Oxidative Dehydrogenation of Propane over Vanadium-Containing Faujasite Zeolite

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
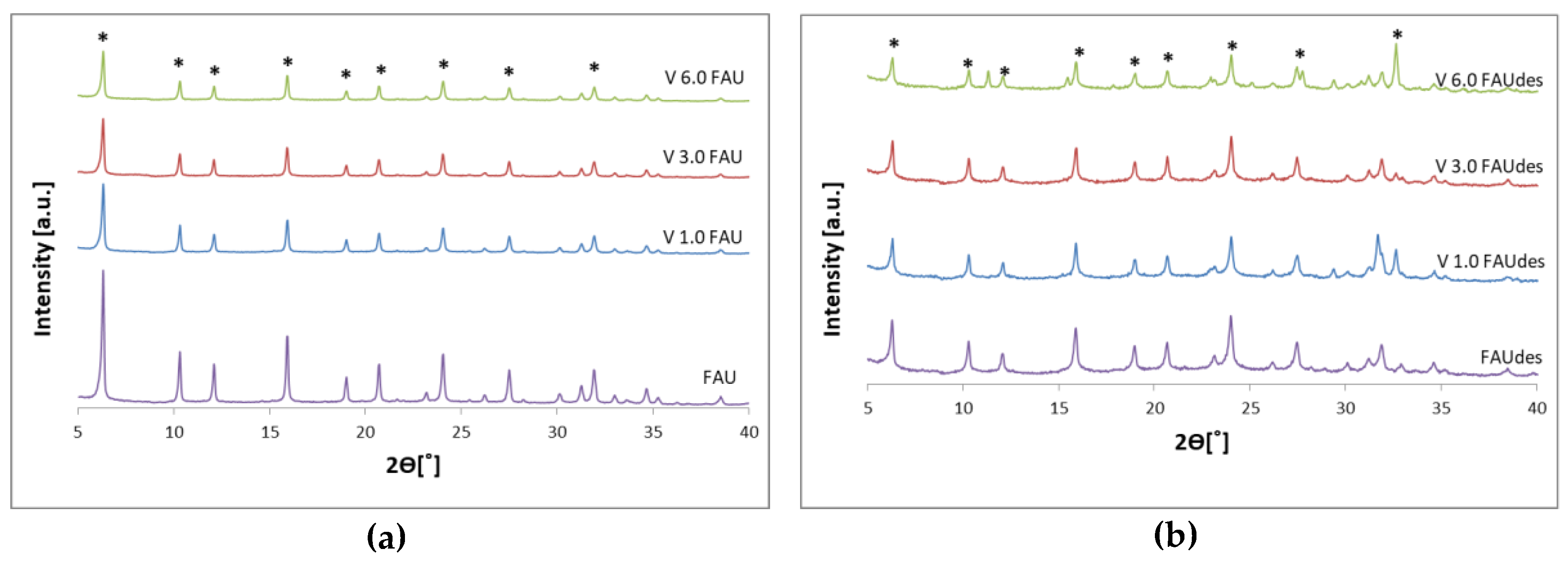
2.1. Structure and Morphology of the Obtained Samples
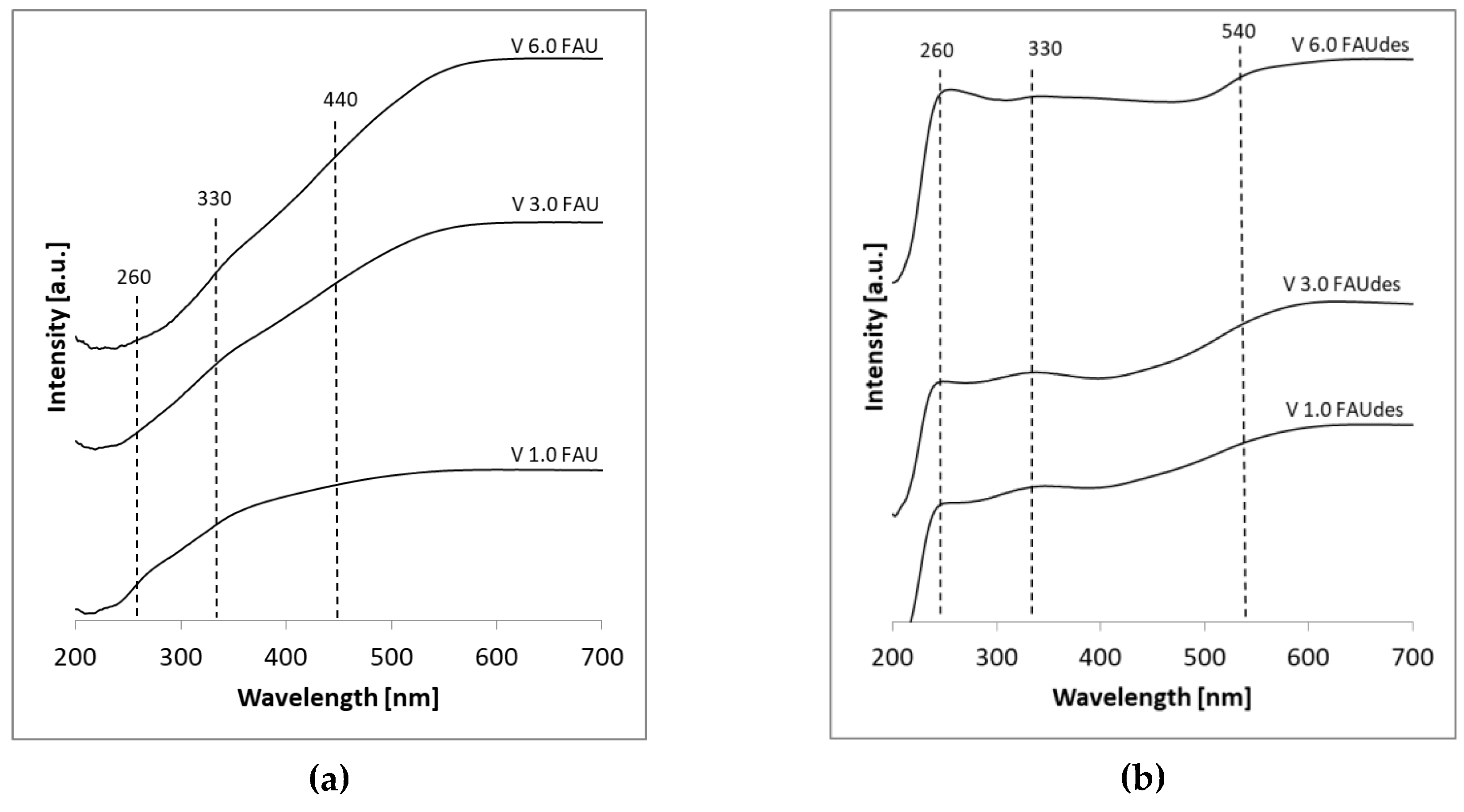
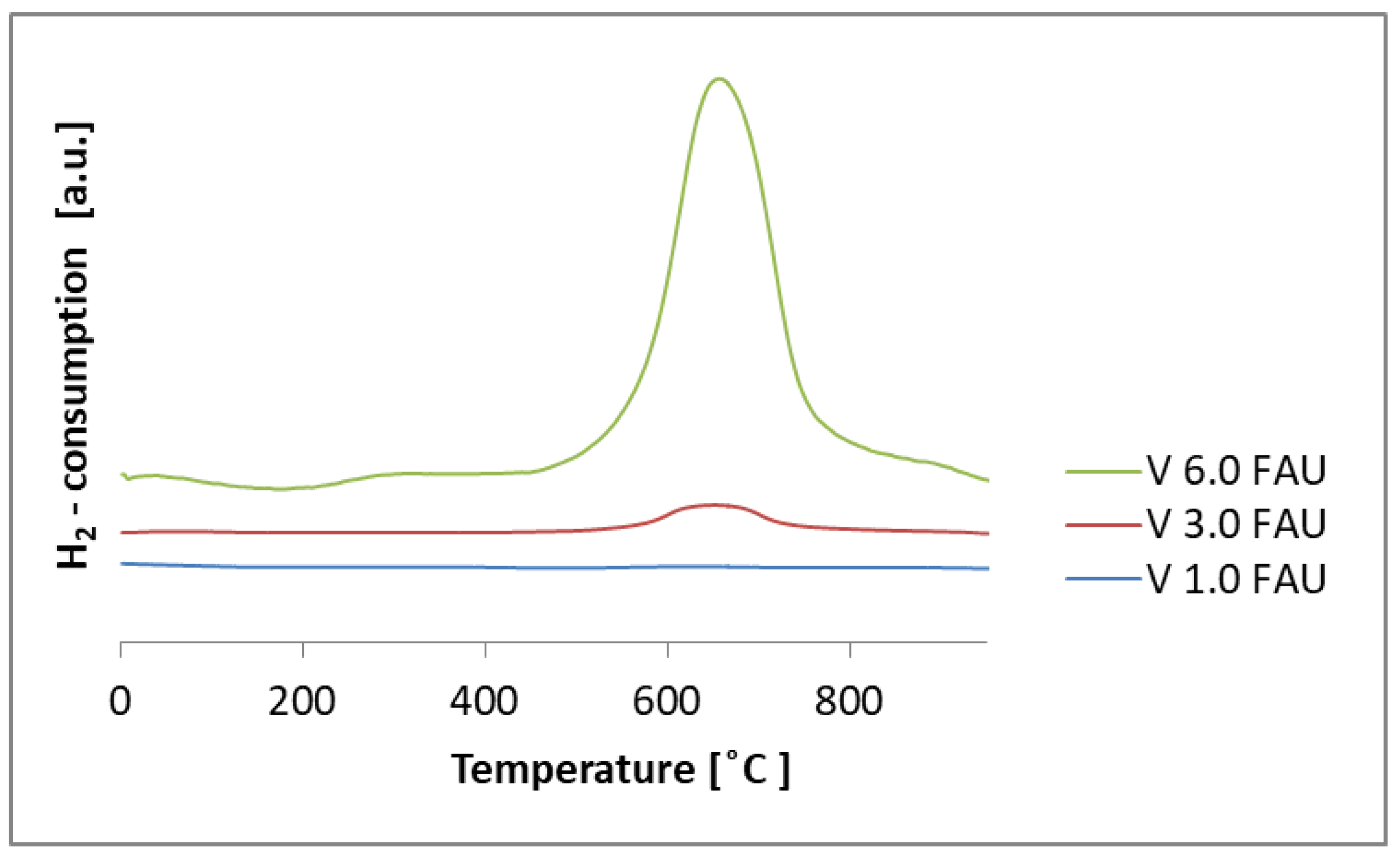
2.2. Physico-Chemical Characterization of the Obtained Samples
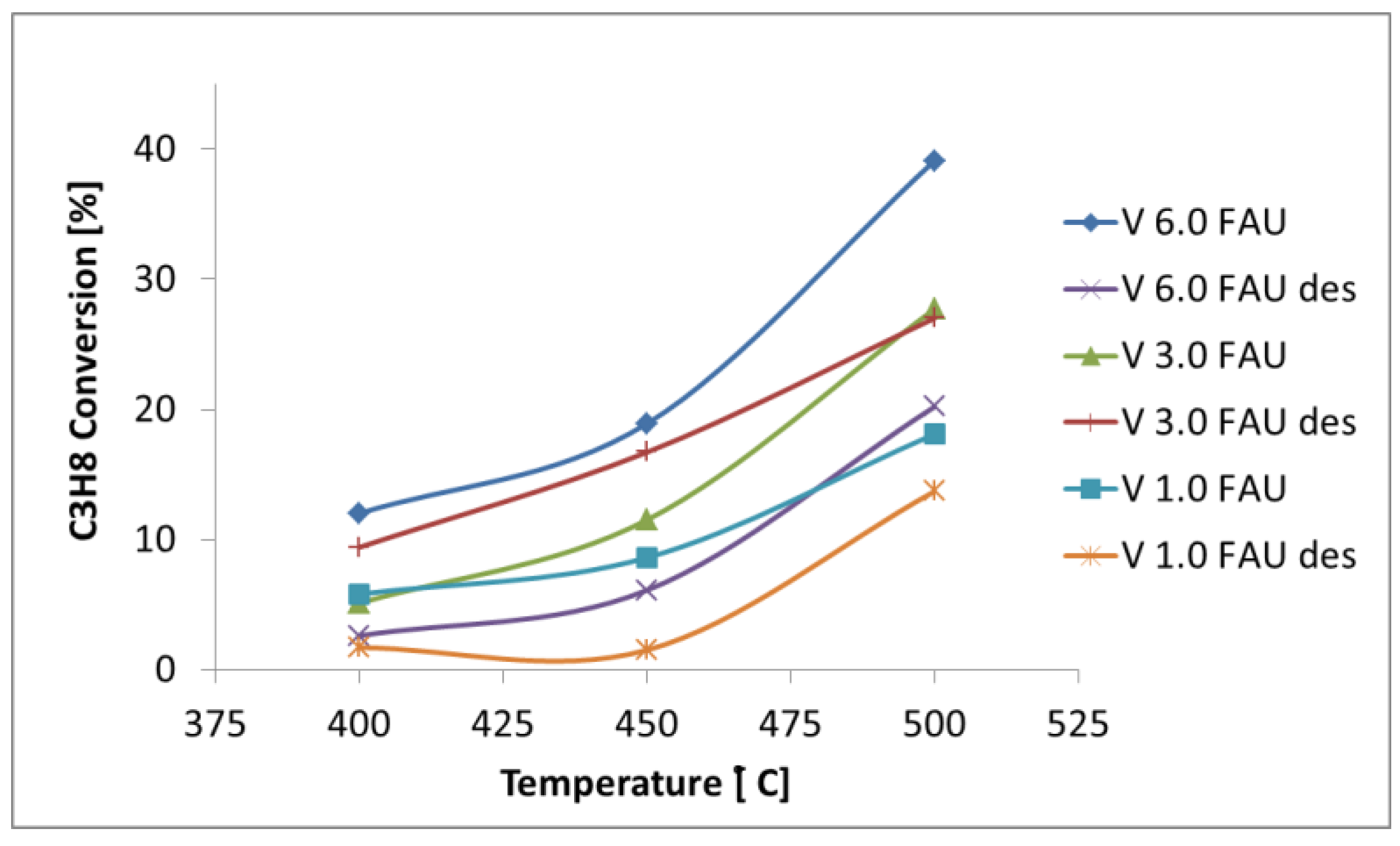
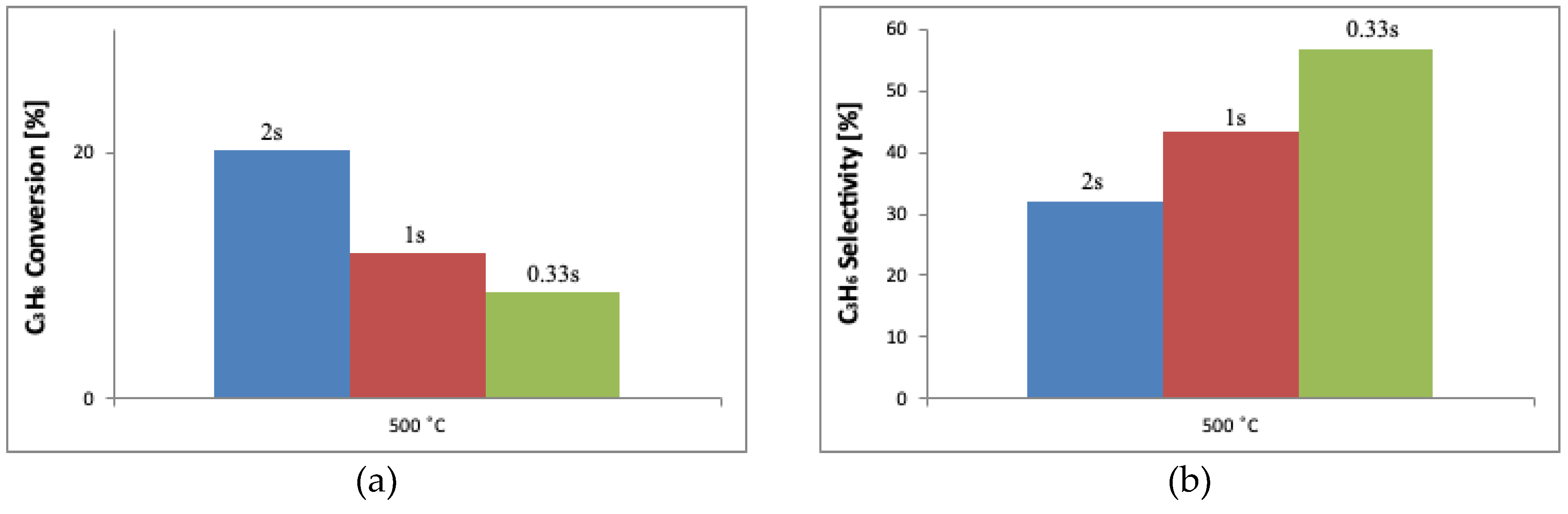
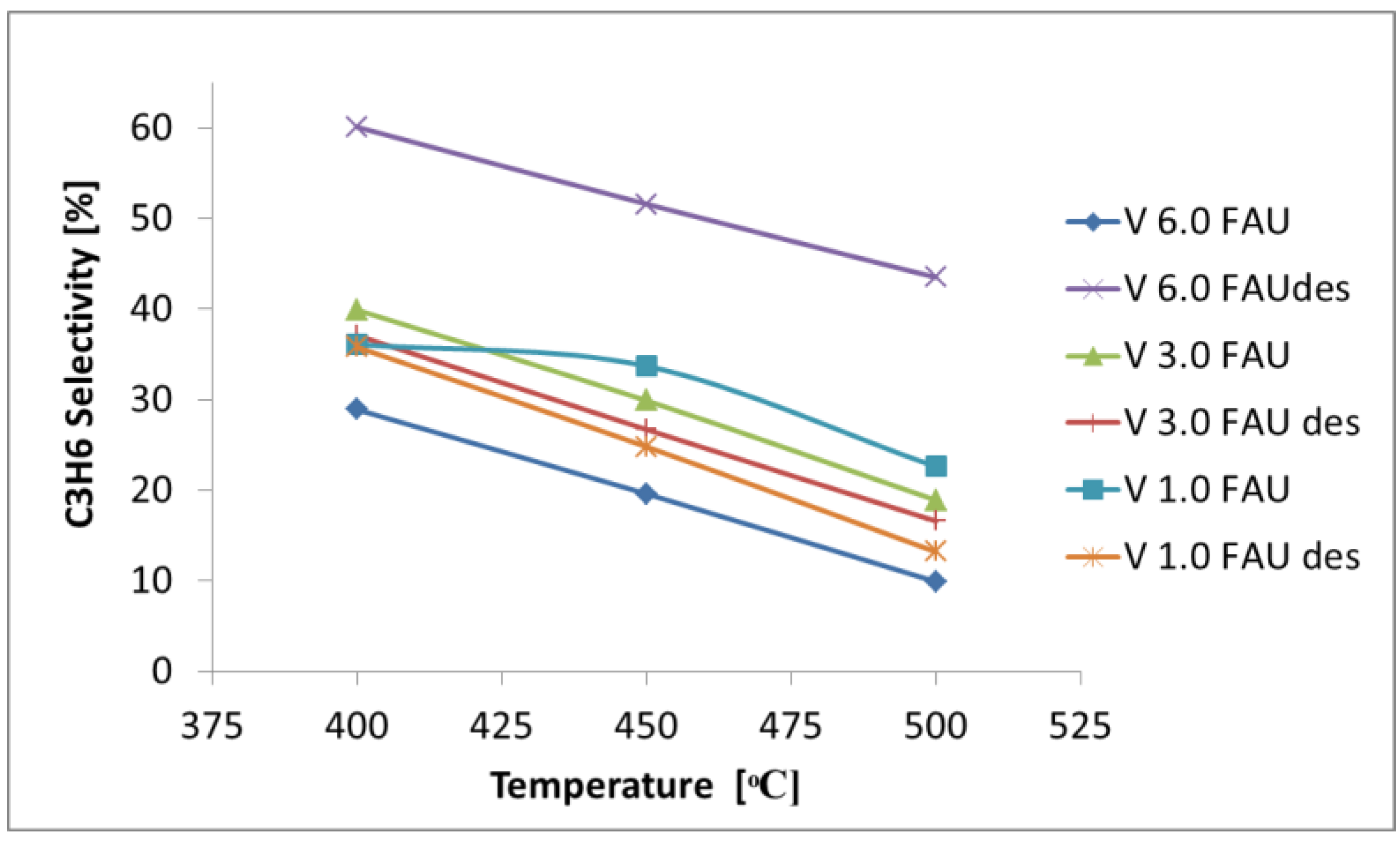
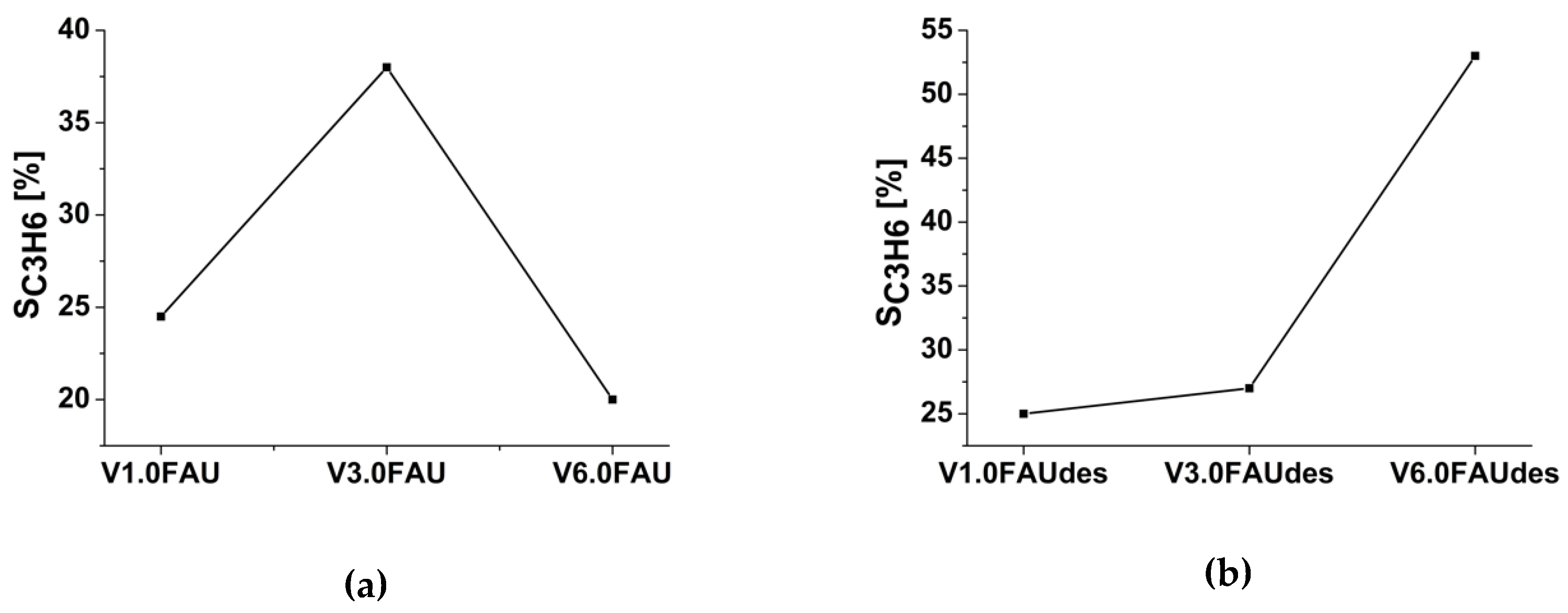
2.3. Determination of the Catalytic Properties in the Oxidative Dehydrogenation of Propane
3. Materials and Methods
3.1. Catalyst Preparation
3.2. X-ray Diffraction (XRD) Analysis
3.3. Specific Surface Area and Porosity Studies
3.4. Determination of Surface Morphology and Composition by Scanning Electron Microscopy (SEM) with the Energy-Dispersive X-ray Spectroscopy (EDS) Method
3.5. Determination of Elemental Composition by the X-ray Fluorescence (XRF) Method
3.6. Determination of Vanadium Coordination by the Ultra-Violet Visible Spectroscopy (UV-VIS) Method
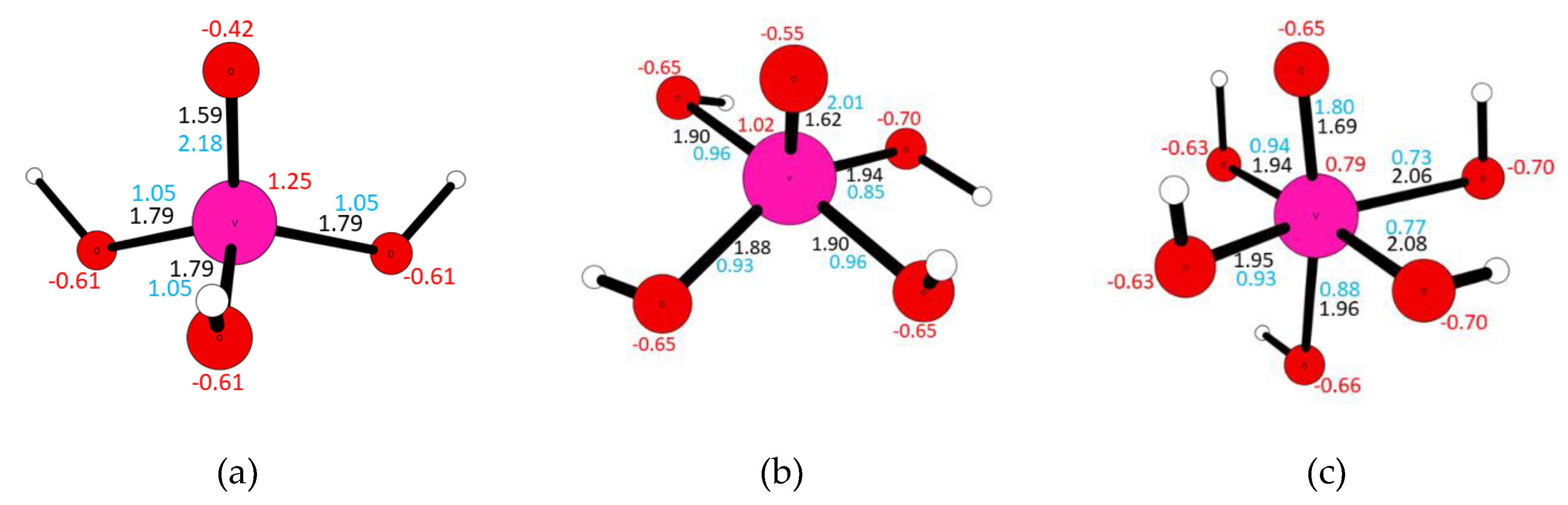
3.7. Quantum Chemical Calculations
3.8. Determination of Reducibility by Temperature-Programmed Reduction (TPR-H2)
3.9. Determination of Acidity by the Temperature-Programmed Desorption of Ammonia (TPD-NH3)
