
Functionalization of 2-Mercapto-5-methyl-1,3,4-thiadiazole: 2-(ω-Haloalkylthio) Thiadiazoles vs. Symmetrical Bis-Thiadiazoles

Abstract
:
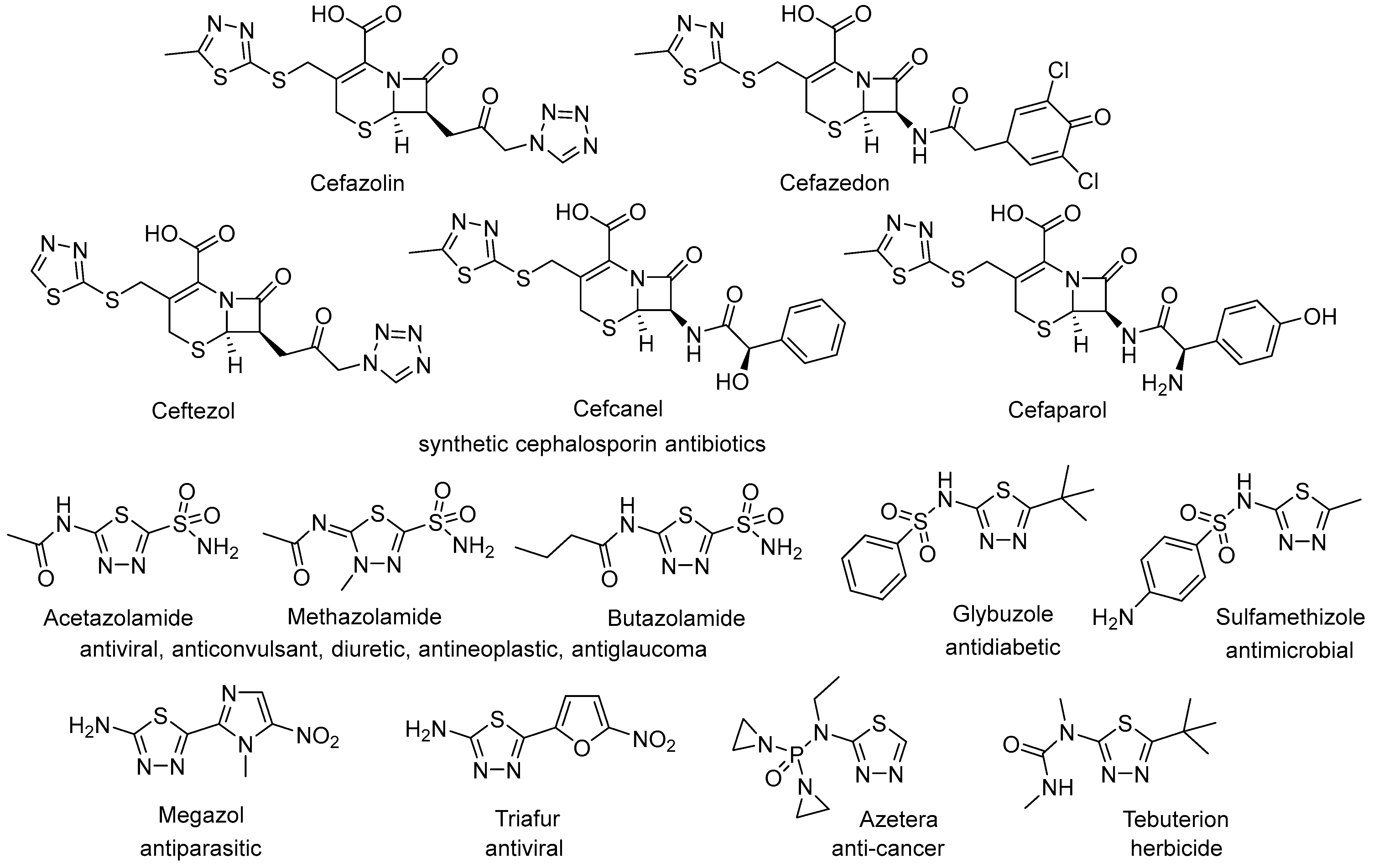
1. Introduction
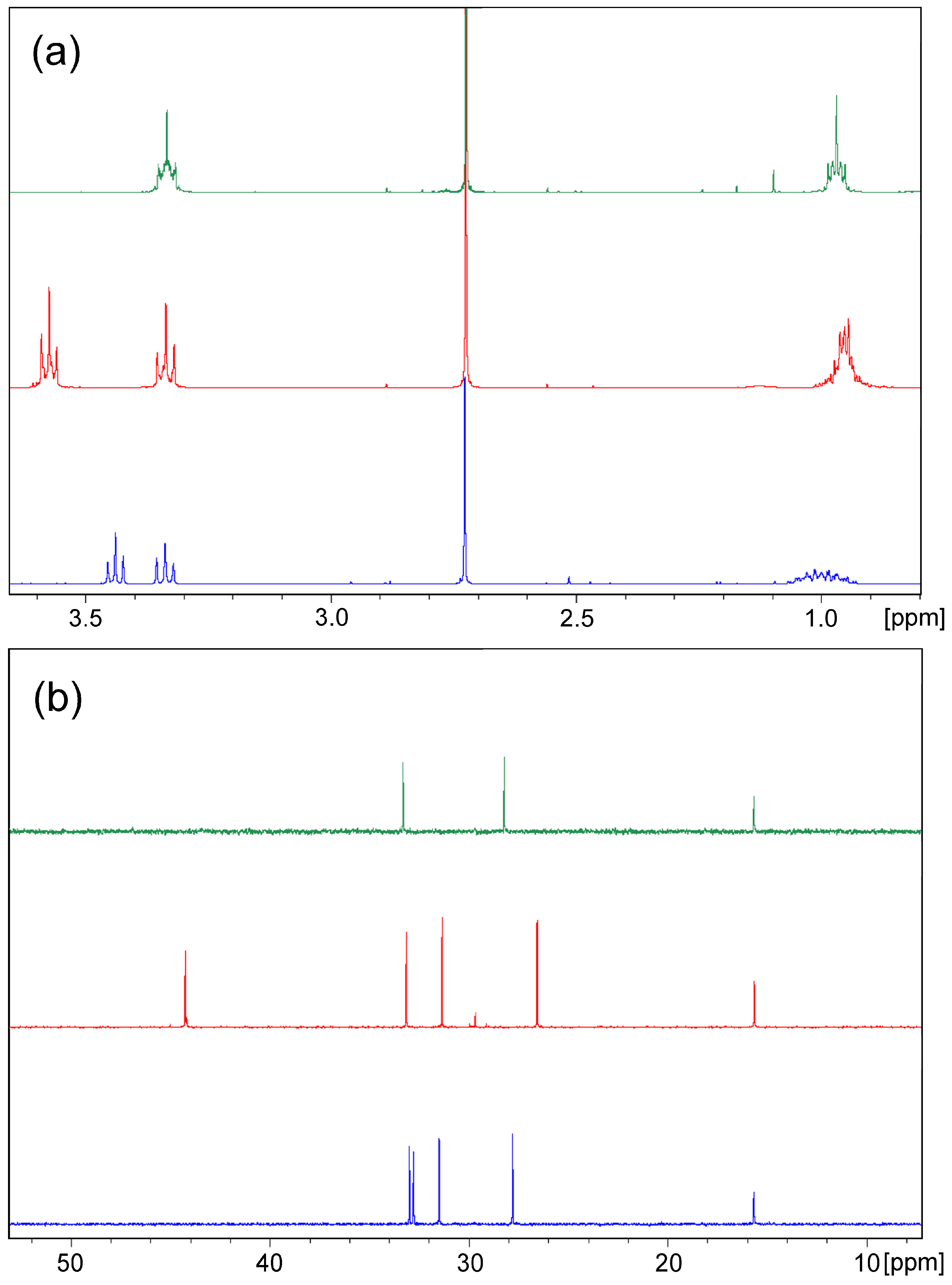
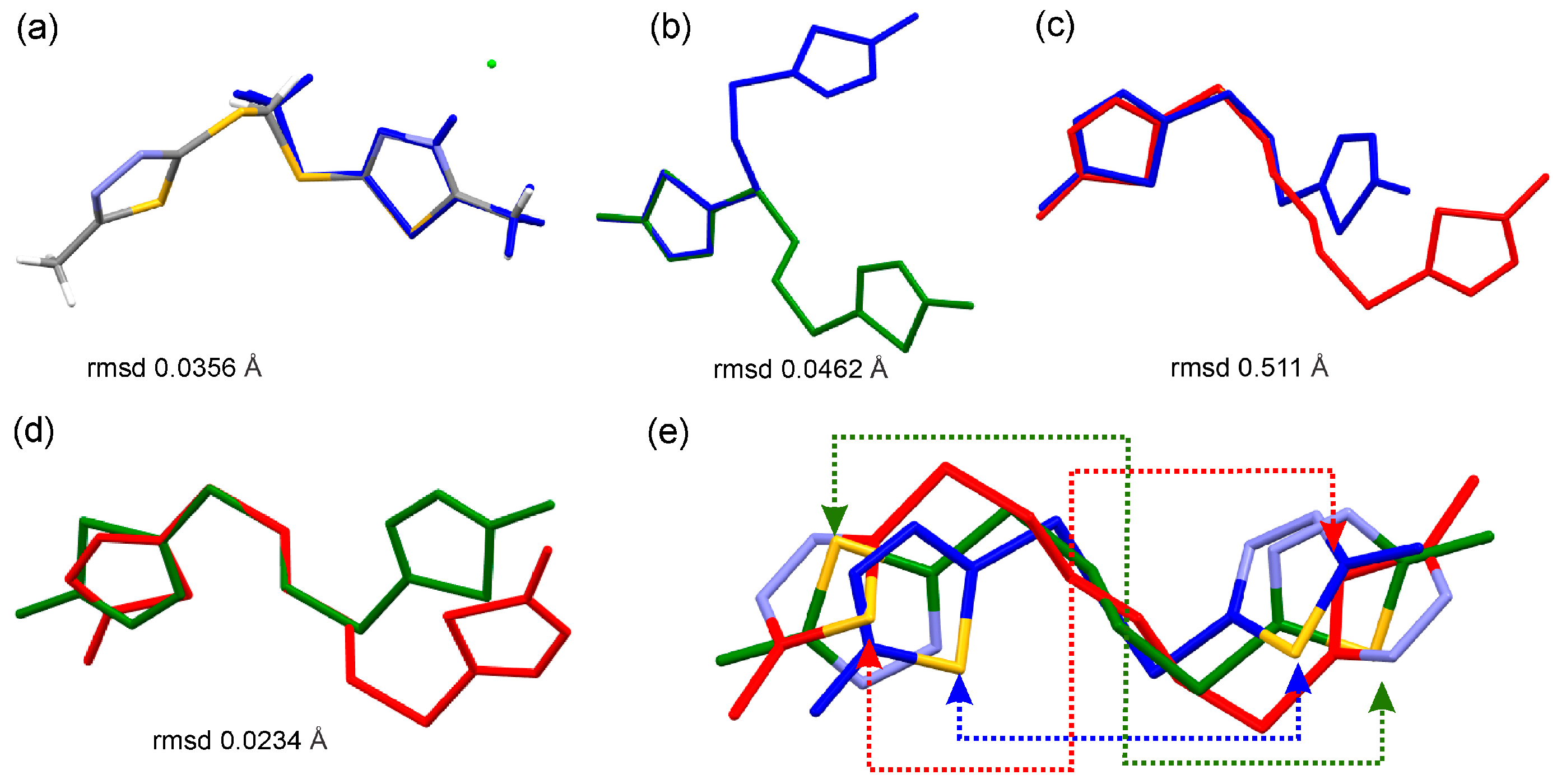
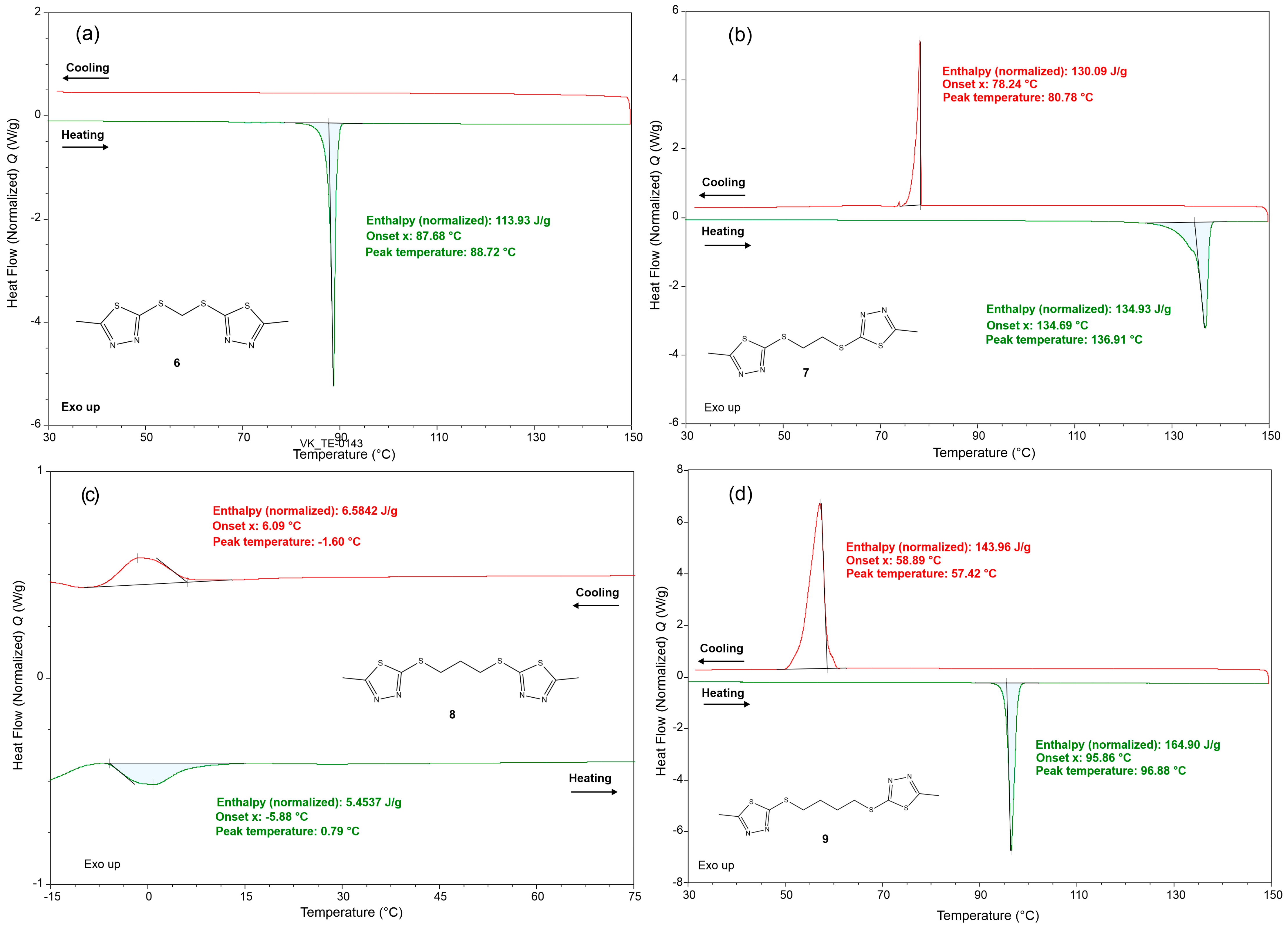
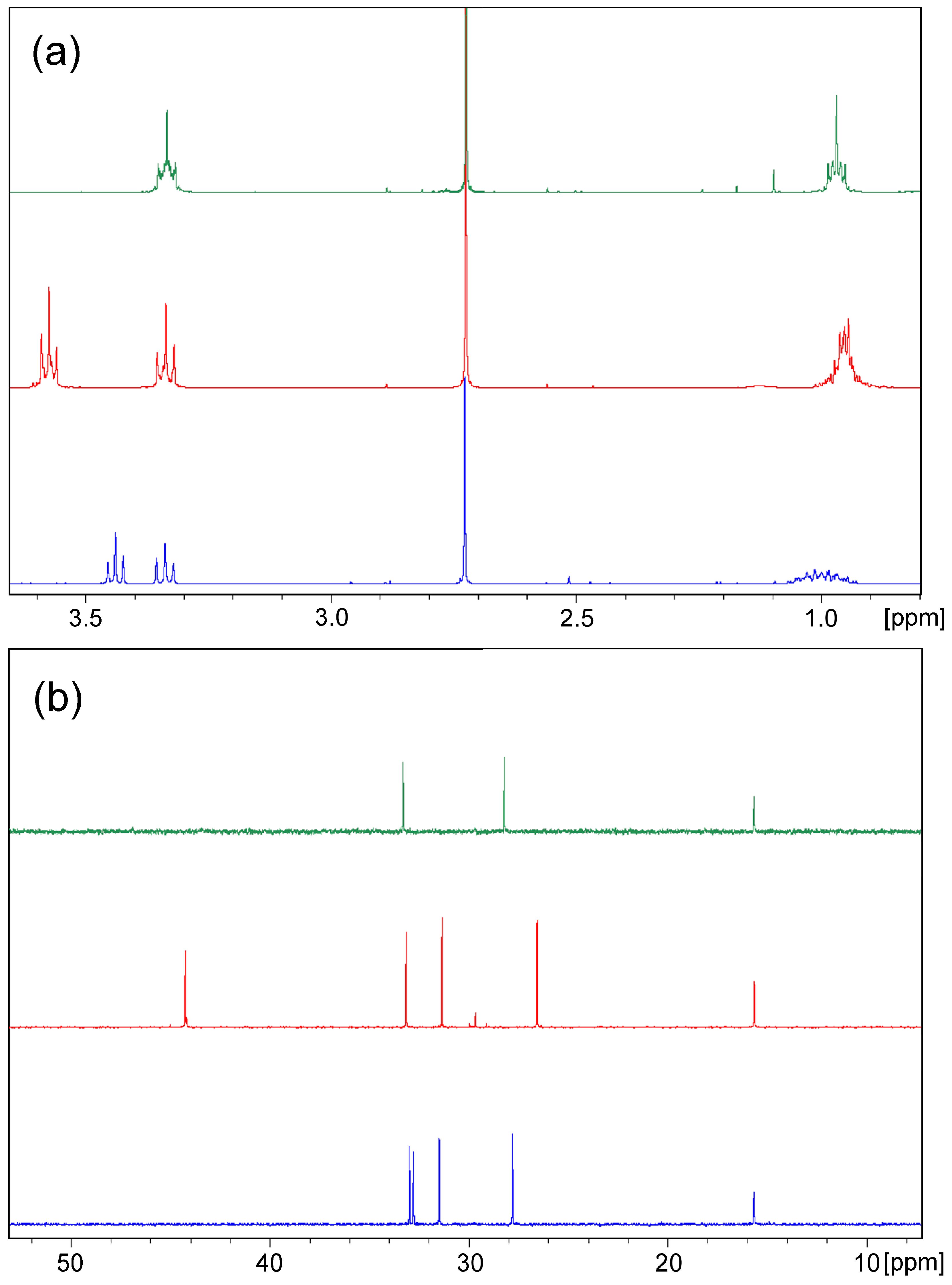
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Reaction with α,ω-Dibromoalkanes
3.3. Reaction with α,ω-Dichloroalkanes
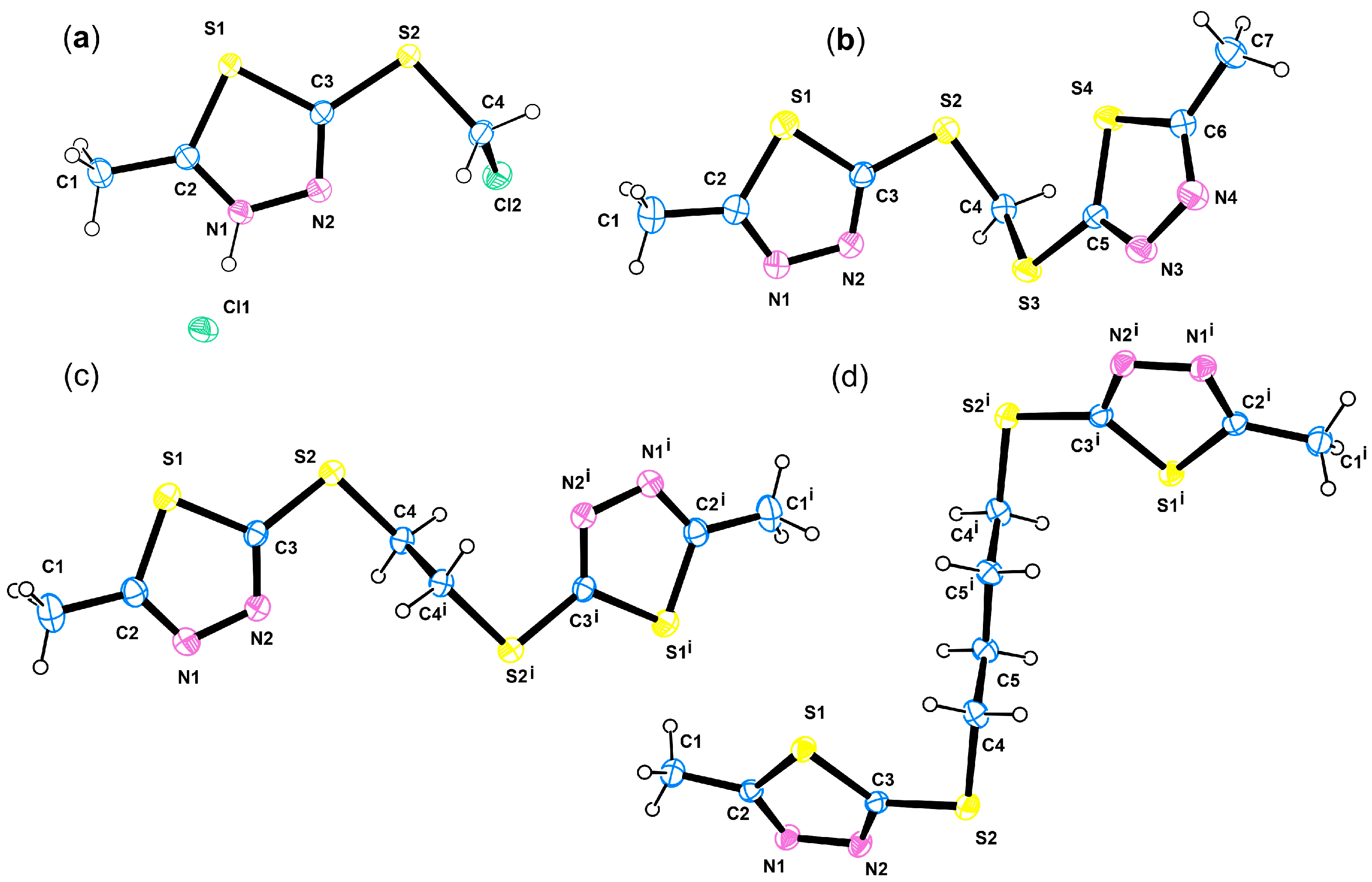
3.4. Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jain, A.K.; Sharma, S.; Vaidya, A.; Ravichandran, V.; Agrawal, R.K. 1,3,4-Thiadiazole and its Derivatives: A Review on Recent Progress in Biological Activities. Chem. Biol. Drug Des. 2013, 81, 557–576. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, C.-Y.; Wang, X.-M.; Yang, Y.-H.; Zhu, H.-L. 1,3,4-Thiadiazole: Synthesis, reactions, and applications in medicinal, agricultural, and materials chemistry. Chem. Rev. 2014, 114, 5572–5610. [Google Scholar] [CrossRef] [PubMed]
- Khalilullah, H.; Khan, M.U.; Mahmood, D.; Akhtar, J.; Osman, G. 1,3,4-Thiadiazole: A biologically active scaffold. Int. J. Pharm. Pharm. Sci. 2014, 6, 8–15. [Google Scholar]
- Matysiak, J. Biological and pharmacological activities of 1,3,4-thiadiazole based compounds. Mini Rev. Med. Chem. 2015, 15, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.; George, M.; Mathews, P. A review on various biological activities of 1,3,4- thiadiazole derivatives. J. Pharm. Chem. Biol. Sci. 2015, 3, 329–345. [Google Scholar]
- Han, X.; Yu, Y.L.; Hu, Y.S.; Liu, X.H. 1,3,4-thiadiazole: A privileged scaffold for drug design and development. Curr. Top. Med. Chem. 2021, 21, 2546–2573. [Google Scholar] [CrossRef] [PubMed]
- Irfan, A.; Ullah, S.; Anum, A.; Jabeen, N.; Zahoor, A.F.; Kanwal, H.; Kotwica-Mojzych, K.; Mojzych, M. Synthetic transformations and medicinal significance of 1,2,3-thiadiazoles derivatives: An update. Appl. Sci. 2021, 11, 5742. [Google Scholar] [CrossRef]
- Schatz, J.; Gogić, K.; Benkert, T. 1,3,4-Thiadiazoles. In Comprehensive Heterocyclic Chemistry IV, 4th ed.; Elsevier Science: Amsterdam, The Netherlands, 2022; Volume 5, Chapter 5.10; pp. 407–447. [Google Scholar]
- Anthwal, T.; Paliwal, S.; Nain, S. Diverse Biological Activities of 1,3,4-Thiadiazole Scaffold. Chemistry 2022, 4, 1654–1671. [Google Scholar] [CrossRef]
- Aliabadi, A. 1,3,4-Thiadiazole based anticancer agents. Anti-Cancer Agents Med. Chem. 2016, 16, 1301–1314. [Google Scholar] [CrossRef]
- Raj, V.; Rai, A.; Saha, S. Human Cancer cell line based approach of 1,3,4-thiadiazole and its fused ring: A comprehensive review. Anti-Cancer Agents Med. Chem. 2017, 17, 500–523. [Google Scholar] [CrossRef]
- Raj, V.; Rai, A.; Singh, A.K.; Keshari, A.K.; Trivedi, P.; Ghosh, B.; Kumar, U.; Kumar, D.; Saha, S. Discovery of Novel 2-Amino-5-(Substituted)-1,3,4-Thiadiazole Derivatives: New Utilities for Colon Cancer Treatment. Anti-Cancer Agents Med. Chem. 2018, 18, 719–738. [Google Scholar] [CrossRef] [PubMed]
