


O,S-Acetals in a New Modification of oxo-Friedel–Crafts–Bradsher Cyclization—Synthesis of Fluorescent (Hetero)acenes and Mechanistic Considerations

Abstract
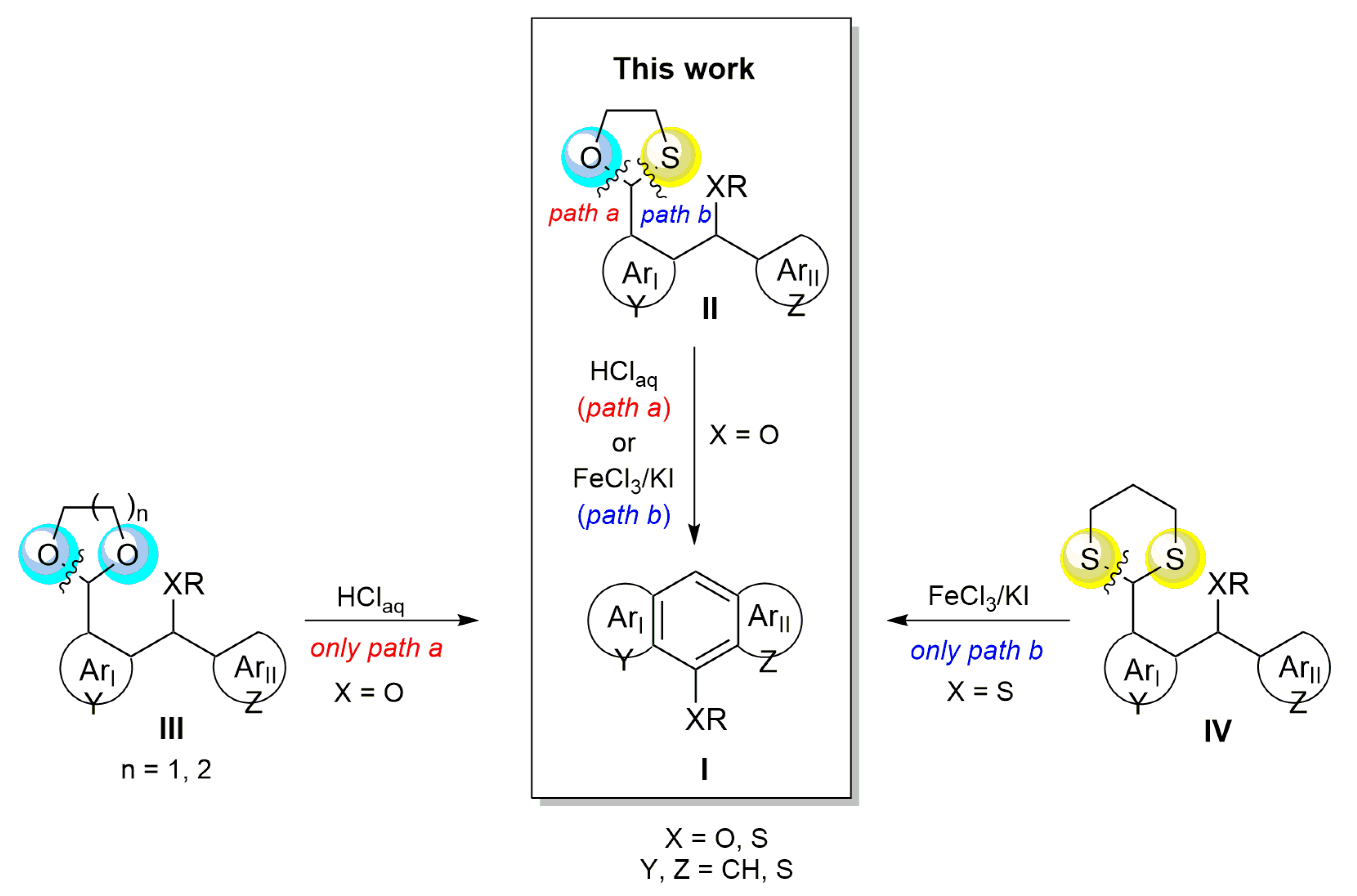
:1. Introduction
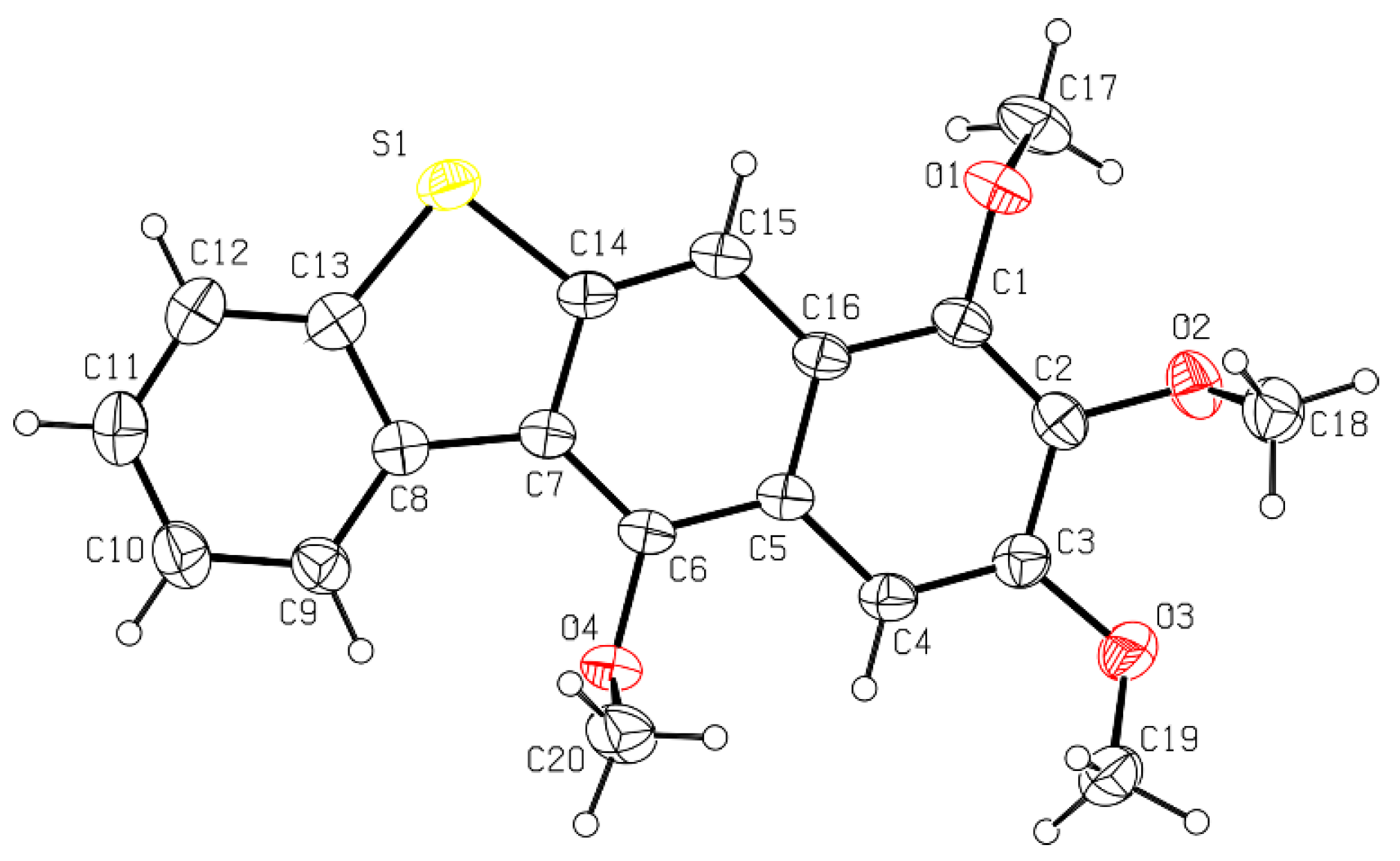
2. Results and Discussion
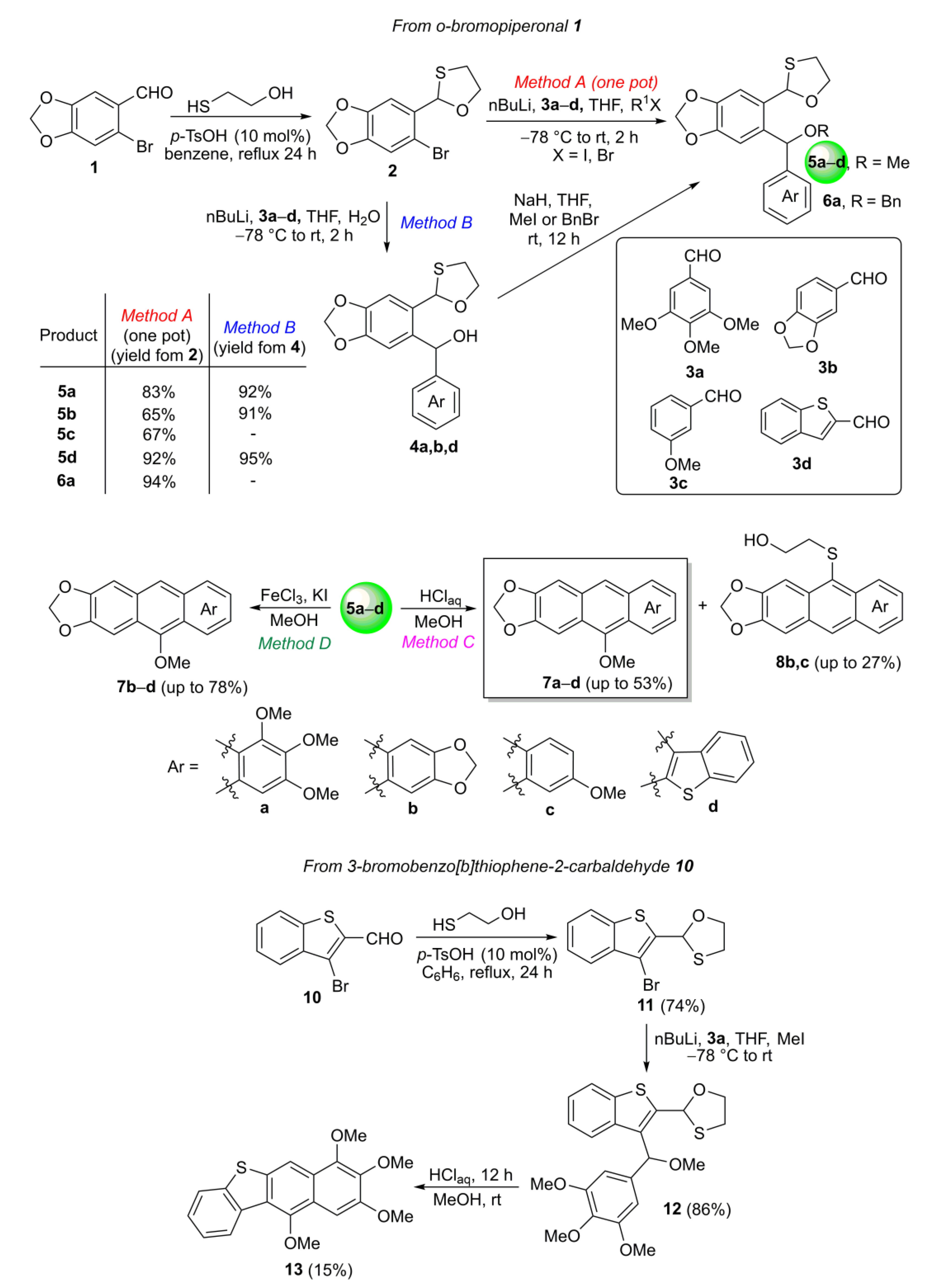
2.1. Synthesis
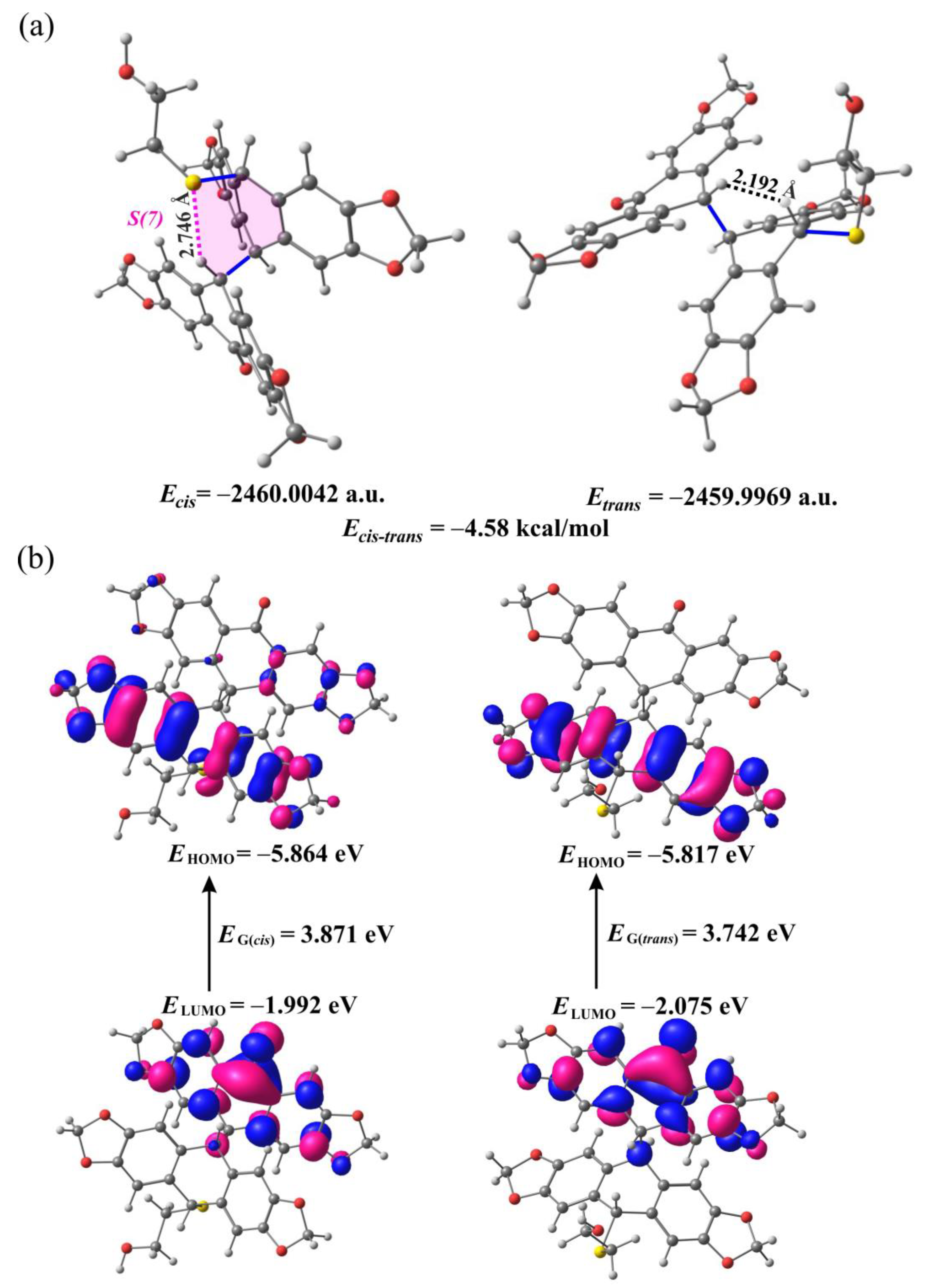
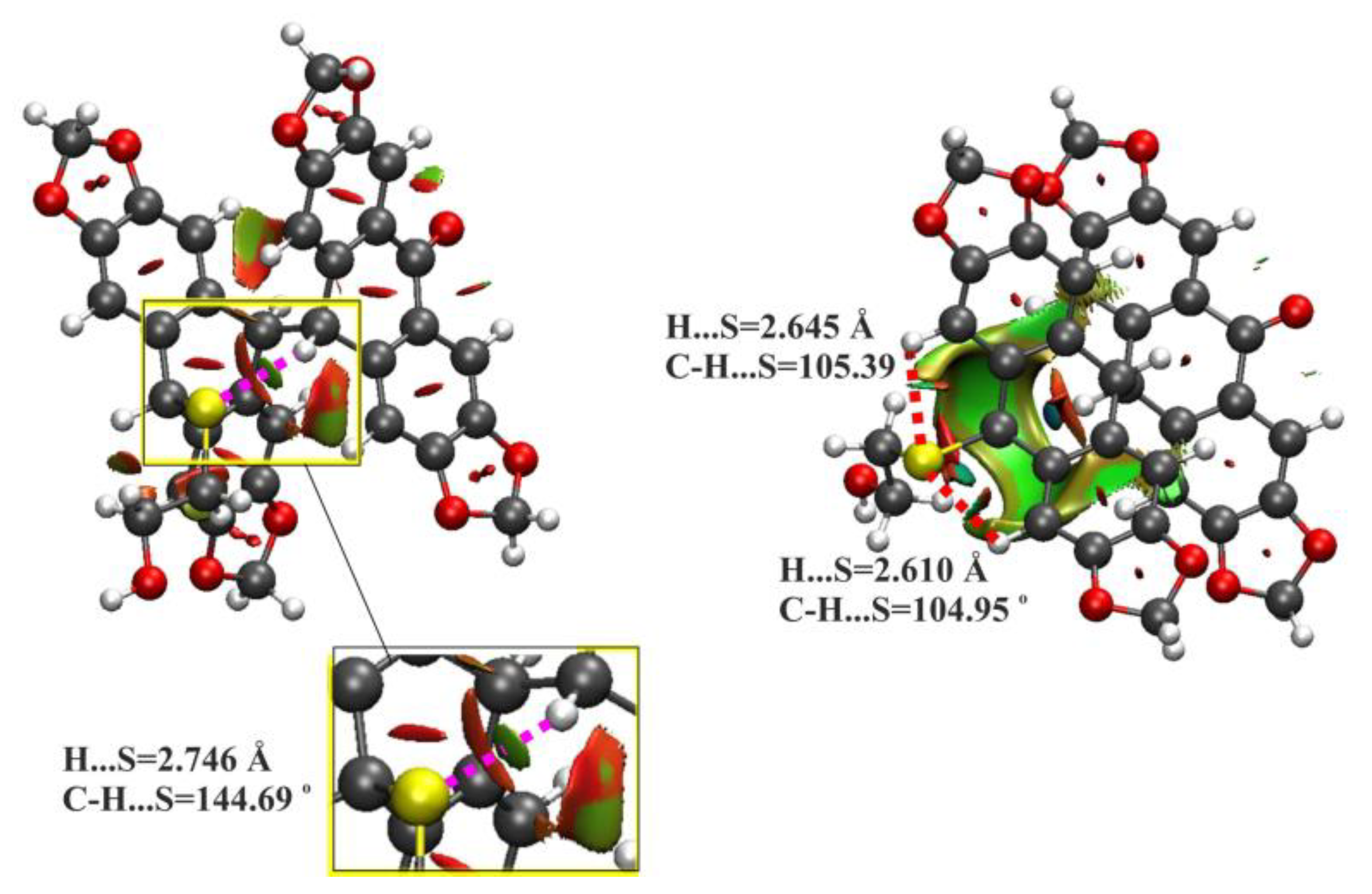
2.2. DFT Calculations for 9a and 9b
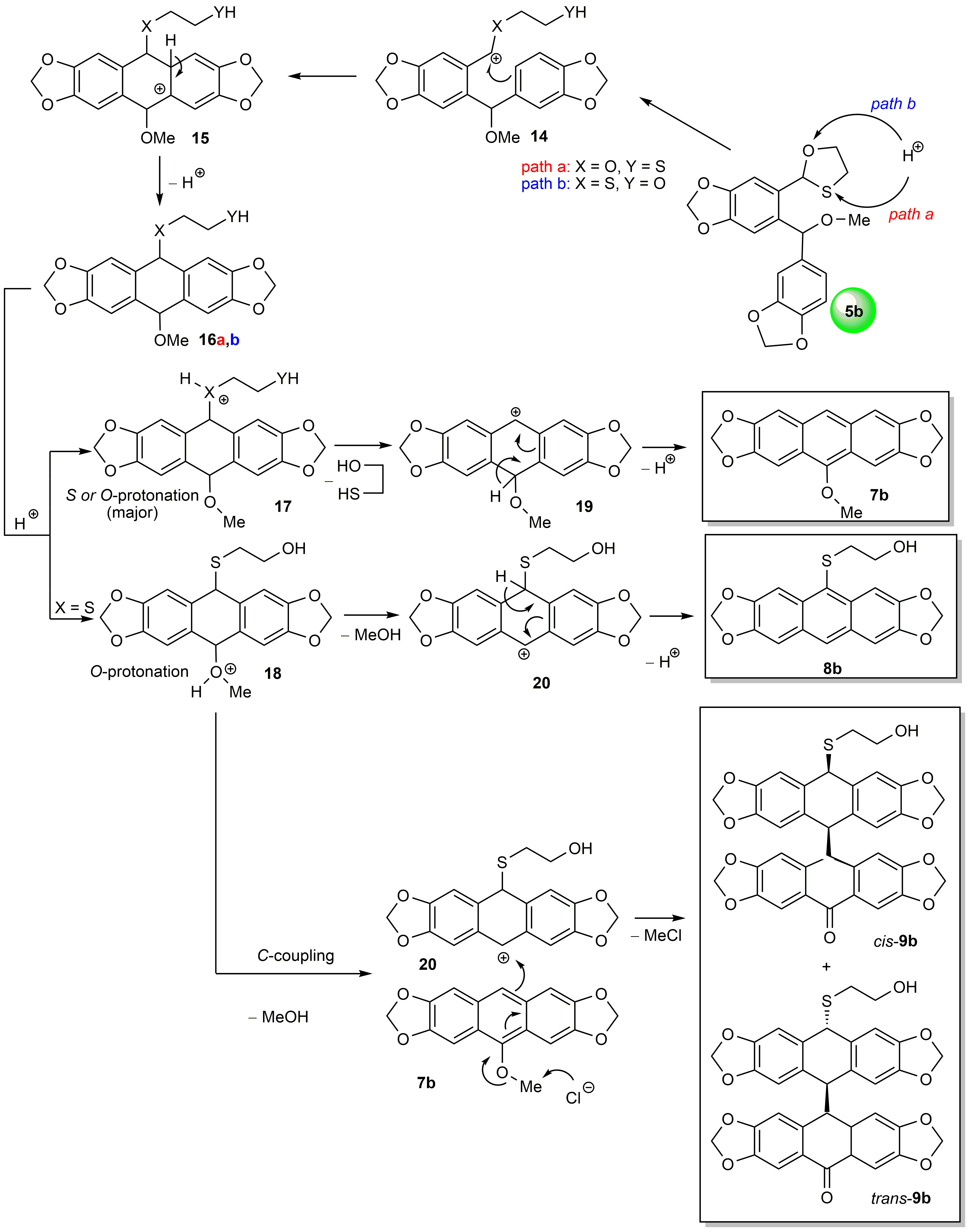
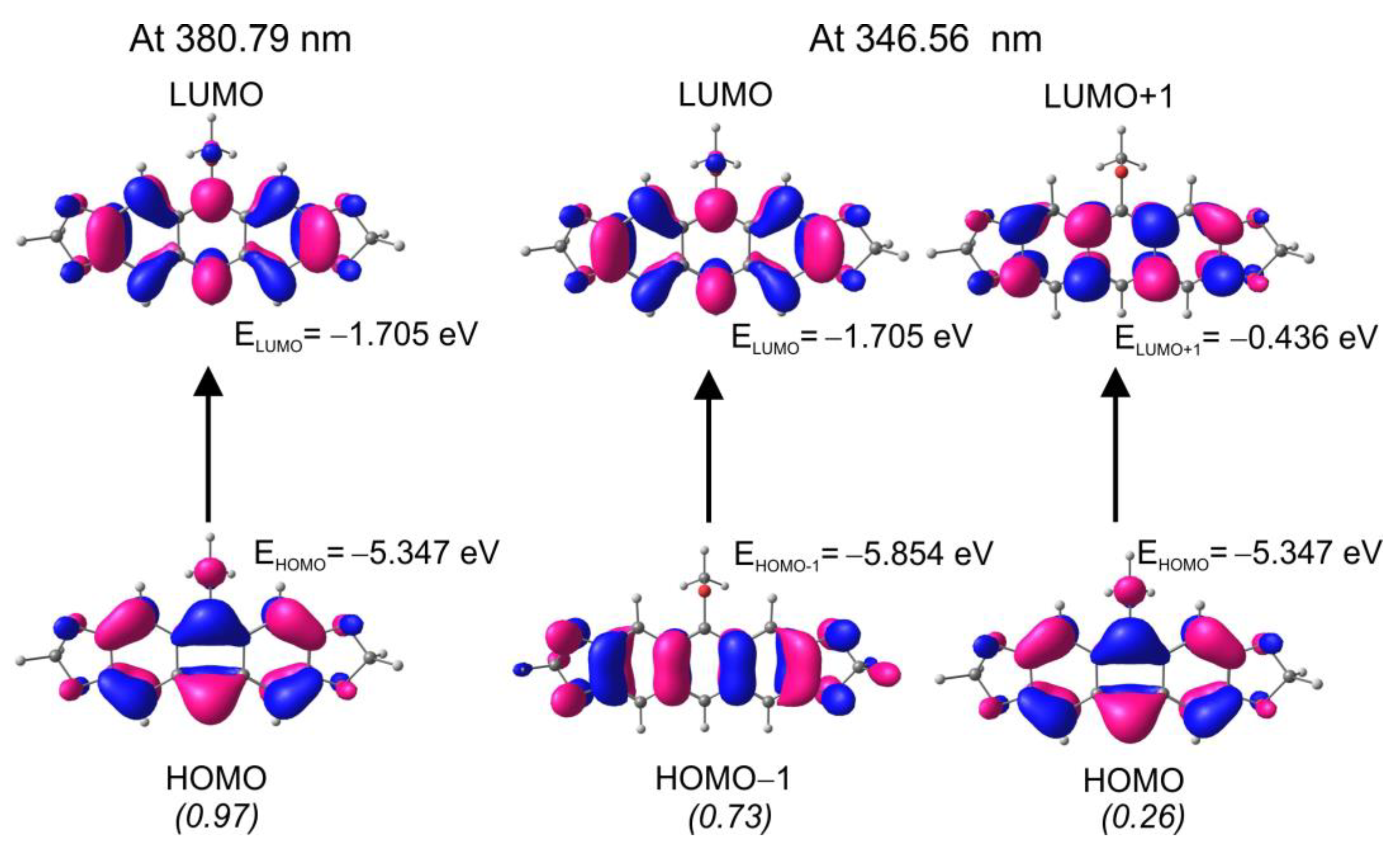
2.3. Mechanistic Considerations and DFT Calculations
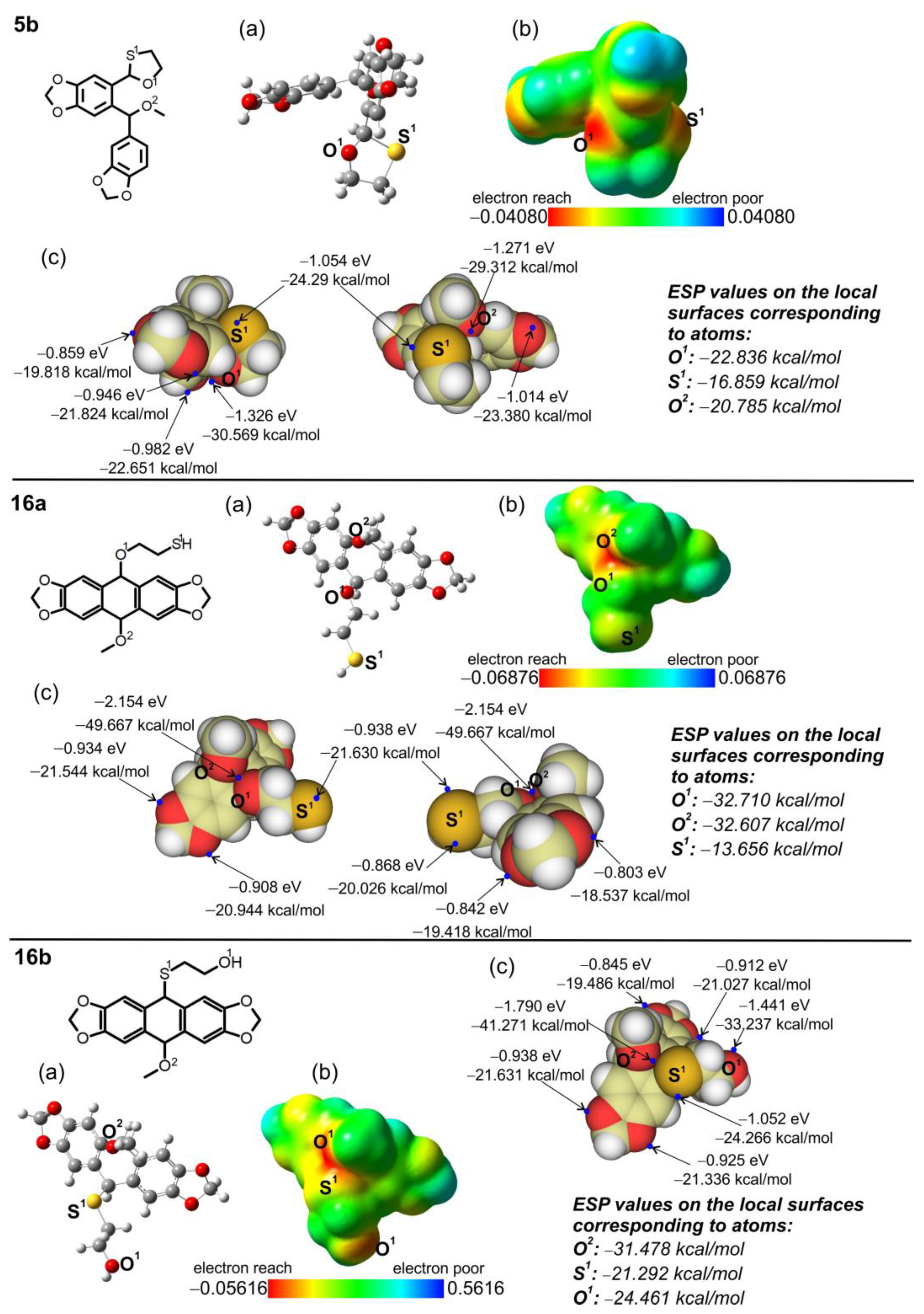
2.4. Electron Character of RO-Acenes 7, 13 and HO(CH2)2S-Acenes 8
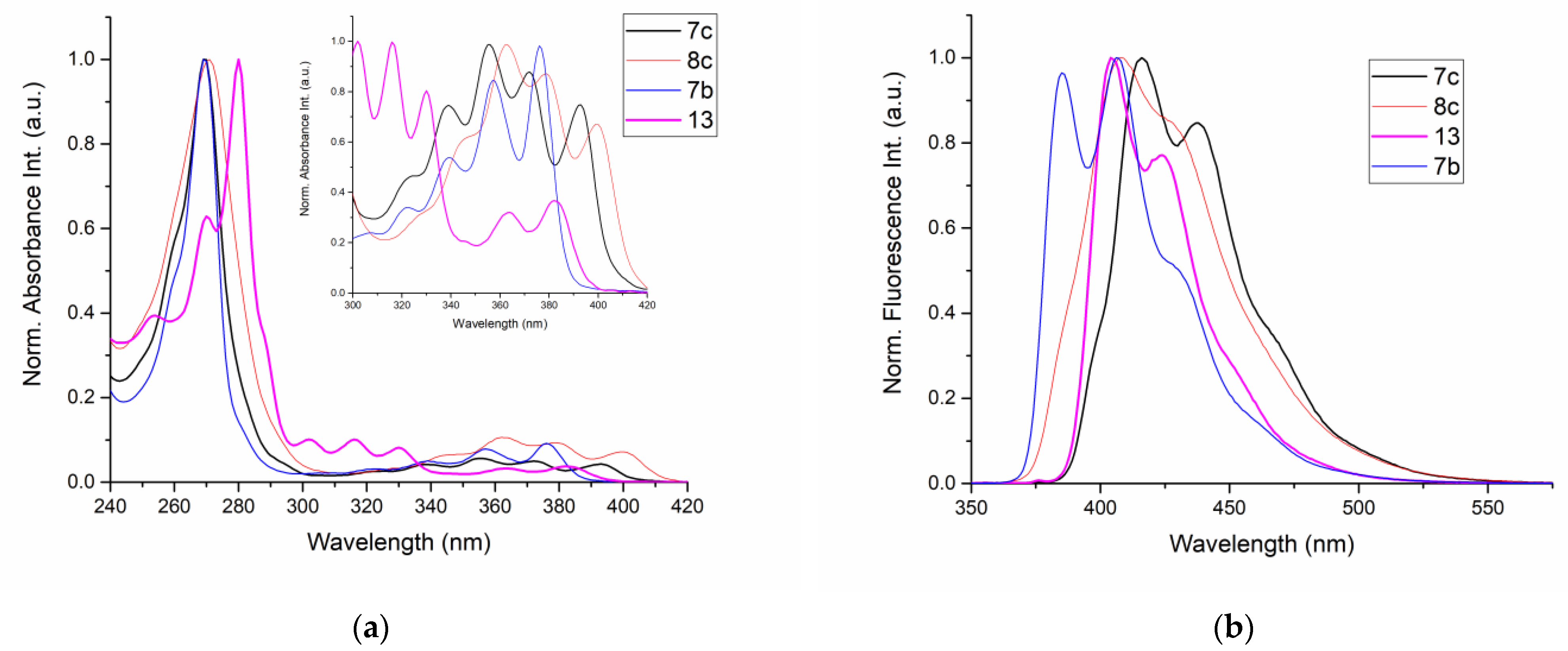
2.5. Photopysical Properties
3. Materials and Methods
3.1. Synthesis of o-Bromopiperonal O,S-acetal 2
3.2. General Procedure for the Synthesis of o-(O,S-acetalaryl)arylmethanols 4
3.3. General Procedure for the One-Pot Synthesis of o-(O,S-acetalaryl)arylmethyl Methyl Ethers 5 from o-Bromopiperonal O,S-acetal 2 (Method A)
3.4. Procedure for the Synthesis of o-(O,S-acetalaryl)arylmethyl Benzyl Ether 6a
3.5. General Procedure for the Synthesis of o-(O,S-acetalaryl)arylmethyl Methyl Ethers 5 from o-(O,S-acetalaryl)arylmethanols 4 (Method B)
3.6. General Procedure for the Synthesis of MeO-Substituted Acenes 7, 13 and 8 Using HClaq (Method C)
3.7. General Procedure for the Synthesis of MeO-Substituted Acene 7 Using FeCl3/KI in MeOH (Method D)
3.8. Synthesis of 3-Bromobenzo[b]thiophene-2-carbaldehyde O,S-acetal 11
3.9. Synthesis of o-(O,S-acetalaryl)arylmethyl Methyl Ether 12
3.10. Synthesis of Acene 13 Using HClaq
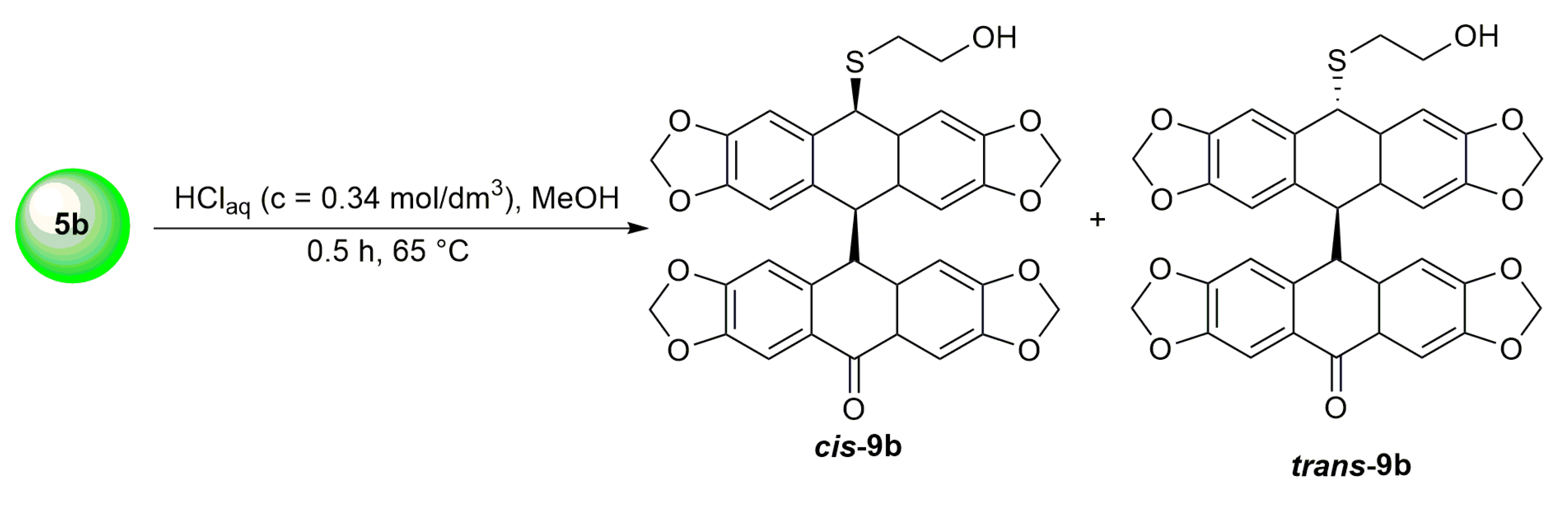
3.11. Synthesis of Dimeric Isomers cis-9b and trans-9b from o-(O,S-acetalaryl)arylmethyl Methyl Ether 5b Using HClaq
