.0519.
Cis-1-(3-fluoro-4-hydroxycyclopent-2-en-1-yl)pyrimidine-2,4(1H,3H)-dione (±)-8. Diisopropyl azodicarboxylate (DIAD, 1.67 mL, 8.51 mmol) was added dropwise, under an argon atmosphere, to a stirred mixture of (±)-7 (361 mg, 1.7 mmol), PPh3 (2.23 g, 8.51 mmol) and benzoic acid (1.09 g, 8.51 mmol) in anhydrous THF (37 mL) at 0 °C. The mixture was allowed to reach room temperature and stirred for 1 h, then the solvents were evaporated under reduced pressure. The residue was dissolved in MeOH (37 mL), K2CO3 (588 mg, 4.25 mmol) was added and the solution was stirred at room temperature for 4 h. The solvents were evaporated under reduced pressure and the crude material was purified by flash chromatography on silica gel (CH2Cl2/MeOH, 93/7) yielding (±)-8 (336 mg, 93%) as a white solid. Rf = 0.39 (CH2Cl2/MeOH, 9/1). 1H-NMR (D2O, 400 MHz): δ 7.70 (d, J = 8.0 Hz, 1H, H6), 5.89 (d, J = 8.0 Hz, 1H, H5), 5.44–5.39 (m, 2H, H2′ and H1′), 4.78–4.76 (m, 1H, H4′), 3.11–3.03 (m, 1H, H5′a), 1.67–1.61 (m, 1H, H5′b). 13C-NMR (D2O, 100 MHz): δ 166.5 (CO), 165.2 (d, J = 286 Hz, C3′), 152.2 (CO), 143.3 (C6), 105.0 (d, J = 13.1 Hz, C2′), 102.1 (C5), 68.6 (d, J = 21.4, C4′), 54.0 (d, J = 11.5 Hz, C1′), 37.8 (d, J = 5.6 Hz, C5′). 19F-NMR (D2O, 376 MHz): δ −123.1. UV (EtOH 95) λmax = 266 nm (εmax = 6400). HRMS (ESI−): calculated for C9H8FN2O3 [M − H]− 211.0519; found: 211.0520.
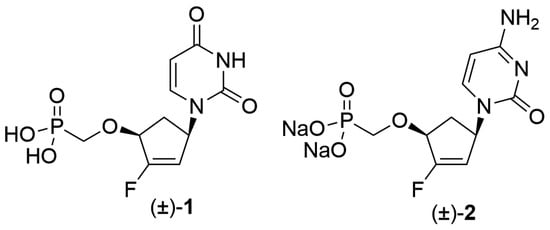
((4-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluorocyclopent-2-en-1-yl)oxy)methylphosphonic acid (±)-1. A solution of (±)-8 (26 mg, 0.123 mmol) in anhydrous DMF (0.5 mL) was added, under an argon atmosphere, to a suspension of NaH (60% in mineral oil, 25 mg, 0.625 mmol) in anhydrous DMF (0.5 mL). The mixture was stirred at room temperature for 15 min then a solution of diethyl-(tosyloxymethyl)phosphonate (47 mg, 0.146 mmol) was added. The reaction mixture was stirred at room temperature for 3 days, then AcOH (50 μL) was added and the solvents were evaporated under reduced pressure to furnish crude (±)-9. This intermediate was dissolved in anhydrous DMF (1.5 mL) and TMSBr (80 μL, 0.617 mmol) was added dropwise at 0 °C under an argon atmosphere. The solution was stirred at room temperature for 16 h until a further aliquot of TMSBr was added (80 μL, 0.617 mmol). After 4 additional hours of stirring, TMSBr (160 μL, 1.3 mmol) was again added and the mixture was stirred for a further 4 h at room temperature then, the reaction was stopped by adding triethylammonium bicarbonate buffer (TEAB 1M, pH 7) and concentrated to dryness under high vacuum. Purification by reverse-phase chromatography on RP-18 (H2O/MeOH, 100/0 to 80/20) followed by column chromatography on silica gel (i-PrOH/NH4OH/H2O, 7/2/1) afforded the phosphonate (±)-1 (9 mg, 24% yield). Rf = 0.15 (i-PrOH/NH4OH/H2O, 7/2/1). 1H-NMR (D2O, 400 MHz): δ 7.77 (d, J = 8.0 Hz, 1H, H6), 5.89 (d, J = 8.0 Hz, 1H, H5), 5.51–5.40 (m, 2H, H1′ and H3′), 4.63 (dd, J = 7.4, 2.8 Hz, 1H, H4′), 3.71 (d, J = 9.2 Hz, 2H, CH2P), 3.00 (ddd, J = 15.9, 7.9, 7.9 Hz, 1H, H5′a), 1.97–1.73 (m, 1H, H5′b). 13C-NMR (D2O, 100 MHz): δ 166.5 (CO), 164.5 (d, J = 286 Hz, C2′), 152.2 (CO), 143.7 (C6), 106.7 (d, J = 12.4 Hz, C3′), 102.2 (C5), 78.5 (C1′), 65.2 (CH2P), 53.8 (d, J = 11.4 Hz, C4′), 34.9 (C5′). 19F NMR (D2O, 376 MHz): δ −120.7. 31P-NMR (162 MHz, D2O): δ 14.7. UV (H2O) λmax = 264 nm (εmax = 8600). HRMS (ESI−): calculated for C10H11FN2O6P [M − H]− 305.0339; found: 305.0341.
