Tricarbonyl(h6-Arene)Chromium(0) Complexes as Chiral Auxiliaries: Asymmetric Synthesis of b-Lactones

Dipartimento di Chimica Organica e Industriale dell’Universita’ di Milano e Centro CNR, Via Venezian, 21-20133 Milano Italy
*
Author to whom correspondence should be addressed.
Molecules 2001, 6(1), 13-20; https://doi.org/10.3390/60100013
Submission received: 24 July 2000
/
Revised: 18 December 2000
/
Accepted: 18 December 2000
/
Published: 16 January 2001
Abstract
:The optically pure tricarbonyl(η6-2-substituted benzaldehydes)chromium are used as chiral auxiliaries in the condensation with a series of doubly lithiated carboxylic acids. The intramolecular ring closure of the obtained Cr(CO)3 complexed β-hydroxyacids affords the optically pure β-lactones in satisfactory yield.
Introduction
The synthesis and reactivity of β-lactones in racemic and enantiopure form have received in the last years a growing interest due to different reasons: i) the 2-oxetanone ring is a basic component in some biologically active natural products [1]; ii) their high reactivity toward a large series of electrophilic and nucleophilic species makes these compounds useful building blocks in the organic synthesis [2]; iii) their use as monomers in the preparation of biodegradable polymers [3].
As a part of our studies on the application of chiral tricarbonyl(arene)chromium derivatives in asymmetric synthesis of products with potential biological activity [4]-[7], we were interested in the application of these organometallic substrates for the synthesis of some optically pure 2-oxetanones.
Scheme.


{kind=link}
| Prod. | R | R1 | R2 | Yield % | m.p. °C | [α]D | Absol. Config.a) | Config. of 1 |
|---|---|---|---|---|---|---|---|---|
| 3a | OMe | Me | Me | 95 | 132 | +36.5 | 3R | (-)-1R |
| 3b | Cl | Me | Me | 86 | 142 | +32.1 | 3R | (-)-1R |
| 3c | OMe | Me | H | 95 (1:1) | 98 d | +126 | 2R3R | (-)-1R |
| 3d | OMe | H | Me | 101 d | +127.9 | 2S3R | (-)-1R | |
| 3e | OMe | CMe3 | H | 96 (1:1) | 110 d | -103.5 | 2R3S | (+)-1S |
| 3f | OMe | H | CMe3 | 107 d | -129.7 | 2S3S | (+)-1S | |
| 4a | OMe | Me | Me | 70 | 118 | +42.8 | 4R | (-)-1R |
| 4b | Cl | Me | Me | 65 | 111 | -20.3 | 4R | (-)-1R |
| 4c | OMe | Me | H | 60 | 67 | +67.1 | 3R4R | (-)-1R |
| 4d | OMe | H | Me | -- | -- | -- | -- | (-)-1R |
| 4e | OMe | CMe3 | H | 60 | 110 | -54 | 3R4S | (+)-1S |
| 4f | OMe | H | CMe3 | 60 | 105 | -63.3 | 3S4S | (+)-1S |
| 5a | OMe | Me | Me | 97 | Oil | +94.7 | 4R | (-)-1R |
| 5b | Cl | Me | Me | 98 | Oil | +74.7 | 4R | (-)-1R |
| 5c | OMe | Me | H | 97 | Oil | +67.1 | 3S4R | (-)-1R |
| 5d | OMe | H | Me | -- | -- | -- | -- | (-)-1R |
| 5e | OMe | CMe3 | H | 98 | Oil | -60.4 | 3R4S | (+)-1S |
| 5f | OMe | H | CMe3 | 98 | Oil | -75.2 | 3S4S | (+)-1S |
a New stereogenic centers
Among the many routes for the access to this class of compunds [8]-[10], we have chosen the intramolecular ring closure of β-hydroxyacids, obtained by addition of aldehydes to doubly lithiated carboxylic acids [11].
In this paper (for preliminary results see [12]) we report the results on the stereoselective synthesis of some Cr(CO)3 complexed β-hydroxyacids 3 and their cyclization to new Cr(CO)3 complexed β- lactones 4 using both the racemic and the optically pure tricarbonyl(2-methoxybenzaldeyde) [13] or (2- chlorobenzaldehyde) chromium [14] 1 and the dianion of the appropriate acid 2 (Scheme).
