The Yin and Yang of VEGF and PEDF: Multifaceted Neurotrophic Factors and Their Potential in the Treatment of Parkinson’s Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Parkinson’s Disease
2.1. Onset and Symptoms
2.2. A Critical and Timely Field of Research
3. Vascular Endothelial Growth Factor—A Versatile Growth Factor with Pathophysiological Implications and Therapeutic Potential
3.1. VEGF Isoforms and Structure
3.2. VEGF Receptors and Co-Receptors
3.3. VEGF Has Various Physiological Roles
3.4. Pathological Roles
3.5. VEGFs, Neuroprotection, and Parkinson’s Disease
4. PEDF: A Widely Expressed, Pleiotropic Molecule with Anti-angiogenic, Anti-tumorigenic, and Neuroprotective Potential
4.1. PEDF Structure
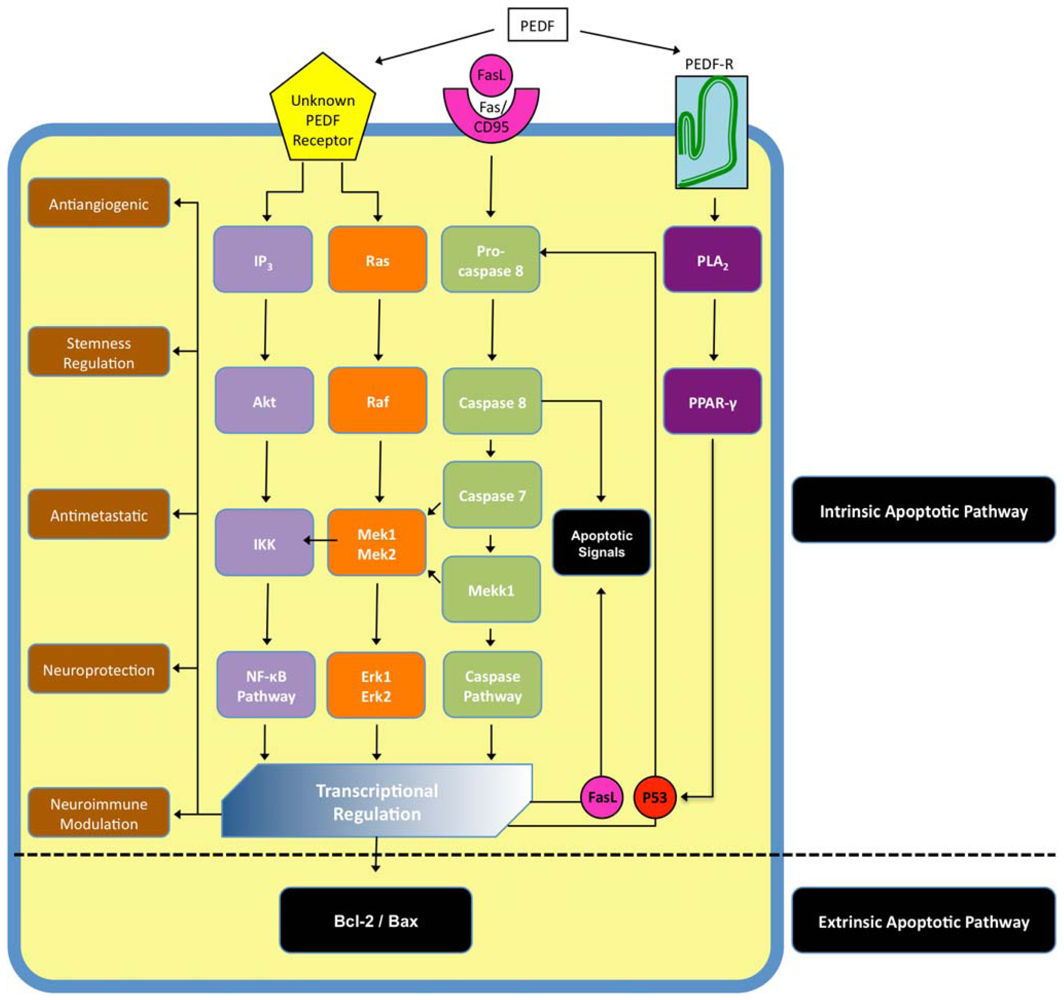
4.2. PEDF Receptors and Cell Signaling
4.3. PEDF, Biological Function, Neuroprotection, and Parkinson’s Disease
5. The Yin and Yang of VEGF and PEDF
6. Conclusion
Acknowledgements
References
- Rezak, M. Current pharmacotherapeutic treatment options in Parkinson's disease. Disease-a- Month 2007, 53, 214–222. [Google Scholar]
- Olanow, CW; Stern, MB; Sethi, K. The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology 2009, 72, S1–S136. [Google Scholar]
- Nutt, JG. Motor fluctuations and dyskinesia in Parkinson's disease. Parkinsonism Relat. Disord 2001, 8, 101–108. [Google Scholar]
- Forno, LS. Neuropathology of Parkinson's disease. J. Neuropathol. Exp. Neurol 1996, 55, 259–272. [Google Scholar]
- Fahn, S; Sulzer, D. Neurodegeneration and neuroprotection in Parkinson disease. NeuroRx 2004, 1, 139–154. [Google Scholar]
- Fernandez-Espejo, E. Pathogenesis of Parkinson's disease: Prospects of neuroprotective and restorative therapies. Mol. Neurobiol 2004, 29, 15–30. [Google Scholar]
- Farrer, MJ. Genetics of Parkinson disease: Paradigm shifts and future prospects. Nat. Rev. Genet 2006, 7, 306–318. [Google Scholar]
- Jankovic, J. Parkinson's disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar]
- Drucker-Colin, R; Verdugo-Diaz, L. Cell transplantation for Parkinson's disease: Present status. Cell Mol. Neurobiol 2004, 24, 301–316. [Google Scholar]
- Kordower, JH. In vivo gene delivery of glial cell line--derived neurotrophic factor for Parkinson's disease. Ann. Neurol 2003, 53, S120–S132, discussion S132–S134. [Google Scholar]
- Lindvall, O; Bjorklund, A. Cell therapy in Parkinson's disease. NeuroRx 2004, 1, 382–393. [Google Scholar]
- Olanow, CW; Goetz, CG; Kordower, JH; Stoessl, AJ; Sossi, V; Brin, MF; Shannon, KM; Nauert, GM; Perl, DP; Godbold, J; Freeman, TB. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson's disease. Ann. Neurol 2003, 54, 403–414. [Google Scholar]
- Kordower, JH; Chu, Y; Hauser, RA; Freeman, TB; Olanow, CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat. Med 2008, 14, 504–506. [Google Scholar]
- Li, JY; Englund, E; Holton, JL; Soulet, D; Hagell, P; Lees, AJ; Lashley, T; Quinn, NP; Rehncrona, S; Bjorklund, A; Widner, H; Revesz, T; Lindvall, O; Brundin, P. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med 2008, 14, 501–503. [Google Scholar]
- Mendez, I; Vinuela, A; Astradsson, A; Mukhida, K; Hallett, P; Robertson, H; Tierney, T; Holness, R; Dagher, A; Trojanowski, JQ; Isacson, O. Dopamine neurons implanted into people with Parkinson's disease survive without pathology for 14 years. Nat. Med 2008, 14, 507–509. [Google Scholar]
- Kirik, D; Georgievska, B; Bjorklund, A. Localized striatal delivery of GDNF as a treatment for Parkinson disease. Nat. Neurosci 2004, 7, 105–110. [Google Scholar]
- Peterson, AL; Nutt, JG. Treatment of Parkinson's disease with trophic factors. Neurotherapeutics 2008, 5, 270–280. [Google Scholar]
- Grothe, C; Timmer, M. The physiological and pharmacological role of basic fibroblast growth factor in the dopaminergic nigrostriatal system. Brain Res. Rev 2007, 54, 80–91. [Google Scholar]