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Yang, Y.; Börjesson, K. Electroactive covalent organic frameworks: A new choice for organic electronics. Trends Chem. 2022, 4, 60–75. [Google Scholar] [CrossRef]
- Koprowski, M.; Owsianik, K.; Knopik, Ł.; Vivek, V.; Romaniuk, A.; Różycka-Sokołowska, E.; Bałczewski, P. Comprehensive Review on Synthesis, Properties, and Applications of Phosphorus (PIII, PIV, PV) Substituted Acenes with More Than Two Fused Benzene Rings. Molecules 2022, 27, 6611. [Google Scholar] [CrossRef]
- Bałczewski, P.; Kowalska, E.; Różycka-Sokołowska, E.; Skalik, J.; Owsianik, K.; Koprowski, M.; Marciniak, B.; Guziejewski, D.; Ciesielski, W. Mono-Aryl/Alkylthio-Substituted (Hetero)acenes of Exceptional Thermal and Photochemical Stability by the Thio-Friedel–Crafts/Bradsher Cyclization Reaction. Chem.—A Eur. J. 2019, 25, 14148–14161. [Google Scholar] [CrossRef]
- Im, Y.; Byun, S.Y.; Kim, J.H.; Lee, D.R.; Oh, C.S.; Yook, K.S.; Lee, J.Y. Recent Progress in High-Efficiency Blue-Light-Emitting Materials for Organic Light-Emitting Diodes. Adv. Funct. Mater. 2017, 27, 1603007. [Google Scholar] [CrossRef]
- Yuan, Q.; Wang, T.; Yu, P.; Zhang, H.; Zhang, H.; Ji, W. A review on the electroluminescence properties of quantum-dot light-emitting diodes. Org. Electron. 2021, 90, 106086. [Google Scholar] [CrossRef]
- Lim, H.; Woo, S.-J.; Ha, Y.H.; Kim, Y.-H.; Kim, J.-J. Breaking the Efficiency Limit of Deep-Blue Fluorescent OLEDs Based on Anthracene Derivatives. Adv. Mater. 2022, 34, 2100161. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, T.; Dong, S.; Wen, Z.; Xu, H.; Miao, Y.; Wang, H.; Yu, J. Anthracene and carbazole based asymmetric fluorescent materials for high-efficiency deep-blue non-doped organic light emitting devices with CIEy=0.06. Dye. Pigment. 2022, 199, 110047. [Google Scholar] [CrossRef]