- Janowska, S.; Paneth, A.; Wujec, M. Cytotoxic properties of 1,3,4-Thiadiazole derivatives—A review. Molecules 2020, 25, 4309. [Google Scholar] [CrossRef] [PubMed]
- Obakachi, V.A.; Kushwaha, B.; Kushwaha, N.D.; Mokoena, S.; Ganai, A.M.; Pathan, T.K.; van Zyl, W.E.; Karpoormath, R. Synthetic and anti-cancer activity aspects of 1,3,4-thiadiazole containing bioactive molecules: A concise review. J. Sulfur Chem. 2021, 42, 670–691. [Google Scholar] [CrossRef]
- Janowska, S.; Khylyuk, D.; Bielawska, A.; Szymanowska, A.; Gornowicz, A.; Bielawski, K.; Noworól, J.; Mandziuk, S.; Wujec, M. New 1,3,4-Thiadiazole Derivatives with Anticancer Activity. Molecules 2022, 27, 1814. [Google Scholar] [CrossRef] [PubMed]
- Serban, G.; Stanasel, O.; Serban, E.; Bota, S. 2-Amino-1,3,4-thiadiazole as a potential scaffold for promising antimicrobial agents. Drug Des. Devel. Ther. 2018, 12, 1545–1566. [Google Scholar] [CrossRef]
- Barbosa, G.A.D.; de Aguiar, A.P. Synthesis of 1,3,4-thiadiazole derivatives and microbiological activities: A review. Rev. Virtual Quim. 2019, 11, 806–848. [Google Scholar] [CrossRef]
- Kumar, D.; Kumar, H.; Kumar, V.; Deep, A.; Sharma, A.; Marwaha, M.G.; Marwaha, R.K. Mechanism-based approaches of 1,3,4 thiadiazole scaffolds as potent enzyme inhibitors for cytotoxicity and antiviral activity. Med. Drug Discov. 2023, 17, 100150. [Google Scholar] [CrossRef]
- Sahoo, B.M.; Dinda, S.C.; Ravi Kumar, B.V.V.; Panda, J.R.; Brahmkshatriya, P.S. Molecular Docking Study, Green Synthesis and Pharmacological Evaluation of 1,3,4-thiadiazole Derivatives as Potential Antiepileptic Agents. Mini-Rev. Med. Chem. 2013, 13, 2076–2081. [Google Scholar] [CrossRef]
- Raj, V.; Rai, A.; Singh, M.; Kumar, R.; Kumar, A.; Kumar, V.; Sharma, S.K. Recent Update on 1,3,4-Thiadiazole Derivatives: As Anticonvulsant Agents. Am. Res. J. Pharm. 2015, 1, 34–61. [Google Scholar]
- Anthwal, T.; Nain, S. 1,3,4-Thiadiazole Scaffold: As Anti-Epileptic Agents. Front. Chem. 2022, 9, 671212. [Google Scholar] [CrossRef]
- Barboiu, M.; Cimpoesu, M.; Guran, C.; Supuran, C. Synthesis and Biological Activity of Metal Complexes of 5-(2-Aminoethyl)-2-Amino-1,3,4-Thiadiazole. Met.-Based Drugs 1996, 3, 227–232. [Google Scholar] [CrossRef]
- Zheng, Y.; Du, M.; Li, J.; Zhang, R.; Bu, X. Tuning the framework formation of silver(i) coordination architectures with heterocyclic thioethers. Dalton Trans. 2003, 8, 1509–1514. [Google Scholar] [CrossRef]
- Zhou, H.; Zheng, J.; Wang, H.; Wang, J.; Song, X.; Cao, Y.; Xiong, C. Preparation of a novel chloromethylated polystyrene-2-mercapto-1,3,4-thiadiazole chelating resin and its adsorption properties and mechanism for separation and recovery of Hg(II) from aqueous solutions. Water Sci. Technol. 2017, 76, 1915–1924. [Google Scholar] [CrossRef]
