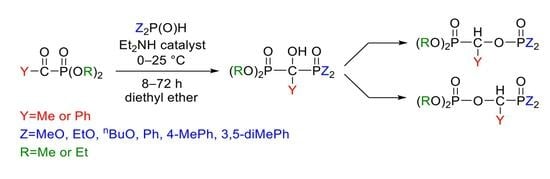
3.3. General Procedure for the Synthesis of Dimethyl 1-Diarylphosphinoyl-1-hydroxy-ethylphosphonate
2.2 mmol (0.33 g) of dimethyl α-oxoethylphosphonate was added dropwise to a mixture of 2.2 mmol diarylphosphine oxide (diphenylphosphine oxide: 0.44 g, bis(4-methylphenyl)phosphine oxide: 0.50 g, bis(3,5-dimethylphenyl)phosphine oxide: 0.56 g) and 0.88 mmol (0.090 mL) of diethylamine in diethyl ether (13 mL) at 0 °C on stirring. After an 8 h reaction time, the precipitated material was removed by filtration, washed with diethyl ether, and the residue recrystallized from acetone. The products were white crystalline compounds.
3.3.1. Dimethyl 1-Diphenylphosphinoyl-1-hydroxy-ethylphosphonate (2d)
Yield: 0.50 g (64%), mp: 131–132 °C; 31P NMR (CDCl3) δP1 23.9 and δP2 29.0 (d, 2JPP = 25.4 Hz); 13C NMR (CDCl3) δ 20.3 (s, CCH3), 53.9 and 54.0 (d, J = 7.4 Hz, 2 OCH3), 74.8 (dd, J1 = 154.6 Hz, J2 = 79.0 Hz, CCH3), 127.8 and 128.2 (d, J = 11.7 Hz, 2 Cγ), 130.4 (dd, J1 = 96.9 Hz, J2 = 5.5 Hz, Cα), 130.8 (d, J = 98.1 Hz, Cα), 131.6 and 131.8 (d, J = 2.8 Hz, 2 Cδ), 132.4 and 132.7 (d, J = 8.6 Hz, 2 Cβ); 1H NMR (CDCl3) δ 1.65 (t, J = 15.6 Hz, 3H, CCH3), 3.44 and 3.70 (d, J = 10.6 Hz, 6H, OCH3), 7.40–7.61 (m, 6H, ArH), 8.09 and 8.18 (dd, J1 = 11.1 Hz, J2 = 6.9 Hz, 4H, ArHβ); [M + H]+ = 355; [M + Na]+found = 377.0681; C16H20O5P2Na required 377.0684.
3.3.2. Dimethyl 1-Bis(4-methylphenyl)phosphinoyl-1-hydroxy-ethylphosphonate (2e)
Yield: 0.52 g (62%), mp: 153–154 °C; 31P NMR (CDCl3) δP1 24.0 and δP2 30.2 (d, 2JPP = 29.0 Hz); 13C NMR (CDCl3) δ 20.6 (s, CCH3), 21.5 (s, 2 ArCH3), 53.9 and 54.0 (d, J = 7.4 Hz, 2 OCH3), 74.6 (dd, J1 = 153.7 Hz, J2 = 76.3 Hz, CCH3), 127.1 (dd, J1 = 99.4 Hz, J2 = 4.9 Hz, Cα), 127.2 (d, J = 100.9 Hz, Cα), 128.7 and 128.9 (d, J = 12.1 Hz, 2 Cγ), 132.4 and 132.7 (d, J = 9.1 Hz, 2 Cβ), 142.1 and 142.3 (d, J = 2.9 Hz, 2 Cδ); 1H NMR (CDCl3) δ 1.61 (t, J = 15.4 Hz, 3H, CCH3), 2.39 (s, 6H, ArCH3), 3.55 and 3.67 (d, J = 10.6 Hz, 6H, OCH3), 7.27–7.29 (m, 4H, ArH), 7.92 and 8.03 (dd, J1 = 11.0 Hz, J2 = 8.0 Hz, 4H, ArHβ); [M + H]+ = 383; [M + Na]+found = 405.1003; C18H24O5P2Na required 405.0997.