3.10. Determination of Catalytic Properties in the ODH (Oxidative Dehydrogenation) of Propane
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- James, O.O.; Mandal, S.; Alele, N.; Chowdhury, B.; Maity, S. Lower alkanes dehydrogenation: Strategies and reaction routes to corresponding alkenes. Fuel Process. Technol. 2016, 149, 239–255. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F. The oxidative dehydrogenation of ethane and propane as an alternative way for the production of light olefins. Catal. Today 1995, 24, 307–313. [Google Scholar] [CrossRef]
- Carrero, C.A.; Schloegl, R.; Wachs, I.E.; Schomaecker, R. Critical literature review of the kinetics for the oxidative dehydrogenation of propane over well-defined supported vanadium oxide catalysts. ACS Catal. 2014, 4, 3357–3380. [Google Scholar] [CrossRef]
- Cavani, F.; Ballarini, N.; Cericola, A. Oxidative dehydrogenation of ethane and propane: How far from commercial implementation? Catal. Today 2007, 127, 113–131. [Google Scholar] [CrossRef]
- Grzybowska-Świerkosz, B. Thirty years in selective oxidation on oxides: What have we learned? Top. Catal. 2000, 11, 23–42. [Google Scholar] [CrossRef]
- Lin, M.M. Selective oxidation of propane to acrylic acid with molecular oxygen. Appl. Catal. A Gen. 2001, 207, 1–16. [Google Scholar] [CrossRef]