Results and Discussion
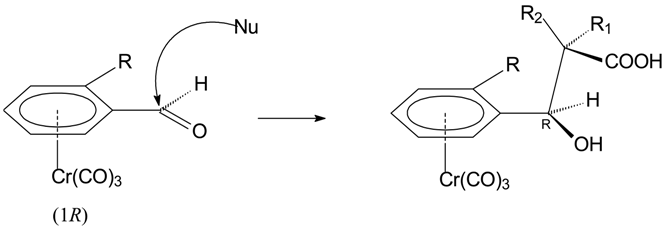
The addition of optically pure benzaldehyde 1 (R=OMe or R=Cl) to a dianion of isobutyric acid (R1=R2=Me), generated with LDA at –50 °C in THF solution, affords in a few minutes and after usual work-up products 3a,b respectively in good yields and each as a single diastereoisomer. When the doubly lithiated propionic (R1=H, R2=Me) or t-butyl propionic (R1=H, R2=CMe3) acids were used, a mixture of threo and erythro hydroxyacids 3c,d and 3e,f were obtained each one as a single diastereoisomer in 1:1 ratio.
The reaction of benzaldehyde with t-butyl propionic acid under kinetic conditions is reported to afford a mixture 60:40 of threo and erythro isomers [15,16]. The small difference in the diastereoselectivity reported there could be due to the steric hindrance of the substituent (OMe) in the ortho position of the complexed aldehyde and/or to the electron withdrawing effect of Cr(CO)3 unit. Therefore, we have repeated the reaction in the usual experimental conditions starting first from the uncomplexed 2-methoxybenzaldehyde and t-butyl propionic acid, obtaining a 60:40 threo/ erythro ratio, exactly the same for the unsubstituted benzaldehyde. It is well known that the Cr(CO)3 unit, responsible for asymmetric induction, is also an electron withdrawing group, similar to NO2 in the para position on the aromatic ring. We have then repeated the condensation with the t-butyl propionic acid and p-NO2 benzaldehyde obtaining the two diastereomeric threo/erythro hydroxyacids in a 54:46 ratio. These results seem to demonstrate that diastereoselection is more influenced by electronic rather than steric factors; the increase in electrophilicity of the carbonyl carbon of the aldehyde could activate the already fast reaction and favour the formation of the less preferred erythro isomer.
The threo and erythro isomers 3c,d and 3e,f were easily separated by column chromatography and the relative stereochemistry of C-2 and C-3 has been assigned on the basis of the 1H-NMR coupling constants of the protons [17].
Treatment of 3a-f with PhSO2Cl in Py [18] affords the corresponding Cr(CO)3 complexed lactones 4a-c,e,f with the exception of the threo 3d that gives rise to the corresponding α-methyl-β-(2- methoxyphenyl) chromium propenic acid (35% yield) by dehydration and the styryl derivative (25% yield) by the known CO2 elimination [19] of the corresponding lactone.
The exposure to air and sunlight of a solution of 4a-c,e,f produces, in nearly quantitative yield, the uncomplexed 2-oxetanones 5a-c,e,f in e.e. greater than 98% (determined by 1H-NMR using chiral shift reagents).
The use of the chiral substrates 1 allows the forecast of the stereochemistry of the new stereogenic centre, created by the attack of the carboxylic acid dianion on the formyl centre. In fact, on the basis of stereochemical model for ortho-substituted benzaldehydes [20], the new centre will be (R) if the optically pure aldehyde is (-)-(R), independent of the nature of the ortho substituent R on the complexed aromatic ring, as the preferred conformation for the aldehyde has the CO group anti with respect to the substituent R. Furthermore, we can assign the absolute configuration to the β-lactones, as the ring closure, obtained by activation of carboxylic function [18], does not change the stereochemistry of the centres of the hydroxyacids.
![Molecules 06 00013 i001]()
Conclusions
Optically pure tricarbonylchromium benzaldehydes, through the condensation with dianions of some carboxylic acids, represent useful alternative chiral auxiliaries in the synthesis of optically pure 2-oxetanones. The absolute configuration of the products can be inferred from the configuration of the starting aldehydes.