3.3.3. Dimethyl 1-Bis(3,5-dimethylphenyl)phosphinoyl-1-hydroxy-ethylphosphonate (2f)
Yield: 0.62 g (69%), mp: 161–162 °C; 31P NMR (CDCl3) δP1 24.3 and δP2 30.0 (d, 2JPP = 29.0 Hz); 13C NMR (CDCl3) δ 20.7 (s, CCH3), 21.3 (s, 4 ArCH3), 53.8 and 54.0 (d, J = 7.4 Hz, 2 OCH3), 74.7 (dd, J1 = 153.6 Hz, J2 = 76.3 Hz, CCH3), 129.9 and 130.1 (d, J = 8.7 Hz, 2 Cβ) 130.2 (dd, J1 = 95.7 Hz, J2 = 5.1 Hz, Cα), 130.5 (d, J = 96.0 Hz, Cα), 133.4 and 133.5 (d, J = 3.0 Hz, 2 Cδ), 137.4 and 137.7 (d, J = 12.4 Hz, 2 Cγ); 1H NMR (CDCl3) δ 1.65 (t, J = 14.7 Hz, 3H, CCH3), 2.36 (d, J = 5.5 Hz, 12H, ArCH3), 3.52 and 3.70 (d, J = 10.6 Hz, 6H, OCH3), 7.14 (s, 2H, ArHδ), 7.67 and 7.77 (d, J = 11.3 Hz, 4H, ArHβ); [M + H]+ = 411; [M + Na]+found = 433.1312; C20H28O5P2Na required 433.1310.
3.4. General Procedure for the Synthesis of Dialkyl 1-(Dialkylphosphonoylethyl)phosphate
2.2 mmol (0.33 g) of dimethyl α-oxoethylphosphonate was added dropwise to a mixture of 2.2 mmol dialkyl phosphite (dimethyl phosphite: 0.20 mL, diethyl phosphite: 0.30 mL, dibutyl phosphite: 0.43 mL) and 0.88 mmol (0.090 mL) of diethylamine in diethyl ether (13 mL) at 0 °C on stirring. After 8–72 h reaction time, the solvent was evaporated and the crude product obtained was purified by column chromatography (using DCM–MeOH 97:3 as the eluent on silica gel).
3.4.1. Dimethyl 1-(Dimethylphosphonoylethyl)phosphate (3a)
Yield: 0.43 g (75%),
31P NMR (CDCl
3) δ
P1 1.1 and δ
P2 22.5 (d,
3JPP = 30.1 Hz), Ref [
34] δ
P1 0.4 and δ
P2 21.9 (d,
3JPP = 29.3 Hz);
13C NMR (CDCl
3) δ 16.6 (s, C
CH
3), 53.4 and 53.6 (dd,
J1 = 6.8 Hz,
J2 = 3.8 Hz, 2 OCH
3); 54.4 and 54.6 (dd,
J1 = 6.3 Hz,
J2 = 3.6 Hz, 2 OCH
3), 69.1 (dd,
J1 = 174.1 Hz,
J2 = 6.9 Hz, CH);
1H NMR (CDCl
3) δ 1.61 (dd,
J1 = 16.7 Hz,
J2 = 7.1 Hz, 3H, CCH
3), 3.77–3.84 (m, 12H, OCH
3), 4.63–4.91 (m, 1H, CH); [M + H]
+ = 263; [M + Na]
+found = 285.0268; C
6H
16O
7P
2Na required 285.0269.
3.4.2. Dimethyl 1-(Diethylphosphonoylethyl)phosphate (3b-1) and Diethyl 1-(Dimethylphosphonoylethyl)phosphate (3b-2)
Yield: 0.56 g (87%), major (83%): 31P NMR (CDCl3) δP1 1.0 and δP2 20.0 (3JPP = 31.3 Hz); 13C NMR (CDCl3) δ 16.38 and 16.43 (d, J = 5.5 Hz, 2 CH2CH3), 16.6 (s, CCH3), 54.4 and 54.5 (d, J = 6.2 Hz, 2 OCH3), 63.0 and 63.1 (d, J = 6.5 Hz, 2 OCH2), 69.4 (dd, J1 = 174.5 Hz, J2 = 6.8 Hz, CH); 1H NMR (CDCl3) δ 1.32 (t, J = 7.0 Hz, 6H, CH2CH3), 1.54 (dd, J1 = 16.7 Hz, J2 = 7.0 Hz, 3H, CCH3), 3.75 and 3.77 (d, J = 11.5 Hz, 6H, OCH3), 4.13–4.20 (m, 4H, CH2CH3); 4.62–4.72 (m, 1H, CH); minor (17%): δP1 −1.3 and δP2 22.6 (3JPP = 31.0 Hz); 13C NMR (CDCl3) δ 16.0 (d, J = 6.8 Hz, 2 CH2CH3), 16.6 (s, CCH3), 53.4 and 53.6 (d, J = 6.5 Hz, 2 OCH3), 64.1 and 64.2 (d, J = 6.1 Hz, 2 OCH2), 68.8 (dd, J1 = 174.1 Hz, J2 = 7.0 Hz, CH); 1H NMR (CDCl3) δ 3.80 and 3.81 (d, J = 10.7 Hz, 6H, OCH3). The other signals were common with those of the major isomer; [M + H]+ = 291; [M + Na]+found = 313.0581; C8H20O7P2Na required 313.0582.