- Eidelberg, D. Metabolic brain networks in neurodegenerative disorders: A functional imaging approach. Trends Neurosci 2009, 32, 548–557. [Google Scholar]
- Graeber, MB. Biomarkers for Parkinson's disease. Exp. Neurol 2009, 216, 249–253. [Google Scholar]
- Maetzler, W; Liepelt, I; Berg, D. Progression of Parkinson's disease in the clinical phase: Potential markers. Lancet Neurol 2009, 8, 1158–1171. [Google Scholar]
- Senger, DR; Galli, SJ; Dvorak, AM; Perruzzi, CA; Harvey, VS; Dvorak, HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar]
- Ferrara, N; Henzel, WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun 1989, 161, 851–858. [Google Scholar]
- Ferrara, N; Gerber, HP; LeCouter, J. The biology of VEGF and its receptors. Nat. Med 2003, 9, 669–676. [Google Scholar]
- Houck, KA; Ferrara, N; Winer, J; Cachianes, G; Li, B; Leung, DW. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol. Endocrinol 1991, 5, 1806–1814. [Google Scholar]
- Baldwin, ME; Roufail, S; Halford, MM; Alitalo, K; Stacker, SA; Achen, MG. Multiple forms of mouse vascular endothelial growth factor-D are generated by RNA splicing and proteolysis. J. Biol. Chem 2001, 276, 44307–44314. [Google Scholar]
- Houck, KA; Leung, DW; Rowland, AM; Winer, J; Ferrara, N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem 1992, 267, 26031–26037. [Google Scholar]
- Keyt, BA; Berleau, LT; Nguyen, HV; Chen, H; Heinsohn, H; Vandlen, R; Ferrara, N. The carboxyl-terminal domain (111–165) of vascular endothelial growth factor is critical for its mitogenic potency. J. Biol. Chem 1996, 271, 7788–7795. [Google Scholar]
- Soker, S; Fidder, H; Neufeld, G; Klagsbrun, M. Characterization of novel vascular endothelial growth factor (VEGF) receptors on tumor cells that bind VEGF165 via its exon 7-encoded domain. J. Biol. Chem 1996, 271, 5761–5767. [Google Scholar]
- Shibuya, M; Yamaguchi, S; Yamane, A; Ikeda, T; Tojo, A; Matsushime, H; Sato, M. Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene 1990, 5, 519–524. [Google Scholar]
- Terman, BI; Carrion, ME; Kovacs, E; Rasmussen, BA; Eddy, RL; Shows, TB. Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene 1991, 6, 1677–1683. [Google Scholar]
- de Vries, C; Escobedo, JA; Ueno, H; Houck, K; Ferrara, N; Williams, LT. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992, 255, 989–991. [Google Scholar]
- Davis-Smyth, T; Chen, H; Park, J; Presta, LG; Ferrara, N. The second immunoglobulin-like domain of the VEGF tyrosine kinase receptor Flt-1 determines ligand binding and may initiate a signal transduction cascade. EMBO J 1996, 15, 4919–4927. [Google Scholar]
- Terman, BI; Dougher-Vermazen, M; Carrion, ME; Dimitrov, D; Armellino, DC; Gospodarowicz, D; Bohlen, P. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem. Biophys. Res. Commun 1992, 187, 1579–1586. [Google Scholar]
- Hiratsuka, S; Minowa, O; Kuno, J; Noda, T; Shibuya, M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9349–9354. [Google Scholar]
- Olsson, AK; Dimberg, A; Kreuger, J; Claesson-Welsh, L. VEGF receptor signalling - in control of vascular function. Nat. Rev. Mol. Cell Biol 2006, 7, 359–371. [Google Scholar]
- Neufeld, G; Cohen, T; Shraga, N; Lange, T; Kessler, O; Herzog, Y. The neuropilins: Multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends Cardiovasc. Med 2002, 12, 13–19. [Google Scholar]
- Soker, S; Takashima, S; Miao, HQ; Neufeld, G; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar]
- Ferrara, N; Carver-Moore, K; Chen, H; Dowd, M; Lu, L; O'Shea, KS; Powell-Braxton, L; Hillan, KJ; Moore, MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380, 439–442. [Google Scholar]
- Carmeliet, P; Ferreira, V; Breier, G; Pollefeyt, S; Kieckens, L; Gertsenstein, M; Fahrig, M; Vandenhoeck, A; Harpal, K; Eberhardt, C; Declercq, C; Pawling, J; Moons, L; Collen, D; Risau, W; Nagy, A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380, 435–439. [Google Scholar]