Experimental Section
General
All reactions were performed under nitrogen atmosphere. Thermolysis with hexacarbonylchromium(0) were carried out in a round-bottomed flask, equipped with a Liebig air condenser and a water condenser. All chemicals were used as obtained from commercial sources. Column chromatography and TLC were carried out using respectively silica gel 60 and silica gel 60 F254 pre-coated pathlength. The melting points were measured using a Büchi 510 apparatus and are uncorrected. The IR spectra were recorded using a 1725X FTIR spectrometer. NMR spectra were recorded in CDCl3 using a Varian XL 300, Bruker AC 300 and AMX 300 spectrometers. Evaluation of enantiomeric excess was performed using Eu(hfc)3,(tris-[3-(heptafluoropropyl-hydroxymethylene)-(+)- camphorato]europium(III) salt. The optical rotations were measured using a Perkin-Elmer 241 Polarimeter, with a 1 dm pathlength at 25 °C. The racemic compounds were prepared as previously reported [13]-[14] The optically pure complexed benzaldehydes were obtained by resolution of the corresponding racemic substrate using a known procedure [13]-[14] Elemental analysis for all compounds is consistent with the proposed structure.
General procedure for preparation of the β-hydroxy acids 3. A solution of 3.3 mmol of the appropriate carboxylic acid 2 in THF (3 mL) was added at –50 °C to a solution of 6.6 mmol of LDA, generated in THF (15 mL) following the usual procedure. The temperature was allowed to raise to 30 °C and the mixture was stirred for 1h. After cooling again the mixture to –50 °C, a solution of complexed benzaldehyde 1 [13]-[14] (1.1 mmol in 2 mL of THF) was added dropwise. The red colour of the aldehyde immediately disappeared and TLC shows the end of the reaction. Cold water (25 mL) was then added and the mixture was extracted first with diethyl ether (2 x 15 mL) and then with AcOEt (2 x 25 mL). The combined organic solvents were washed with water (3 x 30 mL), dried and evaporated under reduced pressure. The crude yellow oil becomes solid upon treatment with petroleum ether. Physical data and spectroscopic results are collected in Table 1 and Table 2. Elemental analyses: 3a Anal. calcd. for C15H16CrO7 (360.289) C, 50.01; H, 4.48. Found C, 49.98; H, 4.49. 3b Anal. calcd. for C14H13ClCrO6 (364.707) C, 46.11; H, 3.59. Found C, 46.13; H, 3.60. 3c Anal. calcd. for C14H14CrO7 (346.262) C, 48.56; H, 4.08. Found C, 48.58; H, 4.07. 3d Anal. calcd. for C14H14CrO7 (346.262) C, 48.56; H, 4.08. Found C, 48.54; H, 4.09. 3e Anal. calcd. for C17H20CrO7 (388.343) C, 52.58; H, 5.19. Found C, 52.60; H, 5.17. 3f Anal. calcd. for C17H20CrO7 (388.343) C, 52.58; H, 5.19. Found C, 52.57; H, 5.20.
General procedure for the synthesis of Cr(CO)3 complexed β-lactones 4. To a solution of appropriate hydroxyacid 3 (0.43 mmol) in pyridine freshly distilled from NaOH (1 mL) and cooled to 0 °C, benzenesulfonyl chloride (0.11 mL, 0.86 mmol) was added. The reaction is complete in 10-45 min and the solution is then poured into 4 mL of water/chopped ice. The product was extracted with diethyl ether (3 x 10 mL) and the combined etheral solution was washed with a saturated solution of NaHCO3 and, after evaporation of the solvent, the yellow oil became crystalline upon addition of petroleum ether and was subsequently filtered. Elemental analyses: 4a. Anal. calcd. for C15H14CrO6 (342.273) C, 52.63; H, 4.12. Found C, 52.61; H, 4.13. 4b Anal. calcd. for C14H11ClCrO5 (346.691) C, 48.50; H, 3.20. Found C, 48.53; H, 3.19. 4c Anal. calcd. for C14H12CrO6 (328.246) C, 51.23; H, 3.69. Found C, 51.25; H, 3.70. 4e Anal. calcd. for C17H18CrO6 (370.327) C, 55.14; H, 4.90. Found C, 55.12; H, 4.91. 4f. Anal. calcd. for C17H18CrO6 (370.327) C, 55.14; H, 4.90. Found C, 55.15; H, 4.88.