- Haykir, G.; Aydemir, M.; Han, S.H.; Gumus, S.; Hizal, G.; Lee, J.Y.; Turksoy, F. The investigation of sky-blue emitting anthracene-carbazole derivatives: Synthesis, photophysics and OLED applications. Org. Electron. 2018, 59, 319–329. [Google Scholar] [CrossRef]
- Lim, H.; Cheon, H.J.; Lee, G.S.; Kim, M.; Kim, Y.-H.; Kim, J.-J. Enhanced Triplet–Triplet Annihilation of Blue Fluorescent Organic Light-Emitting Diodes by Generating Excitons in Trapped Charge-Free Regions. ACS Appl. Mater. Interfaces 2019, 11, 48121–48127. [Google Scholar] [CrossRef]
- Song, D.; Yu, Y.; Yue, L.; Zhong, D.; Zhang, Y.; Yang, X.; Sun, Y.; Zhou, G.; Wu, Z. Asymmetric thermally activated delayed fluorescence (TADF) emitters with 5,9-dioxa-13b-boranaphtho[3,2,1-de]anthracene (OBA) as the acceptor and highly efficient blue-emitting OLEDs. J. Mater. Chem. C 2019, 7, 11953–11963. [Google Scholar] [CrossRef]
- Park, J.; Jang, B.; Moon, Y.J.; Lee, H.; Kim, Y.K.; Yoon, S.S. Efficient deep blue organic light emitting diodes based on [2,7,7,13,13-pentamethyl-9-(10-phenylanthracen-9-yl)-7,13-dihydrobenzo[5,6]-s-indaceno[1,2-g]quinoline] derivatives. Mol. Cryst. Liq. Cryst. 2020, 705, 120–126. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, W.; Ye, S.; Zhang, Q.; Duan, Y.; Guo, R.; Wang, L. Molecular engineering of anthracene-based emitters for highly efficient nondoped deep-blue fluorescent OLEDs. J. Mater. Chem. C 2020, 8, 9678–9687. [Google Scholar] [CrossRef]