- Grabowski, R. Kinetics of oxidative dehydrogenation of C2-C3 alkanes on oxide catalysts. Catal. Rev. 2006, 48, 199–268. [Google Scholar] [CrossRef]
- Hu, Z.-P.; Yang, D.; Wang, Z.; Yuan, Z.-Y. State-of-the-art catalysts for direct dehydrogenation of propane to propylene. Chin. J. Catal. 2019, 40, 1233–1254. [Google Scholar] [CrossRef]
- Langeslay, R.R.; Kaphan, D.M.; Marshall, C.L.; Stair, P.C.; Sattelberger, A.P.; Delferro, M. Catalytic applications of vanadium: A mechanistic perspective. Chem. Rev. 2019, 119, 2128–2191. [Google Scholar] [CrossRef]
- Blasco, T.; Nieto, J.M.L. Oxidative dyhydrogenation of short chain alkanes on supported vanadium oxide catalysts. Appl. Catal. A Gen. 1997, 157, 117–142. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, H. Insights into the structural requirements for oxidative dehydrogenation of propane on crystalline Mg-V-O catalysts. Appl. Catal. A Gen. 2018, 568, 1–10. [Google Scholar] [CrossRef]
- Belomestnykh, I.P.; Isaguliants, G.V. V–Mg–O catalysts for oxidative dehydrogenation of alkylpyridines and alkylthiophenes. Catal. Today 2009, 142, 192–195. [Google Scholar] [CrossRef]
- Cortés, I.; Rubio, O.; Herguido, J.; Menéndez, M. Kinetics under dynamic conditions of the oxidative dehydrogenation of butane with doped V/MgO. Catal. Today 2004, 91–92, 281–284. [Google Scholar] [CrossRef]