General procedure for decomplexation of 4 to β-lactones 5. A solution of 4 in CH2Cl2 was exposed to air and sunlight for 4-6h until the complexed substrate disappeared (TLC). The solvent was evaporated and the residue treated with diethyl ether and filtered over a Celite pad. Evaporation of the solvent affords in nearly quantitative yield the β-lactones 5 as colourless oils. (for physical and spectroscopic data see Table 1 and Table 2). Elemental analyses: 5a Anal. calcd. for C12H14O3 (206.244) C, 69.88; H, 6.84. Found C, 69.90; H, 6.85. 5b Anal. calcd. for C11H11ClO2 (210.662) C, 62.72; H, 5.26. Found C, 62.74; H, 5.27. 5c Anal. calcd. for C11H12O3 (192.217) C, 68.74; H, 6.29. Found C, 68.76; H, 6.28. 5e Anal. calcd. for C14H18O3 (234.298) C, 71.77; H, 7.74. Found C, 71.79; H, 7.76. 5f Anal. calcd. for C14H18O3 (234.298) C, 71.77; H, 7.74. Found C, 71.75; H, 7.73.
| Prod. | 1H NMR (ppm) | Prod. | 1H NMR (ppm) |
|---|---|---|---|
| 3aa | 1.18 (s, 3H); 1.22 (s, 3H); 3.75 (s, 3H); 4.93 (t, 1H, J=6.1 Hz); 5.01 (d, 1H, J=6.5 Hz); 5.19 (s, 1H); 5.57 (t, 1H, J=6.1 Hz); 5.84 (d, 1H, J=6.1 Hz) | 4cc | 0.8 (d, 3H, J=7.8 Hz); 3.75 (s, 3H); 4.0 (dq, 1H, J=7.8 and 6.1 Hz); 4.92 (t, 1H, J=6.2 Hz); 5.55 (d, 1H, J=6.3 Hz); 5.56 (d, 1H, J=6.1 Hz); 5.8 (t, 1H, J=6.3 Hz); 6.8 (d, 1H, J=6.2 Hz) |
| 3ba | 1.18 (s, 3H); 1.21 (s, 3H); 5.09 (t, 1H, J=6.2 Hz); 5.19 (s, 1H); 5.38 (d, 1H, J=6.3 Hz); 5.47 (t, 1H, J=6.3 Hz); 5.79 (d, 1H, J=6.2 Hz) | 4ec | 0.95 (s, 9H); 3.75 (d, 1H, J=6.6 Hz); 3.79 (s, 3H); 4.91 (t, 1H, J=6.2 Hz); 5.12 (d, 1H, J=6.3 Hz); 5.55 (t, 1H, J=6.3 Hz); 5.6 (d, 1H, J=6.6 Hz); 5.9 (d, 1H, J=6.2 Hz) |
| 3ca | 0.8 (d, 3H, J=7.2 Hz); 2.4 (dq, 1H, J=1.7 and 7.2 Hz); 3.8 (s, 3H); 4.95 (d, 1H, J=1.7 Hz); 5.25 (t, 1H, J=6.3 Hz); 5.6 (d, 1H, J=6.8 Hz); 5.82 (t, 1H, J=6.8 Hz); 6.0 (d, 1H, J=6.3 Hz) | 4fc | 1.1 (s, 9H); 3.5 (d, 1H, J=4.0 Hz); 4.0 (s, 3H); 4.88 (t, 1H, J=6.2 Hz); 5.02 (d, 1H, J=6.8 Hz); 5.18 (d, 1H, J=4.0 Hz); 5.6 (t, 1H, J=6.8 Hz); 6.71 (d, 1H, J=6.2 Hz) |
| 3da | 1.15 (d, 3H, J=7.1 Hz); 2.25 (dq, 1H, J=3.5 and 7.1 Hz); 3.8 (s, 3H); 4.4 (d, 1H, J=3.5 Hz); 5.25 (t, 1H, J=6.3 Hz); 5.58 (d, 1H, J=6.8 Hz); 5.8 (t, 1H, J=6.8 Hz); 6.98 (d, 1H, J=6.3 Hz) | 5ac | 0.88 (s, 3H); 1.6 (s, 3H); 3.8 (s, 3H); 5.45 (s, 1H); 6.8-7.4 (m, 4H) |
| 3ea | 1.12 (s, 9H); 2.62 (d, 1H, J=7.7 Hz); 3.72 (s, 3H); 4.88 (t, 1H, J=6.2 Hz); 4.96 (d, 1H, J=6.5 Hz); 5.14 (d, 1H, J=7.7 Hz); 5.56 (t, 1H, J=6.5 Hz); 5.92 (d, 1H, J=6.2 Hz) | 5bc | 0.9 (s, 3H); 1.65 (s, 3H); 5.48 (s, 1H); 7.1-7.5 (m, 4H) |
| 3fa | 1.17 (s, 9H); 2.59 (d, 1H, J=2 Hz); 3.79 (s, 3H); 4.88 (t, 1H, J=6.2 Hz); 5.19 (br s, 1H)b; 5.2 (d, 1H, J=6.5 Hz); 5.49 (t, 1H, J=6.5 Hz); 5.76 (d, 1H, J=6.2 Hz) | 5cc | 0.9 (d, 3H, J=7.6 Hz); 3.8 (s, 3H); 4.0 (dq, 1H, J=7.6 and 6.5 Hz); 5.8 (d, 1H, J=6.5 Hz); 6.95-7.4 (m, 4H) |