- Kudelko, A.; Olesiejuk, M.; Luczynski, M.; Swiatkowski, M.; Sieranski, T.; Kruszynski, R. 1,3,4-Thiadiazole-Containing Azo Dyes: Synthesis, Spectroscopic Properties and Molecular Structure. Molecules 2020, 25, 2822. [Google Scholar] [CrossRef]
- Hipler, F.; Fischer, R.A.; Müller, J. Matrix-isolation pyrolysis investigation of mercapto-functionalized 1,3,4-thiadiazoles: Thermal stability of thiadiazole lubricant additives. Phys. Chem. Chem. Phys. 2005, 7, 731–737. [Google Scholar] [CrossRef]
- Kornis, G.I. Comprehensive Heterocyclic Chemistry II; Katritzky, A.R., Scriven, E.F.V., Rees, C.W., Eds.; Elsevier Science Ltd.: Amsterdam, The Netherlands, 1996; Volume 4, Chapter 4.10; pp. 379–408. [Google Scholar]
- Koutentis, P.A.; Constantinides, C.P. Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Scriven, E.F.V., Rees, C.W., Eds.; Elsevier Science Ltd.: Amsterdam, The Netherlands, 2008; Volume 5, Chapter 5.10; pp. 567–605. [Google Scholar]
- Manimaran, T.; Anand Raj, M.; Jishala, M.I.; Gopalasatheeskumar, K. Review on substituted 1, 3, 4 thiadiazole compounds. Int. J. Pharm. Anal. Res. 2017, 6, 222–231. [Google Scholar]
- Ram, V.J.; Sethi, A.; Nath, M.; Pratap, R. Five-Membered Heterocycles, The Chemistry of Heterocycles, Nomenclature and Chemistry of Three-to-Five Membered Heterocycles; Ram, V.J., Sethi, A., Nath, M., Pratap, R., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2019; Chapter 5; pp. 149–478. [Google Scholar]
- Shamroukh, A.H.; Hegab, M.I. A Review on synthesis, therapeutic, and computational studies of substituted 1, 3, 4 thiadiazole derivatives. Egypt. J. Chem. 2020, 63, 4387–4408. [Google Scholar]
- Kumar, D.; Aggarwal, N.; Kumar, V.; Chopra, H.; Marwaha, R.K.; Sharma, R. Emerging synthetic strategies and pharmacological insights of 1,3,4-thiadiazole derivatives: A comprehensive review. Future Med. Chem. 2024, 16, 563–581. [Google Scholar] [CrossRef]
- Kadu, N.S.; Masand, V.H. Synthesis of 1,3,4-thiadiazole derivative using appropriate reaction conditions. Int. J. Res. Appl. Sci. Engin. Technol. 2022, 10, 312–321. [Google Scholar] [CrossRef]
- Chen, Z.; Xu, W.; Liu, K.; Yang, S.; Fan, H.; Bhadury, P.S.; Huang, D.-Y.; Zhang, Y. Synthesis and antiviral activity of 5-(4-chlorophenyl)-1,3,4-thiadiazole sulfonamides. Molecules 2010, 15, 9046–9056. [Google Scholar] [CrossRef]
- Kumar, B.S.; Reddy, P.R.; Ravindranath, L.R.K.R.; Prasad, A.R.G.; Mallika, A. Synthesis, characterization and in vitro santimicrobial evaluation of sulphonyl urea derivatives as potential inhibitors of beta-ketoacyl-acyl carrier protein synthase III (FabH). Acta Univ. 2015, 25, 12–21. [Google Scholar]
- Alam, J.; Alam, O.; Ali, R.; Mohd; Naim, J.; Khan, S.A. Synthesis and anti-inflammatory activity of some new thiadiazole linked pyrazole benzene sulphonamides as cyclooxygenase inhibitors. Orient. J. Chem. 2015, 31, 1873–1885. [Google Scholar] [CrossRef]