- Madeira, L.M.; Martın-Aranda, R.M.; Maldonado-Hódar, F.J.; Fierro, J.L.G.; Portela, M.F. Oxidative dehydrogenation ofn-butane over alkali and alkaline earth-promoted α-NiMoO4Catalysts. J. Catal. 1997, 169, 469–479. [Google Scholar] [CrossRef]
- Madeira, L.M.; Portela, M.F. Mechanistic effects resulting from the cesium-doping of a NiMoO4 catalyst in n-butane oxidative dehydrogenation. Appl. Catal. A Gen. 2005, 281, 179–189. [Google Scholar] [CrossRef]
- Lisi, L.; Ruoppolo, G.; Casaletto, M.P.; Galli, P.; Massucci, M.A.; Patrono, P.; Pinzari, F. Vanadium-metal(IV)phosphates as catalysts for the oxidative dehydrogenation of ethane. J. Mol. Catal. A Chem. 2005, 232, 127–134. [Google Scholar] [CrossRef]
- Casaletto, M.P.; Mattogno, G.; Massucci, M.A. Alumina-supported vanadyl phosphates catalysts for the oxidative dehydrogenation of ethane: An XPS characterisation. Appl. Surf. Sci. 2003, 211, 216–226. [Google Scholar] [CrossRef]
- Venegas, J.M.; Grant, J.T.; McDermott, W.P.; Burt, S.P.; Micka, J.; Carrero, C.A.; Hermans, I. Selective oxidation of n-butane and isobutane catalyzed by boron nitride. ChemCatChem 2017, 9, 2118–2127. [Google Scholar] [CrossRef]
- Grasselli, R.K. Fundamental principles of selective heterogeneous oxidation catalysis. Top. Catal. 2002, 21, 79–88. [Google Scholar] [CrossRef]