- Desai, N.K.; Kolekar, G.B.; Patil, S.R. Fabrication and Characterization of Anthracene Doped P-terphenyl Thin Films By Spin Coating Technique; Investigation of Fluorescence Properties. J. Fluoresc. 2020, 30, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Sun, M.; Xu, L.; Wang, R.; Zhou, H.; Pan, Y.; Zhang, S.; Sun, Q.; Xue, S.; Yang, W. Highly efficient non-doped blue fluorescent OLEDs with low efficiency roll-off based on hybridized local and charge transfer excited state emitters. Chem. Sci. 2020, 11, 5058–5065. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.-S.; Ha, Y.H.; Kwon, S.-K.; Kim, Y.-H.; Kim, J.-J. Design Strategy of Anthracene-Based Fluorophores toward High-Efficiency Deep Blue Organic Light-Emitting Diodes Utilizing Triplet–Triplet Fusion. ACS Appl. Mater. Interfaces 2020, 12, 15422–15429. [Google Scholar] [CrossRef]
- Wu, Z.; Zhu, X.; Li, Y.; Chen, H.; Zhuang, Z.; Shen, P.; Zeng, J.; Chi, J.; Ma, D.; Zhao, Z.; et al. High-Performance Hybrid White OLEDs with Ultra-Stable Emission Color and Small Efficiency Roll-Off Achieved by Incorporating a Deep-Blue Fluorescent Neat Film. Adv. Opt. Mater. 2021, 9, 2100298. [Google Scholar] [CrossRef]
- Kang, S.; Kwon, H.; Jeong, J.; Kim, Y.-C.; Park, J. Synthesis and Electroluminescence Properties of New Blue Emitting Polymer Based on Dual-Core Type for Solution Process OLEDs. Macromol. Res. 2022, 30, 454–459. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, G.; Luo, X.; Tian, X.; Zhang, D.; Guo, S.; Zhou, H.; Miao, Y.; Huang, J.; Wang, H. Anthracene-based blue fluorescence materials utilized in non-doped OLEDs with high luminance and a low efficiency roll-off. Dye. Pigment. 2022, 204, 110391. [Google Scholar] [CrossRef]
- Lee, J.H.; Lin, H.-Y.; Chen, C.-H.; Lee, Y.-T.; Chiu, T.-L.; Lee, J.-H.; Chen, C.-T.; Adachi, C. Deep Blue Fluorescent Material with an Extremely High Ratio of Horizontal Orientation to Enhance Light Outcoupling Efficiency (44%) and External Quantum Efficiency in Doped and Non-Doped Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces 2021, 13, 34605–34615. [Google Scholar] [CrossRef]