Trans-4-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-fluorocyclopent-2-en-1-yl acetate (±)-10. To a stirred solution of (±)-7 (370 mg, 1.75 mmol) in anhydrous THF (7 mL) were added NEt3 (0.37 mL, 2.65 mmol), DMAP (10 mg, 0.08 mmol) and Ac2O (0.2 mL, 2.12 mmol) under an argon atmosphere. The mixture was stirred at room temperature for 1 hour before concentration to dryness. The residue was dissolved in water (50 mL) and extracted with EtOAc (3 × 25 mL). The combined organic layers were washed with brine (50 mL), dried (MgSO4) and concentrated in vacuo to afford (±)-10 (340 mg, 77%) as a white powder. Rf = 0.64 (CH2Cl2/MeOH, 9/1). 1H-NMR (MeOD, 400 MHz): δ 7.52 (d, J = 8.0 Hz, 1H, H6), 5.90–5.87 (m, 1H, H4′), 5.68 (d, J = 8 Hz, 1H, H5), 566–5.61 (m, 1H, H1′), 5.61 (d, J = 2.3 Hz, 1H, H3′), 2.48–2.41 (m, H5′a), 2.36 (ddd, J = 15.2, 7.6, 3.9 Hz, 1H, H5′b), 2.09 (s, 3H, CH3). 13C- NMR (MeOD, 100 MHz): δ 172.0 (C=O), 166.3 (C=O), 164.3 (d, J = 285.9 Hz, C3′), 152.6 (C=O), 143.4 (C6), 109.5 (d, J = 12.3 Hz, C2′), 102.9 (C5), 74.1 (d, J = 21.5 Hz, C4′), 57.7 (d, J = 11.4 Hz, C1′), 37.5 (d, J = 4.3 Hz, C5′), 20.7 (CH3). 19F-NMR (MeOD, 376 MHz): δ −123.3. UV (EtOH) λmax = 265 (εmax = 7600). HRMS (ESI+): calculated for C11H12N2O4F [M + H]+ 255.0781; Found 255.0780.