- Shalaby, F; Rossant, J; Yamaguchi, TP; Gertsenstein, M; Wu, XF; Breitman, ML; Schuh, AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376, 62–66. [Google Scholar]
- Gerber, HP. VEGF is required for growth and survival in neonatal mice. Development 1998, 126, 1149–1159. [Google Scholar]
- Gerber, HP; Malik, AK; Solar, GP; Sherman, D; Liang, XH; Meng, G; Hong, K; Marsters, JC; Ferrara, N. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 2002, 417, 954–958. [Google Scholar]
- Zelzer, E; Mamluk, R; Ferrara, N; Johnson, RS; Schipani, E; Olsen, BR. VEGFA is necessary for chondrocyte survival during bone development. Development 2004, 131, 2161–2171. [Google Scholar]
- Inai, T; Mancuso, M; Hashizume, H; Baffert, F; Haskell, A; Baluk, P; Hu-Lowe, DD; Shalinsky, DR; Thurston, G; Yancopoulos, GD; McDonald, DM. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am. J. Pathol 2004, 165, 35–52. [Google Scholar]
- Jain, RK. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med 2001, 7, 987–989. [Google Scholar]
- Kamba, T; Tam, BY; Hashizume, H; Haskell, A; Sennino, B; Mancuso, MR; Norberg, SM; O'Brien, SM; Davis, RB; Gowen, LC; Anderson, KD; Thurston, G; Joho, S; Springer, ML; Kuo, CJ; McDonald, DM. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am. J. Physiol. Heart Circ. Physiol 2006, 290, H560–H576. [Google Scholar]
- Bicknell, R; Harris, AL. Anticancer strategies involving the vasculature: Vascular targeting and the inhibition of angiogenesis. Semin Cancer Biol 1992, 3, 399–407. [Google Scholar]
- Augustin, HG. Antiangiogenic tumour therapy: Will it work? Trends Pharmacol Sci 1998, 19, 216–222. [Google Scholar]
- Ho, QT; Kuo, CJ. Vascular endothelial growth factor: Biology and therapeutic applications. Int. J. Biochem. Cell Biol 2007, 39, 1349–1357. [Google Scholar]
- Zhang, F; Tang, Z; Hou, X; Lennartsson, J; Li, Y; Koch, AW; Scotney, P; Lee, C; Arjunan, P; Dong, L; Kumar, A; Rissanen, TT; Wang, B; Nagai, N; Fons, P; Fariss, R; Zhang, Y; Wawrousek, E; Tansey, G; Raber, J; Fong, GH; Ding, H; Greenberg, DA; Becker, KG; Herbert, JM; Nash, A; Yla-Herttuala, S; Cao, Y; Watts, RJ; Li, X. VEGF-B is dispensable for blood vessel growth but critical for their survival, and VEGF-B targeting inhibits pathological angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 6152–6157. [Google Scholar]
- Casella, I; Feccia, T; Chelucci, C; Samoggia, P; Castelli, G; Guerriero, R; Parolini, I; Petrucci, E; Pelosi, E; Morsilli, O; Gabbianelli, M; Testa, U; Peschle, C. Autocrine-paracrine VEGF loops potentiate the maturation of megakaryocytic precursors through Flt1 receptor. Blood 2003, 101, 1316–1323. [Google Scholar]
- Jin, K; Zhu, Y; Sun, Y; Mao, XO; Xie, L; Greenberg, DA. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11946–11950. [Google Scholar]
- Sun, Y; Jin, K; Childs, JT; Xie, L; Mao, XO; Greenberg, DA. Vascular endothelial growth factor-B (VEGFB) stimulates neurogenesis: Evidence from knockout mice and growth factor administration. Dev. Biol 2006, 289, 329–335. [Google Scholar]
- Belgore, F; Blann, A; Neil, D; Ahmed, AS; Lip, GY. Localisation of members of the vascular endothelial growth factor (VEGF) family and their receptors in human atherosclerotic arteries. J. Clin. Pathol 2004, 57, 266–272. [Google Scholar]
- Hedman, M; Muona, K; Hedman, A; Kivela, A; Syvanne, M; Eranen, J; Rantala, A; Stjernvall, J; Nieminen, MS; Hartikainen, J; Yla-Herttuala, S. Eight-year safety follow-up of coronary artery disease patients after local intracoronary VEGF gene transfer. Gene Ther 2009, 16, 629–634. [Google Scholar]
- Dvorak, HF; Brown, LF; Detmar, M; Dvorak, AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol 1995, 146, 1029–1039. [Google Scholar]