- Callahan, J.L.; Grasselli, R.K. A selectivity factor in vapor-phase hydrocarbon oxidation catalysis. AIChE J. 1963, 9, 755–760. [Google Scholar] [CrossRef]
- Dźwigaj, S.; Gressel, I.; Grzybowska, B.; Samson, K. Oxidative dehydrogenation of propane on VSiβ catalysts. Catal. Today 2006, 114, 237–241. [Google Scholar] [CrossRef]
- Kubacka, A.; Włoch, E.; Sulikowski, B.; Valenzuela, R.X.; Cortés Corberán, V. Oxidative dehydrogenation of propane on zeolite catalysts. Catal. Today 2000, 61, 343–352. [Google Scholar] [CrossRef]
- Chalupka, K.; Thomas, C.; Millot, Y.; Averseng, F.; Dzwigaj, S. Mononuclear pseudo-tetrahedral V species of VSiBEA zeolite as the active sites of the selective oxidative dehydrogenation of propane. J. Catal. 2013, 305, 46–55. [Google Scholar] [CrossRef]
- Centi, G.; Trifiro, F. Catalytic behavior of V-containing zeolites in the transformation of propane in the presence of oxygen. Appl. Catal. A Gen. 1996, 143, 3–16. [Google Scholar] [CrossRef]
- Teixeira-Neto, A.A.; Marchese, L.; Landi, G.; Lisi, L.; Pastore, H.O. [V,Al]-MCM-22 catalyst in the oxidative dehydrogenation of propane. Catal. Today 2008, 133–135, 1–6. [Google Scholar] [CrossRef]