Trans-4-amino-1-(3-fluoro-4-hydroxycyclopent-2-en-1-yl)pyrimidin-2(1H)-one (±)-11. Lawesson’s reagent (870 mg, 2.15 mmol) was added to a suspension of (±)-10 (340 mg, 1.34 mmol) in anhydrous freshly distilled 1,2-dichloroethane (65 mL), at room temperature under an argon atmosphere. The mixture was refluxed for 20 h, then cooled to room temperature, concentrated and the residue was filtrated through a pad of silica gel (CH2Cl2/MeOH, 9/1). The filtrate was concentrated to vacuum and the resulting crude material was dissolved in NH3/MeOH (7N, 25 mL) before being heated in a Parr high pressure reactor at 100 °C for 4 h. Concentration to dryness and purification by column chromatography on silica gel (CH2Cl2/MeOH, 9/1) afforded (±)-11 (220 mg, 78%) as a colorless oil. Rf = 0.33 (CH2Cl2/MeOH, 8/2). 1H-NMR (MeOD, 400 MHz): δ 7.49 (d, J = 7.4 Hz, 1H, H6), 5.88 (d, J = 7.4 Hz, 1H, H5), 5.72–5.66 (m, 1H, H1′), 5.29 (d, J = 2.4 Hz, 1H, H2′), H4′ signal in H2O peak, 2.38 (dddd, J = 14.6, 8.1, 3.2, 1.2 Hz, 1H, H5′a), 2.13 (ddd, J = 14.7, 7.7, 3.6 Hz, 1H, H5′b). 13C-NMR (MeOD, 100 MHz): δ 168.2 (d, J = 287.7 Hz, C3′), 167.5 (CN), 158.9 (CO), 143.2 (C6), 106.2 (d, J = 12.5 Hz, C2′), 96.3 (C5), 71.1 (d, J = 21.5 Hz, C4′), 58.1 (d, J = 11.7 Hz, C1′), 40.7 (d, J = 5.6 Hz, C5′). 19F-NMR (MeOD, 376 MHz): δ −123.78. UV (EtOH) λmax = 276 nm (εmax = 6400). HRMS (ESI−): calculated for C9H11N3O2F [M + H]+ 212.0835; Found 212.0837.
Cis-4-amino-1-(3-fluoro-4-hydroxycyclopent-2-en-1-yl)pyrimidin-2(1H)-one (±)-12. Diisopropyl azodicarboxylate (DIAD, 0.98 mL, 5.00 mmol) was added dropwise to a stirred solution of (±)-11 (210 mg, 1.00 mmol), PPh3 (1.31 g, 5.00 mmol) and benzoic acid (610 mg, 5.00 mmol) in anhydrous THF (20 mL) at 0 °C under an argon atmosphere. The mixture was allowed to reach room temperature and stirred for 2 h before concentration to dryness under reduced pressure. The residue was dissolved in MeOH (20 mL) before K2CO3 (276 mg, 2.00 mmol) was added. The mixture was stirred at room temperature for 2 h then the solvents were evaporated under reduced pressure. Purification by flash chromatography on silica gel (CH2Cl2/MeOH, 9/1) afforded partially purified (±)-12 (125 mg, 60%) as a colourless oil. Compound (±)-12 was used in the next step without further purification. Rf = 0.23 (CH2Cl2/MeOH, 8/2). 1H-NMR (MeOD, 400 MHz): δ 7.65 (d, J = 7.4 Hz, 1H, H6), 5.91 (d, J = 7.4 Hz, 1H, H5), 5.52–5.47 (m, 1H, H1′), 5.29 (d, J = 2.6 Hz, 1H, H2′), 4.64 (dd, J = 7.8, 3.5 Hz, 1H, H4′), 4.45 (br s, 1H, NH), 3.04–2.96 (m, 1H, H5′a), 1.53 (ddd, J = 14.7, 6.0, 3.5 Hz, 1H, H5′b). 13C-NMR (MeOD, 100 MHz): δ 167.8 (d, J = 286.6 Hz, C3′), 167.5 (CN), 158.9 (CO), 143.6 (C6), 106.2 (d, J = 12.2 Hz, C2′), 96.5 (C5), 70.1 (d, J = 21.7 Hz, C4′), 55.5 (d, J = 10.8 Hz, C1′), 40.3 (d, J = 5.7 Hz, C5′). 19F-NMR (MeOD, 376 MHz): δ −124.87. UV (EtOH) λmax = 275nm (εmax = 4300). HRMS (ESI+): calculated for C9H11N3O2F [M + H]+ 212.0835; Found 212.0836.