- Berger, DP; Herbstritt, L; Dengler, WA; Marme, D; Mertelsmann, R; Fiebig, HH. Vascular endothelial growth factor (VEGF) mRNA expression in human tumor models of different histologies. Ann. Oncol 1995, 6, 817–825. [Google Scholar]
- Wood, JM; Bold, G; Buchdunger, E; Cozens, R; Ferrari, S; Frei, J; Hofmann, F; Mestan, J; Mett, H; O'Reilly, T; Persohn, E; Rosel, J; Schnell, C; Stover, D; Theuer, A; Towbin, H; Wenger, F; Woods-Cook, K; Menrad, A; Siemeister, G; Schirner, M; Thierauch, KH; Schneider, MR; Drevs, J; Martiny-Baron, G; Totzke, F. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res 2000, 60, 2178–2189. [Google Scholar]
- Holash, J; Davis, S; Papadopoulos, N; Croll, SD; Ho, L; Russell, M; Boland, P; Leidich, R; Hylton, D; Burova, E; Ioffe, E; Huang, T; Radziejewski, C; Bailey, K; Fandl, JP; Daly, T; Wiegand, SJ; Yancopoulos, GD; Rudge, JS. VEGF-Trap: A VEGF blocker with potent antitumor effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar]
- Herbst, RS. Therapeutic options to target angiogenesis in human malignancies. Expert Opin. Emerg. Drugs 2006, 11, 635–650. [Google Scholar]
- Boer, K; Troost, D; Spliet, WG; van Rijen, PC; Gorter, JA; Aronica, E. Cellular distribution of vascular endothelial growth factor A (VEGFA) and B (VEGFB) and VEGF receptors 1 and 2 in focal cortical dysplasia type IIB. Acta Neuropathol 2008, 115, 683–696. [Google Scholar]
- Poesen, K; Lambrechts, D; Van Damme, P; Dhondt, J; Bender, F; Frank, N; Bogaert, E; Claes, B; Heylen, L; Verheyen, A; Raes, K; Tjwa, M; Eriksson, U; Shibuya, M; Nuydens, R; Van Den Bosch, L; Meert, T; D'Hooge, R; Sendtner, M; Robberecht, W; Carmeliet, P. Novel role for vascular endothelial growth factor (VEGF) receptor-1 and its ligand VEGF-B in motor neuron degeneration. J. Neurosci 2008, 28, 10451–10459. [Google Scholar]
- Wada, K; Arai, H; Takanashi, M; Fukae, J; Oizumi, H; Yasuda, T; Mizuno, Y; Mochizuki, H. Expression levels of vascular endothelial growth factor and its receptors in Parkinson's disease. Neuroreport 2006, 17, 705–709. [Google Scholar]
- Falk, T; Zhang, S; Sherman, SJ. Vascular endothelial growth factor B (VEGF-B) is upregulated and exogenous VEGF-B is neuroprotective in a culture model of Parkinson's disease. Mol. Neurodegener 2009, 4, 49. [Google Scholar]
- Ryu, JK; Cho, T; Choi, HB; Wang, YT; McLarnon, JG. Microglial VEGF receptor response is an integral chemotactic component in Alzheimer's disease pathology. J. Neurosci 2009, 29, 3–13. [Google Scholar]
- Bogaert, E; Van Damme, P; Poesen, K; Dhondt, J; Hersmus, N; Kiraly, D; Scheveneels, W; Robberecht, W; van Den Bosch, L. VEGF protects motor neurons against excitotoxicity by upregulation of GluR2. Neurobiol. Aging 2009. [Google Scholar] [CrossRef]
- Hoglinger, GU; Widmer, HR; Spenger, C; Meyer, M; Seiler, RW; Oertel, WH; Sautter, J. Influence of time in culture and BDNF pretreatment on survival and function of grafted embryonic rat ventral mesencephalon in the 6-OHDA rat model of Parkinson's disease. Exp. Neurol 2001, 167, 148–157. [Google Scholar]
- Lin, LF; Doherty, DH; Lile, JD; Bektesh, S; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar]
- Casper, D; Roboz, GJ; Blum, M. Epidermal growth factor and basic fibroblast growth factor have independent actions on mesencephalic dopamine neurons in culture. J. Neurochem 1994, 62, 2166–2177. [Google Scholar]
- Pitzer, MR; Sortwell, CE; Daley, BF; McGuire, SO; Marchionini, D; Fleming, M; Collier, TJ. Angiogenic and neurotrophic effects of vascular endothelial growth factor (VEGF165): Studies of grafted and cultured embryonic ventral mesencephalic cells. Exp. Neurol 2003, 182, 435–445. [Google Scholar]
- Silverman, WF; Krum, JM; Mani, N; Rosenstein, JM. Vascular, glial and neuronal effects of vascular endothelial growth factor in mesencephalic explant cultures. Neuroscience 1999, 90, 1529–1541. [Google Scholar]