3.4.3. Dimethyl 1-(Dibutylphosphonoylethyl)phosphate (3c-1) and Dibutyl 1-(Dimethylphosphonoylethyl)phosphate (3c-2)
Yield: 0.53 g (70%), major (81%) 31P NMR (CDCl3) δP1 1.1 and δP2 20.0 (3JPP = 31.6 Hz); 13C NMR (CDCl3) δ 13.5 (s, 2 CH2CH3), 16.7 (s, CCH3), 18.6 (s, 2 CH2CH3), 32.5 and 32.6 (d, J = 3.2 Hz, 2 OCH2CH2), 54.3–54.4 and 54.5–54.6 (m, 2 OCH3), 66.4–66.9 (m, 2 OCH2), 69.5 (dd, J1 = 174.6 Hz, J2 = 6.9 Hz, CH); 1H NMR (CDCl3) δ 0.96 (t, J = 7.9 Hz, 6H, CH2CH3), 1.40–1.47 (m, 4H, CH2CH3), 1.60 (dd, J1 = 16.5 Hz, J2 = 7.1 Hz, 3H, CCH3), 1.67–1.72 (m, 4H, OCH2CH2), 3.80 and 3.83 (d, J = 11.5 Hz, 6H, OCH3), 4.04–4.20 (m, 4H, OCH2), 4.66–4.78 (m, 1H, CH); minor (19%) 31P NMR (CDCl3) δP1 –0.9 and δP2 22.8 (3JPP = 31.4 Hz); 13C NMR (CDCl3) δ 68.8 (dd, J1 = 174.1 Hz, J2 = 7.1 Hz, CH). The other signals are common with those of the major isomer; 1H NMR (CDCl3) δ 3.85 and 3.86 (d, J = 10.5 Hz, 6H, OCH3). The other signals are common with those of the major isomer; [M + H]+ = 347; [M + Na]+found = 369.1201; C12H28O7P2Na required 369.1208.
3.5. General Procedure for Diethyl (Diarylphosphinoyloxybenzyl)phosphonate and Diethyl (Diarylphosphinoylbenzyl)phosphate
1.5 mmol (0.36 g) of diethyl α-oxobenzylphosphonate was added slowly to a mixture of 1.5 mmol (bis(4-methylphenyl)phosphine oxide: 0.35 g, bis(3,5-dimethylphenyl)phosphine oxide: 0.40 g) and 0.60 mmol (0.060 mL) of diethylamine in diethyl ether (13 mL) at 0 °C on stirring. After an 8 h reaction time, the solvent was evaporated, and the crude product obtained was purified with column chromatography (using ethyl acetate as the eluent on silica gel).
3.5.1. Diethyl (Diphenylphosphinoylbenzyl)phosphate (5d-1) and Diethyl (Diphenylphosphinoyloxybenzyl)phosphonate (5d-2)
Yield: 0.47 g (70%), major (60%): 31P NMR (CDCl3) δP1 –1.5 and δP2 28.6 (3JPP = 31.3 Hz); 13C NMR (CDCl3) δ 15.6 and 15.8 (d, J = 7.4 Hz, 2 CH2CH3), 63.8 and 63.9 (d, J = 6.0 Hz, 2 OCH2), 77.4 (dd, J1 = 85.7 Hz, J2 = 7.9 Hz, CH). The aromatic range was rather complex between δ 128.0–132.6; 1H NMR (CDCl3) δ 0.90 and 0.96 (t, J = 7.1 Hz, 6H, CH2CH3), 3.41–3.70 (m, 4H, OCH2), 6.06 (dd, J1 = 9.7 Hz, J2 = 4.4 Hz, 1H, CH), aromatic region: 7.15–7.98 (m, 15H, ArH); minor (40%): 31P NMR (CDCl3) δP1 17.2 and δP2 34.7 (3JPP = 26.7 Hz); 13C NMR (CDCl3) δ 16.2 and 16.3 (d, J = 5.8 Hz, 2 CH2CH3), 63.3 and 63.5 (d, J = 6.9 Hz, 2 OCH2), 72.0 (dd, J1 = 172.6 Hz, J2 = 7.0 Hz, CH). The aromatic range was rather complex between δ 128.0–132.6; 1H NMR (CDCl3) δ 1.09 and 1.18 (t, J = 7.1 Hz, 6H, CH2CH3), 3.78–4.15 (m, 4H, OCH2), 5.63 (dd, J1 = 13.5 Hz, J2 = 11.2 Hz, 1H, CH), aromatic region: 7.15–7.98 (m, 15H, ArH); [M + H]+ = 445; [M + Na]+found = 467.1154; C23H26O5P2Na required 467.1153.