N-(1-(3-Fluoro-4-hydroxycyclopent-2-en-1-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)benzamide (±)-13. To a stirred solution of (±)-12 (125 mg, 0.592 mmol) in anhydrous DMF (2.5 mL) was added benzoic anhydride (127 mg, 0.561 mmol) under an argon atmosphere. The reaction mixture was stirred at room temperature for 24 h and then concentrated to dryness. The solid residue was washed with Et2O, then purified by flash chromatography on silica gel (CH2Cl2 to CH2Cl2/MeOH, 9/1) to afford (±)-13 (81 mg, 44%) as a white solid. Rf = 0.46 (CH2Cl2/MeOH, 9/1). 1H-NMR (MeOD, 400 MHz): δ 8.11 (d, J = 7.4 Hz, 1H, CHAr), 7.98–7.95 (m, 2H, CHAr and H6), 7.64–7.60 (m, 2H, CHAr), 7.55–7.50 (m, 2H, CHAr and H5), 5.58 (ddd, J = 8.8, 6.1, 3.1 Hz, 1H, H1′), 5.34 (d, J = 2.6 Hz, 1H, H2′), 4.67 (dd, J = 7.8, 3.3 Hz, 1H, H4′), 3.08 (ddd, J = 14.8, 8.1, 8.1 Hz, 1H, H5′a), 1.64 (ddd, J = 14.9, 5.8, 3.3 Hz, 1H, H5′b). 13C-NMR (MeOD, 100 MHz): δ 168.1 (d, J = 287.8 Hz, C3′), 164.1 (CN), 158.2 (CO), 147.2 (C6), 134.2 (CAr), 133.9 (CHAr), 129.5 (CHAr), 128.9 (CHAr), 105.4 (d, J = 13.2 Hz, C2′), 99.1 (C5), 69.8 (d, J = 21.7 Hz, C4′), 56.6 (d, J = 10.8 Hz, C1′), 40.0 (d, J = 5.8 Hz, C5′). 19F-NMR (MeOD, 376 MHz): δ −122.4. UV (EtOH) λmax = 260 nm (εmax = 16900). HRMS (ESI+): calculated for C16H15N3O3F [M + H]+ 316.1097; Found 316.1097.
Sodium-(((4-(4-amino-2-oxopyrimidin-1(2H)-yl)-2-fluorocyclopent-2-en-1-yl)oxy)methyl) phosphonate (±)-2. (±)-13 (78 mg, 0.25 mmol) and diethyl-(tosyloxymethyl)phosphonate (321 mg, 0.80 mmol) were dissolved in anhydrous DMF under an argon atmosphere and the solution was heated at 40 °C for 30 min. LiOtBu (2.2M in THF, 0.27 mL, 0.59 mmol) was added and the reaction mixture was heated at 40 °C for a further 30 min. Concentration to dryness and purification by flash chromatography afforded the desired intermediate phosphonate contaminated with tosylate. This mixture was used in the next step without further purification. To a solution of intermediate phosphonate in anhydrous DMF (4 mL) at 0 °C and under an argon atmosphere was added dropwise TMSBr (0.32 mL, 2.5 mmol). The solution was stirred at room temperature for 24 h, then neutralized by adding triethylammonium bicarbonate buffer (TEAB 1 M, pH 7) and concentrated to dryness under high vacuum. The residue was dissolved in NH3/MeOH (7 M) and heated in a Parr high pressure reactor at 50 °C for 4 h. After concentration to dryness, purification by reverse-phase chromatography on RP-18 (H2O to H2O/MeOH, 50/50) followed by an ion exchange on DOWEX 50WX2 (Na+ form) afforded (±)-2 (59 mg, 67%) as a white powder. 1H-NMR (D2O, 400 MHz): δ 7.74 (d, J = 7.4 Hz, 1H, H6), 6.06 (d, J = 7.4 Hz, 1H, H5), 5.49–5.45 (m, 2H, H3′ and H4′), 4.64–4.61 (m, 1H, H1′), 3.68 (d, J = 9.4 Hz, 2H, OCH2P), 3.03–2.95 (m, 1H, H5′a), 1.81–1.77 (m, 1H, H5′b). 13C-NMR (D2O, 100 MHz): δ 165.9 (CN), 164.2 (d, J = 286.2 Hz, C2′), 158.2 (CO), 143.3 (C6), 107.2 (d, J = 12.1 Hz, C3′), 96.4 (C5), 78.3 (d, J = 20.4 Hz, C1′), 65.8 (d, J = 155.6 Hz, CH2P), 54.3 (d, J = 11.0 Hz, C4′), 35.3 (d, J = 5.3 Hz, C5′). 19F-NMR (D2O, 376 MHz): δ −121.35. 31P-NMR (D2O, 162 MHz): δ 14.6. UV (H2O) λmax = 274 nm (εmax = 6500). HRMS (ESI−): calculated for C10H14FN3O5P [M + H]+ 306.0655; Found 306.0656.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