- Jin, KL; Mao, XO; Greenberg, DA. Vascular endothelial growth factor: Direct neuroprotective effect in in vitro ischemia. Proc. Natl. Acad. Sci. USA 2000, 97, 10242–10247. [Google Scholar]
- Yasuhara, T; Shingo, T; Kobayashi, K; Takeuchi, A; Yano, A; Muraoka, K; Matsui, T; Miyoshi, Y; Hamada, H; Date, I. Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson's disease. Eur. J. Neurosci 2004, 19, 1494–1504. [Google Scholar]
- Yasuhara, T; Shingo, T; Muraoka, K; Yuan, W; Kameda, M; Takeuchi, A; Yano, A; Nishio, S; Matsui, T; Miyoshi, Y; Hamada, H; Date, I. The differences between high and low-dose administration of VEGF to dopaminergic neurons of in vitro and in vivo Parkinson's disease model. Brain Res 2005, 1038, 1–10. [Google Scholar]
- Yasuhara, T; Shingo, T; Muraoka, K; Kameda, M; Agari, T; Wen Ji, Y; Hayase, H; Hamada, H; Borlongan, CV; Date, I. Neurorescue effects of VEGF on a rat model of Parkinson's disease. Brain Res 2005, 1053, 10–18. [Google Scholar]
- Barleon, B; Sozzani, S; Zhou, D; Weich, HA; Mantovani, A; Marme, D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 1996, 87, 3336–3343. [Google Scholar]
- Harrigan, MR; Ennis, SR; Masada, T; Keep, RF. Intraventricular infusion of vascular endothelial growth factor promotes cerebral angiogenesis with minimal brain edema. Neurosurgery 2002, 50, 589–598. [Google Scholar]
- Schoch, HJ; Fischer, S; Marti, HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain 2002, 125, 2549–2557. [Google Scholar]
- Olofsson, B; Pajusola, K; von Euler, G; Chilov, D; Alitalo, K; Eriksson, U. Genomic organization of the mouse and human genes for vascular endothelial growth factor B (VEGF-B) and characterization of a second splice isoform. J. Biol. Chem 1996, 271, 19310–19317. [Google Scholar]
- Li, Y; Zhang, F; Nagai, N; Tang, Z; Zhang, S; Scotney, P; Lennartsson, J; Zhu, C; Qu, Y; Fang, C; Hua, J; Matsuo, O; Fong, GH; Ding, H; Cao, Y; Becker, KG; Nash, A; Heldin, CH; Li, X. VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. J. Clin. Invest 2008, 118, 913–923. [Google Scholar]
- Mould, AW; Greco, SA; Cahill, MM; Tonks, ID; Bellomo, D; Patterson, C; Zournazi, A; Nash, A; Scotney, P; Hayward, NK; Kay, GF. Transgenic overexpression of vascular endothelial growth factor-B isoforms by endothelial cells potentiates postnatal vessel growth in vivo and in vitro. Circ. Res 2005, 97, e60–70. [Google Scholar]
- Silvestre, JS; Tamarat, R; Ebrahimian, TG; Le-Roux, A; Clergue, M; Emmanuel, F; Duriez, M; Schwartz, B; Branellec, D; Levy, BI. Vascular endothelial growth factor-B promotes in vivo angiogenesis. Circ. Res 2003, 93, 114–123. [Google Scholar]
- Aase, K; von Euler, G; Li, X; Ponten, A; Thoren, P; Cao, R; Cao, Y; Olofsson, B; Gebre-Medhin, S; Pekny, M; Alitalo, K; Betsholtz, C; Eriksson, U. Vascular endothelial growth factor-B-deficient mice display an atrial conduction defect. Circulation 2001, 104, 358–364. [Google Scholar]
- Reichelt, M; Shi, S; Hayes, M; Kay, G; Batch, J; Gole, GA; Browning, J. Vascular endothelial growth factor-B and retinal vascular development in the mouse. Clin. Exp. Ophthalmol 2003, 31, 61–65. [Google Scholar]
- Rissanen, TT; Markkanen, JE; Gruchala, M; Heikura, T; Puranen, A; Kettunen, MI; Kholova, I; Kauppinen, RA; Achen, MG; Stacker, SA; Alitalo, K; Yla-Herttuala, S. VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ. Res 2003, 92, 1098–1106. [Google Scholar]
- Yue, X; Falk, T; Zhang, S; Sherman, SJ. Vascular endothelial growth factor-B-186 improves motor behavior in vivo in a rat model of Parkinson's disease. Mov. Disord 2010, 25, S254. [Google Scholar]
- Ek, ET; Dass, CR; Choong, PF. PEDF: A potential molecular therapeutic target with multiple anti-cancer activities. Trends Mol. Med 2006, 12, 497–502. [Google Scholar]
- Tombran-Tink, J; Chader, GG; Johnson, LV. PEDF: A pigment epithelium-derived factor with potent neuronal differentiative activity. Exp. Eye Res 1991, 53, 411–414. [Google Scholar]