- Julbe, A.; Farrusseng, D.; Jalibert, J.C.; Mirodatos, C.; Guizard, C. Characteristics and performance in the oxidative dehydrogenation of propane of MFI and V-MFI zeolite membranes. Catal. Today 2000, 56, 199–209. [Google Scholar] [CrossRef]
- Gackowski, M.; Tarach, K.; Kuterasiński, Ł.; Podobiński, J.; Jarczewski, S.; Kuśtrowski, P.; Datka, J. Hierarchical zeolites Y obtained by desilication: Porosity, acidity and catalytic properties. Microporous Mesoporous Mater. 2018, 263, 282–288. [Google Scholar] [CrossRef]
- Gackowski, M.; Tarach, K.; Kuterasiński, Ł.; Podobiński, J.; Sulikowski, B.; Datka, J. Spectroscopic IR and NMR studies of hierarchical zeolites obtained by desilication of zeolite Y: Optimization of the desilication route. Microporous Mesoporous Mater. 2019, 281, 134–141. [Google Scholar] [CrossRef]
- Klisińska, A.; Loridant, S.; Grzybowska, B.; Stoch, J.; Gressel, I. Effect of additives on properties of V2O5/SiO2 and V2O5/MgO catalysts: II. Structure and physicochemical properties of the catalysts and their correlations with oxidative dehydrogenation of propane and ethane. Appl. Catal. A Gen. 2006, 309, 17–27. [Google Scholar] [CrossRef]
- Hunger, B.; Heuchel, M.; Clark, L.A.; Snurr, R.Q. Characterization of acidic OH groups in zeolites of different types: An interpretation of NH3-TPD results in the light of confinement effects. J. Phys. Chem. B 2002, 106, 3882–3889. [Google Scholar] [CrossRef]