- Cristina, A.; Leonte, D.; Vlase, L.; Csaba Bencze, L.; Imre, S.; Marc, G.; Apan, B.; Mogoșan, C.; Zaharia, V. Heterocycles 48. Synthesis, characterization and biological evaluation of imidazo[2,1-b][1,3,4]thiadiazole derivatives as anti-inflammatory agents. Molecules 2018, 23, 2425. [Google Scholar] [CrossRef]
- Shafique, M.; Hameed, S.; Naseer, M.M.; Al-Masoudi, N.A. Synthesis of new chiral 1,3,4-thiadiazole-based di- and tri-arylsulfonamide residues and evaluation of in vitro anti-HIV activity and cytotoxicity. Mol. Divers. 2018, 22, 957–968. [Google Scholar] [CrossRef]
- Almandil, N.B.; Taha, M.; Gollapalli, M.; Rahim, F.; Ibrahim, M.; Mosaddik, A.; Anouar, E.H. Indole bearing thiadiazole analogs: Synthesis, β-glucuronidase inhibition and molecular docking study. BMC Chem. 2019, 13, 14. [Google Scholar] [CrossRef]
- Gomha, S.M.; Edrees, M.M.; Muhammad, Z.A.; El-Reedy, A.A.M. 5-(Thiophen-2-yl)-1,3,4-thiadiazole derivatives: Synthesis, molecular docking and in vitro cytotoxicity evaluation as potential anticancer agents. Drug Des. Devel. Ther. 2018, 12, 1511–1523. [Google Scholar] [CrossRef]
- Qurban, J.; Gouda, M.A. Synthesis and cytotoxic activity of some new sulfa drugs containing thiadiazole. Indian J. Heterocycl. Chem. 2021, 31, 559–564. [Google Scholar]
- Vardanyan, R.S.; Hruby, V.J. Antiepileptic Drugs. In Synthesis of Essential Drugs; Elsevier B.V.: Amsterdam, The Netherlands, 2006; Chapter 9; pp. 125–133. [Google Scholar]
- Vardanyan, R.S.; Hruby, V.J. Diuretics. In Synthesis of Essential Drugs; Elsevier B.V.: Amsterdam, The Netherlands, 2006; Chapter 21; pp. 277–293. [Google Scholar]
- Vardanyan, R.S.; Hruby, V.J. Antimicrobial Drugs. In Synthesis of Essential Drugs; Elsevier B.V.: Amsterdam, The Netherlands, 2006; Chapter 33; pp. 499–523. [Google Scholar]
- Chhajed, M.; Shrivastava, A.K.; Taile, V. Synthesis of 5-arylidine amino-1,3,4-thiadiazol-2-[(N-substituted benzyol)]sulphonamides endowed with potent antioxidants and anticancer activity induces growth inhibition in HEK293, BT474 and NCI-H226 cells. Med. Chem. Res. 2014, 23, 3049–3064. [Google Scholar] [CrossRef]
- El-Gazzar, M.G.; Zaher, N.H.; El-Tablawy, S.Y. Morphological changes of some pathogenic microbial strains induced by novel thiadiazole derivatives. Med. Chem. Res. 2014, 23, 1844–1854. [Google Scholar] [CrossRef]
- Çevik, U.A.; Celik, I.; İnce, U.; Maryam, Z.; Ahmad, I.; Patel, H.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis, biological evaluation, and molecular modeling studies of new 1,3,4-thiadiazole derivatives as potent antimicrobial agents. Chem. Biodivers. 2023, 20, e202201146. [Google Scholar]
- Kariyone, K.; Harada, H.; Kurita, M.; Takano, T. Cefazolin, a new semisynthetic cephalosporin antibiotic. I. synthesis and chemical properties of cefazolin. J. Antibiot. 1970, 23, 131–136. [Google Scholar] [CrossRef]
- Hussain, S.A.; Mahmood, T.; Chawala, N. Improved and economical synthesis of cefazolin sodium. Pak. J. Pharm. 2012, 25, 23–26. [Google Scholar]