- Bałczewski, P.; Kowalska, E.; Różycka-Sokołowska, E.; Uznański, P.; Wilk, J.; Koprowski, M.; Owsianik, K.; Marciniak, B. Organosulfur Materials with High Photo- and Photo-Oxidation Stability: 10-Anthryl Sulfoxides and Sulfones and Their Photophysical Properties Dependent on the Sulfur Oxidation State. Materials 2021, 14, 3506. [Google Scholar] [CrossRef]
- Newman, M.S.; Hussain, N.S. Synthesis of nuclear monobromobenz[a]anthracenes. J. Org. Chem. 1982, 47, 2837–2840. [Google Scholar] [CrossRef]
- Andrus, M.B.; Ye, Z.; Zhang, J. Highly selective glycine phase-transfer catalysis using fluoroanthracenylmethyl cinchonidine catalysts. Tetrahedron Lett. 2005, 46, 3839–3842. [Google Scholar] [CrossRef]
- Harvey, R.G.; Cortez, C.; Sugiyama, T.; Ito, Y.; Sawyer, T.W.; DiGiovanni, J. Biologically active dihydrodiol metabolites of polycyclic aromatic hydrocarbons structurally related to the potent carcinogenic hydrocarbon 7,12-dimethylbenz[a]anthracene. J. Med. Chem. 1988, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Hallman, J.L.; Bartsch, R.A. Synthesis of naphtho[f]ninhydrin. J. Org. Chem. 1991, 56, 6243–6245. [Google Scholar] [CrossRef]
- Platt, K.L.; Oesch, F. Reductive cyclization of keto acids to polycyclic aromatic hydrocarbons by hydroiodic acid-red phosphorus. J. Org. Chem. 1981, 46, 2601–2603. [Google Scholar] [CrossRef]
- Ihmels, H.; Meiswinkel, A.; Mohrschladt, C.J.; Otto, D.; Waidelich, M.; Towler, M.; White, R.; Albrecht, M.; Schnurpfeil, A. Anthryl-Substituted Heterocycles as Acid-Sensitive Fluorescence Probes. J. Org. Chem. 2005, 70, 3929–3938. [Google Scholar] [CrossRef]