- Gil, B.; Broclawik, E.; Datka, J.; Klinowski, J. Acidic hydroxyl groups in zeolites X and Y: A correlation between infrared and solid-state NMR spectra. J. Phys. Chem. 1994, 98, 930–933. [Google Scholar] [CrossRef]
- Gackowski, M.; Datka, J. Acid properties of hierarchical zeolites Y. Molecules 2020, 25, 1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klisińska, A.; Samson, K.; Gressel, I.; Grzybowska, B. Effect of additives on properties of V2O5/SiO2 and V2O5/MgO catalysts: I. Oxidative dehydrogenation of propane and ethane. Appl. Catal. A Gen. 2006, 309, 10–16. [Google Scholar] [CrossRef]
- Al-Ghamdi, S.A. Oxygen-Free Propane Oxidative Dehydrogenation Over Vanadium Oxide Catalysts: Reactivity and Kinetic Modelling; Monograph, The University of Western Ontario: London, ON, Canada, 2013. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirac, P.A.M. Quantum mechanics of many-electron systems. Proc. R. Soc. Lond. Ser. A 1929, 123, 714–733. [Google Scholar]
- Slater, J.C. A simplification of the hartree-fock method. Phys. Rev. 1951, 81, 385–390. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- TURBOMOLE V6.3 2011, a. d. o. U. o. K. a.; Forschungszentrum Karlsruhe GmbH, -.; TURBOMOLE GmbH, s. a. f. Available online: http://www.turbomole.com (accessed on 2 July 2011).