- Zhao, M. Method for Preparing Cefazedone. CN104230958, 24 December 2014. [Google Scholar]
- Minghua, L.; Zhangwei, G.; Yanxiao, F.; Qingli, L.; Yuqing, Z. Synthesis Technology of Cefazedone. CN110437256A, 12 November 2019. [Google Scholar]
- Ludescher, J.; Wieser, J. Cephalosporin Derivative. EP0463553A1, 2 January 1992. [Google Scholar]
- Dunn, G.L.; Hoover, J.R.E.; Berges, D.A.; Taggart, J.J.; Davis, L.D.; Dietz, E.M.; Jakas, D.R.; Yim, N.; Actor, P.; Uri, J.V.; et al. Orally active 7-phenylglycyl cephalosporins—Structure-activity studies related to cefatrizine (SK AND F 60771). J. Antibiot. 1976, 29, 65–80. [Google Scholar] [CrossRef]
- Ali, M.A.; Livingstone, S.E. Metal-complexes of sulphur-nitrogen chelating-agents. Coord. Chem. Rev. 1974, 13, 101–132. [Google Scholar]
- Mews, R.; Watson, P.G.; Lork, E. Three-coordinate sulphur(VI)-nitrogen species: An attempt to breathe some new life into an old topic. Coord. Chem. Rev. 1997, 158, 233–273. [Google Scholar] [CrossRef]
- Mensforth, E.J.; Hill, M.R.; Batten, S.R. Coordination polymers of sulphur-donor ligands. Inorg. Chim. Acta 2013, 403, 9–24. [Google Scholar] [CrossRef]
- Zhang, W.H.; Ren, Z.G.; Lang, J.P. Rational construction of functional molybdenum (tungsten)-copper-sulfur coordination oligomers and polymers from preformed cluster precursors. Chem. Soc. Rev. 2016, 45, 4995–5019. [Google Scholar] [CrossRef]
- Aarabi, F.; Naake, T.; Fernie, A.R.; Hoefgen, R. Coordinating sulfur pools under sulfate deprivation. Trends Plant Sci. 2020, 25, 1227–1239. [Google Scholar] [CrossRef]
- Haiduc, I. Inverse coordination metal complexes with oxalate and sulfur, selenium and nitrogen analogues as coordination centers. Topology and systematization. J. Coord. Chem. 2020, 73, 1619–1700. [Google Scholar] [CrossRef]
- Kamakura, Y.; Tanaka, D. Metal-organic frameworks and coordination polymers composed of sulfur-based nodes. Chem. Lett. 2021, 50, 523–533. [Google Scholar] [CrossRef]
- Seregin, I.V.; Kozhevnikova, A.D. Phytochelatins: Sulfur-containing metal(loid)-chelating ligands in plants. Int. J. Mol. Sci. 2023, 24, 2430. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.K.; Almeida, R.M.; Moura, I.; Moura, J.J.G. Rubredoxins derivatives: Simple sulphur-rich coordination metal sites and its relevance for biology and chemistry. Coord. Chem. Rev. 2017, 352, 379–397. [Google Scholar] [CrossRef]
- Colovic, M.B.; Vasic, V.M.; Djuric, D.M.; Krstic, D.Z. Sulphur-containing amino acids: Protective role against free radicals and heavy metals. Curr. Med. Chem. 2018, 25, 324–335. [Google Scholar] [CrossRef]
- Tanifuji, K.; Ohki, Y. Metal-Sulfur Compounds in N2Reduction and Nitrogenase-Related Chemistry. Chem. Rev. 2020, 120, 5194–5251. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, V.; Capaccio, V.; Lamparelli, D.H.; Capacchione, C. Metal complexes bearing sulfur-containing ligands as catalysts in the reaction of CO2 with epoxides. Catalysts 2020, 10, 825. [Google Scholar] [CrossRef]