- Sangaiah, R.; Gold, A.; Toney, G.E. Synthesis of a series of novel polycyclic aromatic systems: Isomers of benz[a]anthracene containing a cyclopenta-fused ring. J. Org. Chem. 1983, 48, 1632–1638. [Google Scholar] [CrossRef]
- Newman, M.S.; Prabhu, V.S.; Veeraraghavan, S. Synthesis of nuclear monobromobenz[a]anthracenes. J. Org. Chem. 1983, 48, 2926–2928. [Google Scholar] [CrossRef]
- Bradsher, C.; Sinclair, E. Notes—Cyclodehydrations in Liquid Sulfur Dioxide. J. Org. Chem. 1957, 22, 79–81. [Google Scholar] [CrossRef]
- Yamato, T.; Sakaue, N.; Shinoda, N.; Matsuo, K. Selective preparation of polycyclic aromatic hydrocarbons. Part 4.1 New synthetic route to anthracenes from diphenylmethanes using Friedel–Crafts intramolecular cyclization. J. Chem. Soc. Perkin Trans. 1 1997, 8, 1193–1200. [Google Scholar] [CrossRef]
- Bałczewski, P.; Skalik, J.; Uznański, P.; Guziejewski, D.; Ciesielski, W. Use of isomeric, aromatic dialdehydes in the synthesis of photoactive, positional isomers of higher analogs of o-bromo(hetero)acenaldehydes. RSC Adv. 2015, 5, 24700–24704. [Google Scholar] [CrossRef]
- Bodzioch, A.; Marciniak, B.; Różycka-Sokołowska, E.; Jeszka, J.K.; Uznański, P.; Kania, S.; Kuliński, J.; Bałczewski, P. Synthesis and Optoelectronic Properties of Hexahydroxylated 10-O-R-Substituted Anthracenes via a New Modification of the Friedel–Crafts Reaction Using O-Protected ortho-Acetal Diarylmethanols. Chem.—A Eur. J. 2012, 18, 4866–4876. [Google Scholar] [CrossRef] [PubMed]
- Bałczewski, P.; Bodzioch, A.; Różycka-Sokołowska, E.; Marciniak, B.; Uznański, P. First Approach to Nitrogen-Containing Fused Aromatic Hydrocarbons as Targets for Organoelectronics Utilizing a New Transformation of O-Protected Diaryl Methanols. Chem.—A Eur. J. 2010, 16, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Bałczewski, P.; Kowalska, E.; Skalik, J.; Koprowski, M.; Owsianik, K.; Różycka-Sokołowska, E. Ultrasound-assisted synthesis of RO- and RS-substituted (hetero)acenes via oxo- and thio-Friedel-Crafts/Bradsher reactions. Ultrason. Sonochem. 2019, 58, 104640. [Google Scholar] [CrossRef]