Sample Availability: Samples of the studied compounds are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | V Content (wt.%) |
|---|---|
| V1.0 FAU | 1.07 |
| V3.0FAU | 3.55 |
| V6.0FAU | 8.13 |
| V1.0FAUdes | 1.26 |
| V3.0FAUdes | 4.45 |
| V6.0FAUdes | 6.62 |
| Sample | SSA (m2/g) | Pore Volume (cm3/g) | Micropore Pore Volume (cm3/g) | Mesopore Pore Volume (cm3/g) | SSA Micropores (m2/g) | SSA Mesopores (m2/g) |
|---|---|---|---|---|---|---|
| FAU | 883 | 0.52 | 0.30 | 0.22 | 724 | 158 |
| V1.0 FAU | 750 | 0.47 | 0.27 | 0.20 | 625 | 125 |
| V3.0FAU | 704 | 0.45 | 0.27 | 0.18 | 580 | 124 |
| V6.0FAU | 670 | 0.42 | 0.24 | 0.18 | 565 | 105 |
| FAUdes | 688 | 0.75 | 0.12 | 0.63 | 265 | 423 |
| V1.0FAUdes | 669 | 0.73 | 0.13 | 0.60 | 314 | 355 |
| V3.0FAUdes | 669 | 0.83 | 0.07 | 0.76 | 151 | 518 |
| V6.0FAUdes | 603 | 0.65 | 0.12 | 0.53 | 285 | 318 |
| Nature of Species | Peak Position (nm) | Remarks | References |
|---|---|---|---|
| Tetrahedral monomeric (isolated VO4) | 240–290 280 240 253, 294 | Compound: Na3VO4 (ortho-vanadate) | [22] [23] [24] [25] |
| 215, 225, 250, 291 | Compound: VO(OH)3 TD-DFT results | This work | |
| Tetrahedral 1D chains (polymerized VO4) | 270–290 280, 340 288, 363 281, 353 | Compound: NH4VO3; NaVO3 (meta-vanadate) | [22] [26] [27] [28] |
| Square pyramidal | 410 | Compound: α-VPO5 | [22] |
| 234, 284, 424 | Compound: VO(OH)4− TD-DFT results | This work | |
| Octahedral multilayer (polymerized VO5/VO6) | 470 330–500 480 330, 460 334, 481 | Compound: V2O5 | [22] [23] [24] [26] [29] |
| 218, 232, 263, 320, 393, 452 | Compound: VO(OH)52− TD-DFT results | This work |
| Sample | Weak (mmol NH3/g) 150–250 °C | Medium (mmol NH3/g) 250–350 °C | Strong (mmol NH3/g) 350–500 °C | V. Strong (mmol NH3/g) 500–700 °C | Total Acidity (mmol NH3/g) |
|---|---|---|---|---|---|
| FAU | 4.4 | 13.3 | 0 | 0 | 17.7 |
| V1.0FAU | 8.6 | 0 | 3.0 | 0 | 11.6 |
| V3.0FAU | 0 | 4.2 | 0 | 0 | 4.2 |
| V6.0FAU | 1.9 | 14.7 | 0 | 0 | 16.6 |
| V1.0FAUdes | 0.6 | 1.6 | 0 | 0 | 2.2 |
| V3.0FAUdes | 4.7 | 9.3 | 0 | 0 | 14.0 |
| V6.0FAUdes | 1.2 | 0.8 | 0 | 0 | 2.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smoliło, M.; Samson, K.; Zhou, T.; Duraczyńska, D.; Ruggiero-Mikołajczyk, M.; Drzewiecka-Matuszek, A.; Rutkowska-Zbik, D. Oxidative Dehydrogenation of Propane over Vanadium-Containing Faujasite Zeolite. Molecules 2020, 25, 1961. https://doi.org/10.3390/molecules25081961
Smoliło M, Samson K, Zhou T, Duraczyńska D, Ruggiero-Mikołajczyk M, Drzewiecka-Matuszek A, Rutkowska-Zbik D. Oxidative Dehydrogenation of Propane over Vanadium-Containing Faujasite Zeolite. Molecules. 2020; 25(8):1961. https://doi.org/10.3390/molecules25081961
Chicago/Turabian StyleSmoliło, Małgorzata, Katarzyna Samson, Ting Zhou, Dorota Duraczyńska, Małgorzata Ruggiero-Mikołajczyk, Agnieszka Drzewiecka-Matuszek, and Dorota Rutkowska-Zbik. 2020. "Oxidative Dehydrogenation of Propane over Vanadium-Containing Faujasite Zeolite" Molecules 25, no. 8: 1961. https://doi.org/10.3390/molecules25081961