- Hu, X.; Huang, T.; Zhang, G.; Lin, S.; Chen, R.; Chung, L.-H.; He, J. Metal-organic framework-based catalysts for lithium-sulfur batteries. Coord. Chem. Rev. 2023, 475, 214879. [Google Scholar] [CrossRef]
- Song, Y.; Zou, L.; Wei, C.; Zhou, Y.; Hu, Y. Single-atom electrocatalysts for lithium–sulfur chemistry: Design principle, mechanism, and outlook. Carbon Energy 2023, 5, e286. [Google Scholar] [CrossRef]
- Tanifuji, K.; Ohta, S.; Ohki, Y.; Seino, H. Activation of unsaturated small molecules by bio-relevant multinuclear metal-sulfur clusters. Coord. Chem. Rev. 2023, 475, 214838. [Google Scholar] [CrossRef]
- Egly, J.; Bouché, M.; Chen, W.; Maisse-François, A.; Achard, T.; Bellemin-Laponnaz, S. Synthesis, structural characterization and anti-proliferative activity of (κ1-C)- and (κ2-C,S)-PtII complexes bearing thioether-functionalized N-heterocyclic carbenes. Eur. J. Inorg. Chem. 2018, 2018, 159–166. [Google Scholar] [CrossRef]
- Li, P.; Yang, Y.; Wang, X.; Wu, X. Recent achievements on the agricultural applications of thioether derivatives: A 2010–2020 decade in review. J. Heterocycl. Chem. 2021, 58, 1225–1251. [Google Scholar] [CrossRef]
- Bisht, S.; Kumar, L.; Kaul, G.; Akhir, A.; Saxena, D.; Chopra, S.; Karthik, R.; Goyal, N.; Batra, S. Synthesis and biological evaluation of substituted 3-isoxazolethioethers as antileishmanial and antibacterial agents. ChemistrySelect 2022, 7, e202201664. [Google Scholar] [CrossRef]
- Ozcan, I.; Akkoc, S.; Alici, H.; Capanlar, S.; Sahin, O.; Tahtaci, H. Novel thioether-bridged 2,6-disubstituted and 2,5,6-trisubstituted imidazothiadiazole analogues: Synthesis, antiproliferative activity, ADME, and molecular docking studies. Chem. Biodivers. 2023, 20, e202200884. [Google Scholar] [CrossRef] [PubMed]
- Rezania, H.; Vatanpour, V.; Salehi, E.; Gavari, N.; Shockravi, A.; Ehsani, M. Wholly heterocycles-based polyamide–sulfide containing pyridine and thiazole rings: A super-adsorbent polymer for lead removal. J. Polym. Environ. 2019, 27, 1790–1800. [Google Scholar] [CrossRef]
- Fliedel, C.; Braunstein, P. Thioether-functionalized N-heterocyclic carbenes: Mono- and bis-(S,CNHC) palladium complexes, catalytic C-C coupling, and characterization of a unique Ag4I4(S,CNHC)2 planar cluster. Organometallics 2010, 29, 5614–5626. [Google Scholar] [CrossRef]
- Liu, Y.; Kean, Z.S.; d’Aquino, A.I.; Manrajd, Y.D.; Mendez-Arroyo, J.; Mirkin, C.A. Palladium(II) weak-link approach complexes bearing hemilabile N-heterocyclic carbene-thioether ligands. Inorg. Chem. 2017, 56, 5902–5910. [Google Scholar] [CrossRef]
- Mechrouk, V.; Maisse-François, A.; Bellemin-Laponnaz, S.; Achard, T. β-Alkylation through dehydrogenative coupling of primary alcohols and secondary alcohols catalyzed by thioether-functionalized n-heterocyclic carbene ruthenium complexes. Eur. J. Inorg. Chem. 2023, 26, e202300188. [Google Scholar] [CrossRef]