| 4ac | 1.05 (s, 3H); 1.5 (s, 3H); 3.77 (s, 3H); 4.95 (t, 1H, J=6.3 Hz); 5.07 (d, 1H, J=6.6 Hz); 5.4 (s, 1H); 5.5 (t, 1H, J=6.6 Hz); 5.77 (d, 1H, J=6.3 Hz) | 5ec | 0.81 (s, 9H); 3.5 (d, 1H, J=6.7 Hz); 3.82 (s, 3H); 5.82 (d, 1H, J=6.7 Hz); 6.8-7.5 (m, 4H) |
| 4bc | 1.08 (s, 3H); 1.6 (s, 3H); 5.15 (t, 1H, J=6.2 Hz); 5.31 (s, 1H); 5.41 (t, 1H, J=6.3 Hz); 5.49 (d, 1H, J=6.3 Hz); 5.68 (d, 1H, J=6.2 Hz) | 5fc | 1.1 (s, 9H); 3.4 (d, 1H, J=4.1 Hz); 3.8 (s, 3H); 5.55 (d, 1H, J=4.1 Hz); 6.8-7.3 (m, 4H) |
a) DMSO solutionb) Irradiation produced a sharp singlet for the proton at δ 2.59c) CDCl3 solution
Acknowledgments
Thanks are due to MURST and CNR-Rome for financial support and to Prof. Stefano Maiorana for helpful discussions.
References
- Pommier, A.; Pons, J. M. The synthesis of natural 2-oxetanones. Synthesis 1995, 729–744. [Google Scholar] [CrossRef]
- Pommier, A.; Pons, J.M. Recent advances in β-lactone chemistry. Synthesis 1993, 441–459. [Google Scholar] [CrossRef]
- Abe, H.; Matsubara, I.; Doi, Y. Physical properties and enzymatic degradability of polymer blends of bacterial poly[(R)-3-hydroxybutyrate] and poly[(R,S)-3-hydroxybutyrate] stereoisomers. Macromolecules 1995, 28, 844–853. [Google Scholar] [CrossRef]
- Baldoli, C.; Del Buttero, P.; Licandro, E.; Maiorana, S.; Papagni, A. Tricarbonyl(η6-arene)chromium(0) complexes as chiral auxiliaries. Homochiral β-lactams synthesis “via” [2+2] cycloaddition reaction. Tetrahedron: Asymmetry 1994, 5, 809–812. [Google Scholar] [CrossRef]
- Baldoli, C.; Del Buttero, P.; Licandro, E.; Papagni, A.; Pilati, T. Tricarbonyl(η6-arene)chromium(0) complexes as chiral auxiliaries. Asymmetric Synthesis of β-aminoesters and β- lactams by Reformatsky condensation. Tetrahedron 1996, 52, 4849–4856. [Google Scholar] [CrossRef]
- Baldoli, C.; Del Buttero, P.; Perdicchia, D.; Pilati, T. Stereoselective synthesis of β-sultams using chiral tricarbonyl(η6-arene)chromium(0) complexes. Tetrahedron 1999, 55, 14089–14096. [Google Scholar] [CrossRef]
- Del Buttero, P.; Baldoli, C.; Molteni, G.; Pilati, T. Stereoselective synthesis of a new enantiopure tricyclic β-lactam <<via>> tricarbonyl(η6-arene)chromium(0) complex. Tetrahedron: Asymmetry 2000, 11, 1927–1941. [Google Scholar] [CrossRef]
- Yang, H. W.; Romo, D. Methods for the synthesis of optically active β-lactones (2-oxetanones). Tetrahedron 1999, 55, 6403–6434. [Google Scholar] [CrossRef]
- Case-Green, S. C.; Davies, S. G.; Hedgecock, C. J. R. Asymmetric Synthesis of Homochiral β- lactones via the Iron Chiral Auxiliary. Synlett 1991, 779–780. [Google Scholar] [CrossRef]
- Case-Green, S. C.; Davies, S. G.; Hedgecock, C. J. R. Asymmetric Synthesis of (-)- Tetrahydrolipstatin. Synlett 1991, 781–782. [Google Scholar] [CrossRef]
- Petragnani, N.; Yonashiro, M. The reactions of dianion of carboxylic acids and ester enolates. Synthesis 1982, 521–578. [Google Scholar] [CrossRef]