- Broadhead, ML; Dass, CR; Choong, PF. Cancer cell apoptotic pathways mediated by PEDF: Prospects for therapy. Trends Mol. Med 2009, 15, 461–467. [Google Scholar]
- Tombran-Tink, J; Johnson, LV. Neuronal differentiation of retinoblastoma cells induced by medium conditioned by human RPE cells. Invest Ophthalmol. Vis. Sci 1989, 30, 1700–1707. [Google Scholar]
- Dawson, DW; Volpert, OV; Gillis, P; Crawford, SE; Xu, H; Benedict, W; Bouck, NP. Pigment epithelium-derived factor: A potent inhibitor of angiogenesis. Science 1999, 285, 245–248. [Google Scholar]
- Tombran-Tink, J; Barnstable, CJ. PEDF: A multifaceted neurotrophic factor. Nat. Rev. Neurosci 2003, 4, 628–636. [Google Scholar]
- Sariola, H. The neurotrophic factors in non-neuronal tissues. Cell Mol. Life Sci 2001, 58, 1061–1066. [Google Scholar]
- Becerra, SP. Structure-function studies on PEDF: A noninhibitory serpin with neurotrophic activity. Adv. Exp. Med. Biol 1997, 425, 223–237. [Google Scholar]
- Steele, FR; Chader, GJ; Johnson, LV; Tombran-Tink, J. Pigment epithelium-derived factor: Neurotrophic activity and identification as a member of the serine protease inhibitor gene family. Proc. Natl. Acad. Sci. USA 1993, 90, 1526–1530. [Google Scholar]
- Tombran-Tink, J; Mazuruk, K; Rodriguez, IR; Chung, D; Linker, T; Englander, E; Chader, GJ. Organization, evolutionary conservation, expression and unusual Alu density of the human gene for pigment epithelium-derived factor, a unique neurotrophic serpin. Mol. Vis 1996, 2, 11. [Google Scholar]
- Becerra, SP. Focus on Molecules: Pigment epithelium-derived factor (PEDF). Exp. Eye Res 2006, 82, 739–740. [Google Scholar]
- Alberdi, E; Aymerich, MS; Becerra, SP. Binding of pigment epithelium-derived factor (PEDF) to retinoblastoma cells and cerebellar granule neurons. Evidence for a PEDF receptor. J. Biol. Chem 1999, 274, 31605–31612. [Google Scholar]
- Kozaki, K; Miyaishi, O; Koiwai, O; Yasui, Y; Kashiwai, A; Nishikawa, Y; Shimizu, S; Saga, S. Isolation, purification, and characterization of a collagen-associated serpin, caspin, produced by murine colon adenocarcinoma cells. J. Biol. Chem 1998, 273, 15125–15130. [Google Scholar]
- Meyer, C; Notari, L; Becerra, SP. Mapping the type I collagen-binding site on pigment epithelium-derived factor. Implications for its antiangiogenic activity. J. Biol. Chem 2002, 277, 45400–45407. [Google Scholar]
- Bilak, MM; Becerra, SP; Vincent, AM; Moss, BH; Aymerich, MS; Kuncl, RW. Identification of the neuroprotective molecular region of pigment epithelium-derived factor and its binding sites on motor neurons. J. Neurosci 2002, 22, 9378–9386. [Google Scholar]
- Filleur, S; Volz, K; Nelius, T; Mirochnik, Y; Huang, H; Zaichuk, TA; Aymerich, MS; Becerra, SP; Yap, R; Veliceasa, D; Shroff, EH; Volpert, OV. Two functional epitopes of pigment epithelial-derived factor block angiogenesis and induce differentiation in prostate cancer. Cancer Res 2005, 65, 5144–5152. [Google Scholar]
- Hosomichi, J; Yasui, N; Koide, T; Soma, K; Morita, I. Involvement of the collagen I-binding motif in the anti-angiogenic activity of pigment epithelium-derived factor. Biochem. Biophys. Res. Commun 2005, 335, 756–761. [Google Scholar]
- Notari, L; Baladron, V; Aroca-Aguilar, JD; Balko, N; Heredia, R; Meyer, C; Notario, PM; Saravanamuthu, S; Nueda, ML; Sanchez-Sanchez, F; Escribano, J; Laborda, J; Becerra, SP. Identification of a lipase-linked cell membrane receptor for pigment epithelium-derived factor. J. Biol. Chem 2006, 281, 38022–38037. [Google Scholar]
- Ho, TC; Chen, SL; Yang, YC; Lo, TH; Hsieh, JW; Cheng, HC; Tsao, YP. Cytosolic phospholipase A2-{alpha} is an early apoptotic activator in PEDF-induced endothelial cell apoptosis. Am. J. Physiol. Cell Physiol 2009, 296, C273–284. [Google Scholar]