- Shi, X.; Yi, A.; Liu, Q.; Zhang, Y.; Lin, S.; Lu, X. Nonplanar π-conjugated sulfur heterocyclic quinone polymer cathode for air-rechargeable zinc/organic battery with simultaneously boosted output voltage, rate capability, and cycling life. ACS Nano 2023, 17, 25005–25013. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Wei, L.-L.; Han, Z.; Xu, L.; Li, L.-K. Syntheses, crystal structures, thermal and photoluminescent properties of four coordination complexes with a heterocyclic thioether carboxylate ligand. Z. Anorg. Allg. Chem. 2015, 641, 2490–2497. [Google Scholar] [CrossRef]
- Yoon, M.; Moon, D. New Zr (IV) based metal-organic framework comprising a sulfur-containing ligand: Enhancement of CO2 and H2 storage capacity. Microporous Mesoporous Mat. 2015, 215, 116–122. [Google Scholar] [CrossRef]
- Li, M.-T.; Kong, N.; Lan, Y.Q.; Su, Z.-M. Sulfur-containing bimetallic metal organic frameworks with multi-fold helix as anode of lithium ion batteries. Dalton Trans. 2018, 47, 4827–4832. [Google Scholar] [CrossRef]
- Li, X.; Ma, W.; Li, H.; Zhang, Q.; Liu, H. Sulfur-functionalized metal-organic frameworks: Synthesis and applications as advanced adsorbents. Coord. Chem. Rev. 2020, 408, 213191. [Google Scholar] [CrossRef]
- Dubskikh, V.A.; Kovalenko, K.A.; Nizovtsev, A.S.; Lysova, A.A.; Samsonenko, D.G.; Dybtsev, D.N.; Fedin, V.P. Enhanced adsorption selectivity of carbon dioxide and ethane on porous metal–organic framework functionalized by a sulfur-rich heterocycle. Nanomaterials 2022, 12, 4281. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.E.; Maryanoff, C.A.; McComsey, D.F.; Stanzione, R.C.; Scott, L. The reaction of amines with methylene chloride. Evidence for rapid aminal formation from N-methylenepyrrolidinium chloride and pyrrolidine. J. Org. Chem. 1987, 52, 1857–1859. [Google Scholar] [CrossRef]
- Kurteva, V.; Rusew, R.; Shivachev, B. 4-Methyl-7-((2-((5-methyl-1,3,4-thiadiazol-2-yl)thio)ethyl)thio)-coumarin. Molbank 2022, 2022, M1491. [Google Scholar] [CrossRef]
- Singh, V.N.; Sharma, S. Stereoselective synthesis and characterization of monocyclic cis-β-lactams containing 5-methyl-1,3,4-thiadiazole-2-thiol moiety. J. Heterocycl. Chem. 2021, 58, 2163–2173. [Google Scholar] [CrossRef]
- Al-Mouqdady, O.D.; Al-Janabi, A.S.; Hatshan, M.R.; Al-Jibori, S.A.; Fiahan, A.S.; Wagner, C. Synthesis, characterization, anti-bacterial and anticancer activities of Palladium(II) mixed ligand complexes of 2-mercapto-5-methyl-1,3,4-thiadiazole (HmtzS) and phosphines. Crystal structure of [Pd(mtzS)2(dppf)].H2O.EtOH. J. Mol. Struct. 2022, 1264, 133219. [Google Scholar] [CrossRef]
- Zheng, Q.; Borsley, S.; Nichol, G.S.; Duarte, F.; Cockroft, S.L. The energetic significance of metallophilic interactions. Angew. Chem., Int. Ed. 2019, 131, 12747–12753. [Google Scholar] [CrossRef]
- CrysAlis PRO; Rigaku Oxford Diffraction Ltd.: Yarnton, UK, 2021.