- Del Buttero, P.; Baldoli, C. Arenechromiumtricarbonyl derivatives as chiral auxiliaries: synthesis of enantiomerically pure β-lactones. First International Electronic Conference on Synthetic Organic Chemistry (ECSOC-1), 1997. [Google Scholar]
- Solladié-Cavallo, A.; Solladié, G.; Tsamo, E. Chiral (Arene)tricarbonylchromium Complexes: Resolution of Aldehydes. J. Org. Chem. 1979, 44, 4189–4191. [Google Scholar] [CrossRef]
- Top, S.; Jaouen, G.; Baldoli, C.; Del Buttero, P.; Maiorana, S. Microbial resolution of organometallic planar chirality. Enantioselective reduction of ortho and meta-substituted tricarbonylchromium benzaldhydes by bakers’ yeast. J. Organomet. Chem. 1991, 413, 125–135. [Google Scholar] [CrossRef]
- Mulzer, J.; Segner, J.; Brüntrup, G. Stereoselective synthesis of threo 3-hydroxycarboxylic acids. Stereochemistry of an aldol type addition under kinetic and thermodynamic control. Tetrahedron Lett. 1977, 52, 4651–4654. [Google Scholar] [CrossRef]
- Mulzer, J.; Zippel, M.; Brüntrup, G.; Segner, J.; Finke, J. Stereochemistry of the addition of carboxylic acid dianions to aldehydes under kinetic and thermodynamic control. Synthesis and configurational assignment of 2,3-disubstituted threo- and erythro-3-hydroxycarboxylic acids. Liebigs Ann. Chem. 1980, 1108–1134. [Google Scholar] [CrossRef]
- Canceill, J.; Basselier, J.J.; Jacques, J. Sur la stéréochimie de la réaction de Réformatsky. Spectres IR et spectres de RMN des β-hydroxyesters obtenus. Dosage de leurs mélanges. Bilan des résultats. Bull. Chim. Soc. Fr. 1967, 3, 1024–1030. [Google Scholar]
- Adam, W.; Baeza, J.; Liu, J.C. Stereospecific introduction of double bonds via thermolysis of β- lactones. J. Am. Chem. Soc. 1972, 94, 2000–2006. [Google Scholar] [CrossRef]
- Mulzer, J.; Pointner, A.; Chucholowski, A.; Brüntrup, G. Threo 3-hydroxycarboxylic acids as key intermediates in a highly stereoselective synthesis of (Z) and (E)-olefins and enol ethers. J. Chem. Soc. Chem. Commun. 1979, 52–54, and ref. therein. [Google Scholar] [CrossRef]
- Solladié-Cavallo, A. Chiral arene-chromium-carbonyl complexes in asymmetric synthesis. Adv. Met.-Org. Chem. 1989, 1, 99–133. [Google Scholar]
- Sample Availability: Samples not available.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Buttero, P.D.; Montrasio, D. Tricarbonyl(h6-Arene)Chromium(0) Complexes as Chiral Auxiliaries: Asymmetric Synthesis of b-Lactones. Molecules 2001, 6, 13-20. https://doi.org/10.3390/60100013
AMA Style
Buttero PD, Montrasio D. Tricarbonyl(h6-Arene)Chromium(0) Complexes as Chiral Auxiliaries: Asymmetric Synthesis of b-Lactones. Molecules. 2001; 6(1):13-20. https://doi.org/10.3390/60100013
Chicago/Turabian StyleButtero, Paola Del, and Deborah Montrasio. 2001. "Tricarbonyl(h6-Arene)Chromium(0) Complexes as Chiral Auxiliaries: Asymmetric Synthesis of b-Lactones" Molecules 6, no. 1: 13-20. https://doi.org/10.3390/60100013