- Andreu-Agullo, C; Morante-Redolat, JM; Delgado, AC; Farinas, I. Vascular niche factor PEDF modulates Notch-dependent stemness in the adult subependymal zone. Nat. Neurosci 2009, 12, 1514–1523. [Google Scholar]
- Volpert, OV; Zaichuk, T; Zhou, W; Reiher, F; Ferguson, TA; Stuart, PM; Amin, M; Bouck, NP. Inducer-stimulated Fas targets activated endothelium for destruction by anti-angiogenic thrombospondin-1 and pigment epithelium-derived factor. Nat. Med 2002, 8, 349–357. [Google Scholar]
- Ferreira, JJ; Neutel, D; Mestre, T; Coelho, M; Rosa, MM; Rascol, O; Sampaio, C. Skin cancer and Parkinson's disease. Mov. Disord 2010, 25, 139–148. [Google Scholar]
- Orgaz, JL; Ladhani, O; Hoek, KS; Fernandez-Barral, A; Mihic, D; Aguilera, O; Seftor, EA; Bernad, A; Rodriguez-Peralto, JL; Hendrix, MJ; Volpert, OV; Jimenez, B. 'Loss of pigment epithelium-derived factor enables migration, invasion and metastatic spread of human melanoma'. Oncogene 2009, 28, 4147–4161. [Google Scholar]
- Sasaki, Y; Naishiro, Y; Oshima, Y; Imai, K; Nakamura, Y; Tokino, T. Identification of pigment epithelium-derived factor as a direct target of the p53 family member genes. Oncogene 2005, 24, 5131–5136. [Google Scholar]
- Gao, G; Li, Y; Zhang, D; Gee, S; Crosson, C; Ma, J. Unbalanced expression of VEGF and PEDF in ischemia-induced retinal neovascularization. FEBS Lett 2001, 489, 270–276. [Google Scholar]
- Ohno-Matsui, K; Morita, I; Tombran-Tink, J; Mrazek, D; Onodera, M; Uetama, T; Hayano, M; Murota, SI; Mochizuki, M. Novel mechanism for age-related macular degeneration: An equilibrium shift between the angiogenesis factors VEGF and PEDF. J. Cell Physiol 2001, 189, 323–333. [Google Scholar]
- Funatsu, H; Yamashita, H; Nakamura, S; Mimura, T; Eguchi, S; Noma, H; Hori, S. Vitreous levels of pigment epithelium-derived factor and vascular endothelial growth factor are related to diabetic macular edema. Ophthalmology 2006, 113, 294–301. [Google Scholar]
- Bilak, MM; Corse, AM; Bilak, SR; Lehar, M; Tombran-Tink, J; Kuncl, RW. Pigment epithelium-derived factor (PEDF) protects motor neurons from chronic glutamate-mediated neurodegeneration. J. Neuropathol. Exp. Neurol 1999, 58, 719–728. [Google Scholar]
- DeCoster, MA; Schabelman, E; Tombran-Tink, J; Bazan, NG. Neuroprotection by pigment epithelial-derived factor against glutamate toxicity in developing primary hippocampal neurons. J. Neurosci. Res 1999, 56, 604–610. [Google Scholar]
- Taniwaki, T; Hirashima, N; Becerra, SP; Chader, GJ; Etcheberrigaray, R; Schwartz, JP. Pigment epithelium-derived factor protects cultured cerebellar granule cells against glutamate-induced neurotoxicity. J. Neurochem 1997, 68, 26–32. [Google Scholar]
- Taniwaki, T; Becerra, SP; Chader, GJ; Schwartz, JP. Pigment epithelium-derived factor is a survival factor for cerebellar granule cells in culture. J. Neurochem 1995, 64, 2509–2517. [Google Scholar]
- Houenou, LJ; D'Costa, AP; Li, L; Turgeon, VL; Enyadike, C; Alberdi, E; Becerra, SP. Pigment epithelium-derived factor promotes the survival and differentiation of developing spinal motor neurons. J. Comp. Neurol 1999, 412, 506–514. [Google Scholar]
- Crawford, SE; Stellmach, V; Ranalli, M; Huang, X; Huang, L; Volpert, O; De Vries, GH; Abramson, LP; Bouck, N. Pigment epithelium-derived factor (PEDF) in neuroblastoma: A multifunctional mediator of Schwann cell antitumor activity. J. Cell Sci 2001, 114, 4421–4428. [Google Scholar]
- Kuncl, RW; Bilak, MM; Bilak, SR; Corse, AM; Royal, W; Becerra, SP. Pigment epithelium-derived factor is elevated in CSF of patients with amyotrophic lateral sclerosis. J. Neurochem 2002, 81, 178–184. [Google Scholar]
- McKay, BS; Goodman, B; Falk, T; Sherman, SJ. Retinal pigment epithelial cell transplantation could provide trophic support in Parkinson's disease: Results from an in vitro model system. Exp. Neurol 2006, 201, 234–243. [Google Scholar]