- Bruker, S. APEX3 V2016. 9-0, SAINT V8. 37A; Bruker AXS Inc.: Madison, WI, USA, 2016; Volume 2013, p. 2014. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Section C 2015, 71, 3–8. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
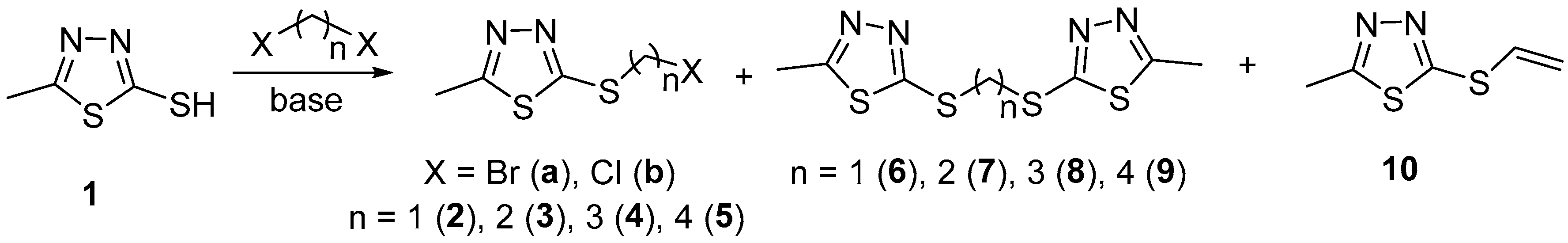

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
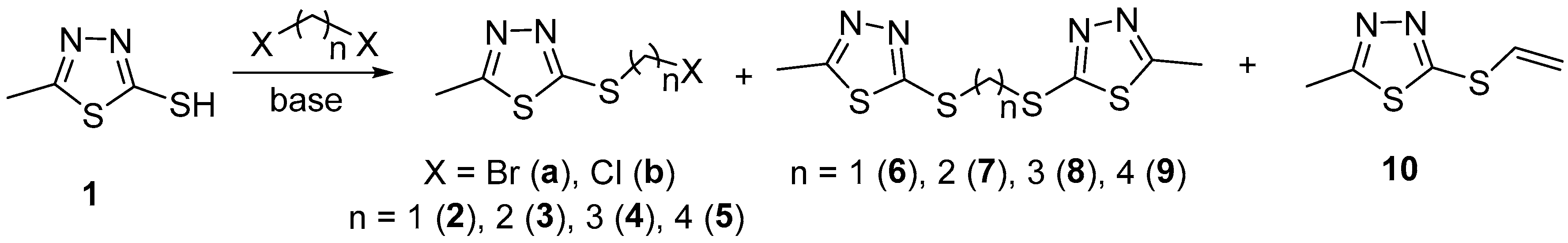{kind=link}
| Entry | Conditions a | Products | ||||
|---|---|---|---|---|---|---|
| Halogenide | Bis-Thiadiazole | Total Yield, % | ||||
| Compd. | Yield, % | Compd. | Yield, % | |||
| 1 | DBM (8; 1:2), K2CO3, EtOH, reflux, 2.5 h | 2a | - | 6 | 76 | 76 |
| 2 | DBM (8; 1:2), Et3N, ether | 2a | - | 6 | 74 | 74 |
| 3 | DBM (2.2; 2:1.1), Et3N, ether | 2a | - | 6 | 75 | 75 |
| 4 | DCM, DIPEA | 2b | 25 | 6 | 71 | 96 |
| 5 | DCM, Et3N | 2b | 18 | 6 | 80 | 98 |
| 6 | DBE (8; 1:2), K2CO3, MeCN | 3a | 13 | 7 | 30 | 55 b |
| 7 | DBE (8; 1:2), Et3N, ether | 3a | 53 | 7 | 40 | 93 |
| 8 | DBE (2.5; 2:1.25), Et3N, ether | 3a | 31 | 7 | 65 | 96 |
| 9 | DBE (2.2; 2:1.1), Et3N, ether | 3a | 28 | 7 | 69 | 97 |
| 10 | DCE, Et3N | 3b | 98 | 7 | - | 98 c |
| 11 | DBP (8; 1:2), Et3N, ether | 4a | 63 | 8 | 33 | 96 |
| 12 | DBP (2.5; 2:1.25), Et3N, ether | 4a | 35 | 8 | 59 | 94 |
| 13 | DBP (2.2; 2:1.1), Et3N, ether | 4a | 25 | 8 | 73 | 98 |
| 14 | DCP, Et3N | 4b | 93 | 8 | - | 93 |
| 15 | DBB (8; 1:2), Et3N, ether | 5a | 66 | 9 | 31 | 97 |
| 16 | DBB (2.5; 2:1.25), Et3N, ether | 5a | 29 | 9 | 58 | 87 |
| 17 | DBB (2.2; 2:1.1), Et3N, ether | 5a | 8 | 9 | 78 | 86 |
| 18 | DCB, Et3N | 5b | 98 | 9 | - | 98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petkova, Z.S.; Rusew, R.I.; Shivachev, B.L.; Kurteva, V.B. Functionalization of 2-Mercapto-5-methyl-1,3,4-thiadiazole: 2-(ω-Haloalkylthio) Thiadiazoles vs. Symmetrical Bis-Thiadiazoles. Molecules 2024, 29, 1938. https://doi.org/10.3390/molecules29091938
Petkova ZS, Rusew RI, Shivachev BL, Kurteva VB. Functionalization of 2-Mercapto-5-methyl-1,3,4-thiadiazole: 2-(ω-Haloalkylthio) Thiadiazoles vs. Symmetrical Bis-Thiadiazoles. Molecules. 2024; 29(9):1938. https://doi.org/10.3390/molecules29091938
Chicago/Turabian StylePetkova, Zhanina S., Rusi I. Rusew, Boris L. Shivachev, and Vanya B. Kurteva. 2024. "Functionalization of 2-Mercapto-5-methyl-1,3,4-thiadiazole: 2-(ω-Haloalkylthio) Thiadiazoles vs. Symmetrical Bis-Thiadiazoles" Molecules 29, no. 9: 1938. https://doi.org/10.3390/molecules29091938