- Mondal, E.; Sahu, P.R.; Khan, A.T. A Useful and Catalytic Method for Protection of Carbonyl Compounds into the Corresponding 1,3-Oxathiolanes and Deprotection to the Parent Carbonyl Compounds. Synlett 2002, 2002, 0463–0467. [Google Scholar] [CrossRef]
- Djerassi, C.; Gorman, M. Studies in Organic Sulfur Compounds. VI.1 Cyclic Ethylene and Trimethylene Hemithioketals. J. Am. Chem. Soc. 1953, 75, 3704–3708. [Google Scholar] [CrossRef]
- Liang, X.; Gao, S.; Yang, J.; He, M. Synthesis of a Novel Strong Brønsted Acidic Ionic Liquid and its Catalytic Activities for the Oxathioacetalization. Catal. Lett. 2008, 125, 396–400. [Google Scholar] [CrossRef]
- Yus, M.; Nájera, C.; Foubelo, F. The role of 1,3-dithianes in natural product synthesis. Tetrahedron 2003, 59, 6147–6212. [Google Scholar] [CrossRef]
- Morton, D.R.; Hobbs, S.J. Facile preparation of cyclic ethylene thioketals and thioacetals with 2-phenyl- and 2-chloro-1,3,2-dithiaborolanes. J. Org. Chem. 1979, 44, 656–658. [Google Scholar] [CrossRef]
- Fuji, K.; Ueda, M.; Sumi, K.; Kajiwara, K.; Fujita, E.; Iwashita, T.; Miura, I. Chemistry of carbanions stabilized by sulfur. 1. Chemistry of 1,3-oxathianes. Synthesis and conformation of 2-substituted 1,3-oxathianes. J. Org. Chem. 1985, 50, 657–661. [Google Scholar] [CrossRef]
- Satchell, D.P.N.; Satchell, R.S. Mechanisms of hydrolysis of thioacetals. Chem. Soc. Rev. 1990, 19, 55–81. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef]
- ACD/Percepta, Version 14.0.0; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2015.