- Watts, RL; Raiser, CD; Stover, NP; Cornfeldt, ML; Schweikert, AW; Allen, RC; Subramanian, T; Doudet, D; Honey, CR; Bakay, RA. Stereotaxic intrastriatal implantation of human retinal pigment epithelial (hRPE) cells attached to gelatin microcarriers: A potential new cell therapy for Parkinson's disease. J. Neural. Transm. Suppl 2003, 65, 215–227. [Google Scholar]
- Ming, M; Li, X; Fan, X; Yang, D; Li, L; Chen, S; Gu, Q; Le, W. Retinal pigment epithelial cells secrete neurotrophic factors and synthesize dopamine: Possible contribution to therapeutic effects of RPE cell transplantation in Parkinson's disease. J. Transl. Med 2009, 7, 53. [Google Scholar]
- Falk, T; Zhang, S; Sherman, SJ. Pigment epithelium derived factor (PEDF) is neuroprotective in two in vitro models of Parkinson's disease. Neurosci. Lett 2009, 458, 49–52. [Google Scholar]
- Watts, RL; Gross, RE; Hauser, RA; Bakay, RAE; Reichmann, H; Weisner, NP; Stover, D; Reissig, E; Steiner-Schulze, H; Fichte, K. The STEPS trial: A phase 2b study evaluating Spheramine® in patients with advanced Parkinson's disease. Mov. Disord 2009, 24, LB-18. [Google Scholar]
- Ogata, N; Nishikawa, M; Nishimura, T; Mitsuma, Y; Matsumura, M. Unbalanced vitreous levels of pigment epithelium-derived factor and vascular endothelial growth factor in diabetic retinopathy. Am. J. Ophthalmol 2002, 134, 348–353. [Google Scholar]
- Ohno-Matsui, K; Yoshida, T; Uetama, T; Mochizuki, M; Morita, I. Vascular endothelial growth factor upregulates pigment epithelium-derived factor expression via VEGFR-1 in human retinal pigment epithelial cells. Biochem. Biophys. Res. Commun 2003, 303, 962–967. [Google Scholar]
- Angayarkanni, N; Selvi, R; Pukhraj, R; Biswas, J; Bhavesh, SJ; Tombran-Tink, J. Ratio of the vitreous vascular endothelial growth factor and pigment epithelial-derived factor in Eales disease. J. Ocul. Biol. Dis. Infor 2009, 2, 20–28. [Google Scholar]
- Elayappan, B; Ravinarayannan, H; Sardar Pasha, SP; Lee, KJ; Gurunathan, S. PEDF inhibits VEGF- and EPO- induced angiogenesis in retinal endothelial cells through interruption of PI3K/Akt phosphorylation. Angiogenesis 2009, 12, 313–324. [Google Scholar]
- Notari, L; Miller, A; Martinez, A; Amaral, J; Ju, M; Robinson, G; Smith, LE; Becerra, SP. Pigment epithelium-derived factor is a substrate for matrix metalloproteinase type 2 and type 9: Implications for downregulation in hypoxia. Invest. Ophthalmol. Vis. Sci 2005, 46, 2736–2747. [Google Scholar]
- Zheng, Z; Chen, H; Zhao, H; Liu, K; Luo, D; Chen, Y; Chen, Y; Yang, X; Gu, Q; Xu, X. Inhibition of JAK2/STAT3-mediated VEGF upregulation under high glucose conditions by PEDF through a mitochondrial ROS pathway in vitro. Invest. Ophthalmol. Vis. Sci 2009, 51, 64–71. [Google Scholar]
- Yasuda, T; Fukuda-Tani, M; Nihira, T; Wada, K; Hattori, N; Mizuno, Y; Mochizuki, H. Correlation between levels of pigment epithelium-derived factor and vascular endothelial growth factor in the striatum of patients with Parkinson's disease. Exp. Neurol 2007, 206, 308–317. [Google Scholar]


© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Falk, T.; Gonzalez, R.T.; Sherman, S.J. The Yin and Yang of VEGF and PEDF: Multifaceted Neurotrophic Factors and Their Potential in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2010, 11, 2875-2900. https://doi.org/10.3390/ijms11082875
Falk T, Gonzalez RT, Sherman SJ. The Yin and Yang of VEGF and PEDF: Multifaceted Neurotrophic Factors and Their Potential in the Treatment of Parkinson’s Disease. International Journal of Molecular Sciences. 2010; 11(8):2875-2900. https://doi.org/10.3390/ijms11082875
Chicago/Turabian StyleFalk, Torsten, Robert T. Gonzalez, and Scott J. Sherman. 2010. "The Yin and Yang of VEGF and PEDF: Multifaceted Neurotrophic Factors and Their Potential in the Treatment of Parkinson’s Disease" International Journal of Molecular Sciences 11, no. 8: 2875-2900. https://doi.org/10.3390/ijms11082875