3.5.2. Diethyl 1-Bis((4-methylphenyl)phosphinoylbenzyl)phosphate (5e-1)
Yield: 0.40 g (65%), 31P NMR (CDCl3) δP1 –1.3 and δP2 29.0 (3JPP = 31.4 Hz); 13C NMR (CDCl3) δ 15.6 and 15.7 (d, J = 7.4 Hz, 2 CH2CH3), 21.6 (d, J = 9.8 Hz, 2 ArCH3), 63.8 and 63.9 (d, J = 5.9 Hz, 2 OCH2), 77.6 (dd, J1 = 85.3 Hz, J2 = 8.0 Hz, CH). The aromatic range was rather complex between δ 124.6–142.9; 1H NMR (CDCl3) δ 0.94 and 1.00 (t, J = 7.3 Hz, 6H, CH2CH3), 2.35 and 2.42 (s, 6H, ArCH3), 3.46–3.74 (m, OCH2), 6.03 (dd, J1 = 9.8 Hz, J2 = 4.5 Hz, 1H, CH), aromatic region: 7.18–7.33 (m, 9H, ArH), 7.55 and 7.83 (dd, J1 = 11.1 Hz J2 = 8.1 Hz, 4H, ArHβ); [M + H]+ = 473; [M + Na]+found = 495.1467; C25H30O5P2Na required 495.1466.
3.5.3. Diethyl 1-Bis((3,5-dimethylphenyl)phosphinoylbenzyl)phosphate (5f-1)
Yield: 0.42 g (72%), 31P NMR (CDCl3) δP1 −1.2 and δP2 29.1 (3JPP = 30.9 Hz); 13C NMR (CDCl3) δ 15.6 and 15.8 (d, J = 7.5 Hz, 2 CH2CH3), 21.2 (d, J = 13.3 Hz, 4 ArCH3), 63.6 and 63.8 (d, J = 5.9 Hz, 2 OCH2), 77.4 (dd, J1 = 84.8 Hz, J2 = 8.0 Hz, CH). The aromatic range was rather complex between δ 128.0–138.2; 1H NMR (CDCl3) δ 0.95 and 1.04 (t, J = 7.4 Hz, 6H, CH2CH3), 2.27 and 2.40 (s, 12H, ArCH3), 3.48–3.76 (m, 4H, OCH2), 6.06 (dd, J1 = 9.7 Hz, J2 = 3.1 Hz, 1H, CH), aromatic region: 7.22–7.34 (m, 9H, ArH), 7.61 (d, J = 11.7 Hz, 2H, ArHβ); [M + H]+ = 501; [M + Na]+found = 523.1771; C27H34O5P2Na required: 523.1779.
3.6. Single Crystal X-ray Diffraction Studies
Single crystals of compound 2d, 2e.0.5 C3H6O and 6 suitable for X-ray diffraction were obtained by slow evaporation of the respective acetone solution. The crystals were introduced into perfluorinated oil and a suitable single crystal was carefully mounted on the top of a thin glass wire. Data collection was performed with an Oxford Xcalibur 3 diffractometer equipped with a Spellman generator (50 kV, 40 mA) and a Kappa CCD detector, operating with Mo-Kα radiation (λ = 0.71071 Ǻ).
Data collection and reduction were performed using CrysAlisPro software [
35]. Absorption correction using the multiscan method [
35] was applied. The structures were solved with SHELXS-97 [
36], refined with SHELXL-97 [
37] and finally checked using PLATON [
38]. Details of the data collection and structure refinement are summarized in
Table 6.
CCDC-2281416, CCDC-2281417 and CCDC-2281418 contain supplementary crystallographic data for compounds
2d,
2e·0.5C3H6O and
6, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via
www.ccdc.cam.ac.uk/data_request/cif (accessed on 13 July 2023).

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