- Míšek, J.; Vargas Jentzsch, A.; Sakurai, S.; Emery, D.; Mareda, J.; Matile, S. A Chiral and Colorful Redox Switch: Enhanced π Acidity in Action. Angew. Chem. Int. Ed. 2010, 49, 7680–7683. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Xing, Z.; Fang, B.; Xie, X.; She, X. Visible light photoredox catalyzed deprotection of 1,3-oxathiolanes. Org. Biom. Chem. 2020, 18, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Björk, M.; Grivas, S. Synthesis of novel 2-aminoimidazo[4,5-b]pyridines, including the thieno analogue of the cooked-food mutagen IFP. J. Heterocycl. Chem. 2006, 43, 101–109. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 1 March 2023).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Reaction Conditions | RO-Acene (Yield) 1 | HO(CH2)2S-Acene (Yield) 1 |
|---|---|---|---|
 5a | HCl (0.17 mol/dm3, 10 equiv.), MeOH, 72 h, rt | 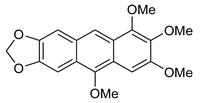 7a (53%) | not observed |
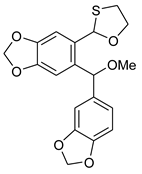 5b | HCl (0.17 mol/dm3, 10 equiv.), MeOH, 72 h, rt | 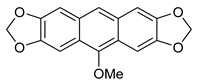 7b (37%) |  8b (12%) |
| HCl (0.17 mol/dm3, 10 equiv.), MeOH, 72 h, rt then 8 h, 65 °C |  7b (8%) |  8b (25%) | |
| HCl (0.34 mol/dm3, 10 equiv.), MeOH, 0.5 h, 65 °C | not observed | 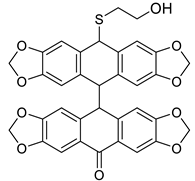 9b (55%) | |
| FeCl3/KI, MeOH, 3 h, 65 °C | 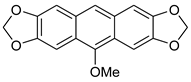 7b (36%) | not observed | |
 5c | HCl (0.17 mol/dm3, 10 equiv.), 2 h, 65 °C then 12 h, rt | 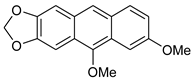 7c (35%) | 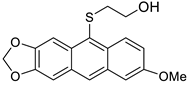 8c (9%) |
| HCl, (0.34 mol/dm3, 5 equiv.), 4 d, rt |  7c (25%) | 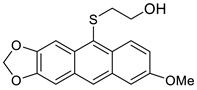 8c (27%) | |
| FeCl3/KI, MeOH, 12 h, 65 °C |  7c (78%) | not observed | |
 5d | HCl (0.17 mol/dm3, 10 equiv.), MeOH, 72 h, rt | 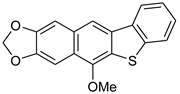 7d (62%) | not observed |
| HCl (0.17 mol/dm3, 10 equiv.), MeOH, 8 h, 65 °C | 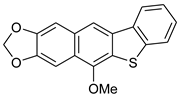 7d (33%) | not observed | |
| HCl (1.78 mol/dm3, 100 equiv.), 3 h, rt | not observed | 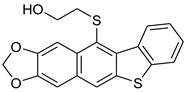 8d (14%) 2 | |
| FeCl3/KI, MeOH, 12 h, 65 °C | 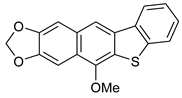 7d (62%) | not observed | |
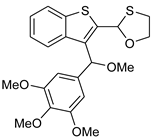 12 | HCl (0.34 mol/dm3, 10 equiv.), MeOH, 12 h, rt | 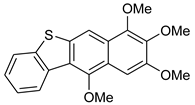 13 (15%) | not observed |
| Compound | Absorption λmax (nm) | Emission 1 λmax (nm) | Stokes Shift (cm−1) |
|---|---|---|---|
| 7b | 269, 322, 339, 357, 376 | 385, 406 | 621 |
| 7c | 270, 323, 339, 356, 372, 393 | 416, 438 | 1472 |
| 8c | 269, 363, 379, 400 | 408, 428 | 553 |
| 13 | 280, 302, 317, 330, 363, 383 | 404, 424 | 1420 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owsianik, K.; Różycka-Sokołowska, E.; Bałczewski, P. O,S-Acetals in a New Modification of oxo-Friedel–Crafts–Bradsher Cyclization—Synthesis of Fluorescent (Hetero)acenes and Mechanistic Considerations. Molecules 2023, 28, 2474. https://doi.org/10.3390/molecules28062474
Owsianik K, Różycka-Sokołowska E, Bałczewski P. O,S-Acetals in a New Modification of oxo-Friedel–Crafts–Bradsher Cyclization—Synthesis of Fluorescent (Hetero)acenes and Mechanistic Considerations. Molecules. 2023; 28(6):2474. https://doi.org/10.3390/molecules28062474
Chicago/Turabian StyleOwsianik, Krzysztof, Ewa Różycka-Sokołowska, and Piotr Bałczewski. 2023. "O,S-Acetals in a New Modification of oxo-Friedel–Crafts–Bradsher Cyclization—Synthesis of Fluorescent (Hetero)acenes and Mechanistic Considerations" Molecules 28, no. 6: 2474. https://doi.org/10.3390/molecules28062474