Molecular Mechanisms of Oligodendrocyte Injury in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
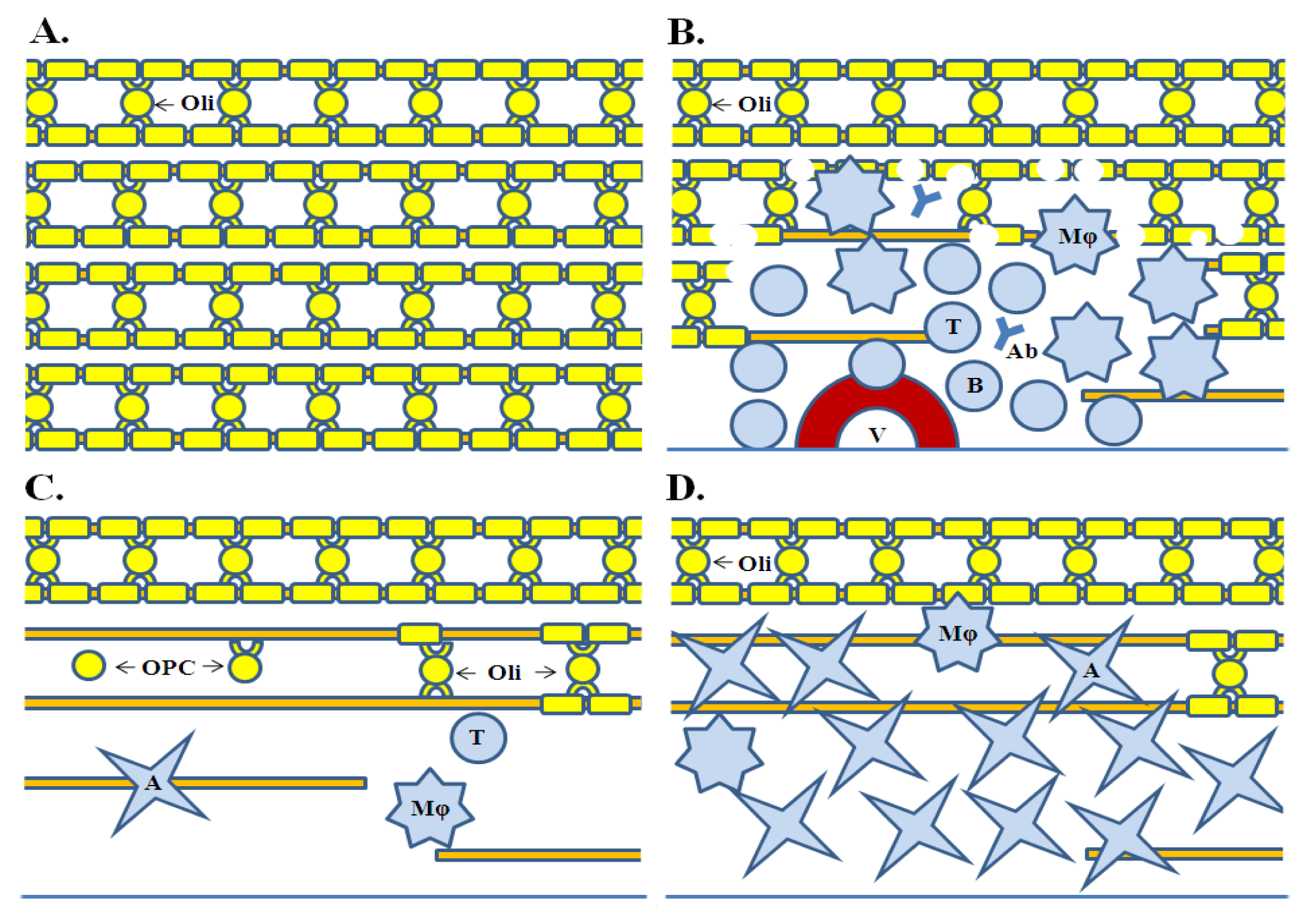
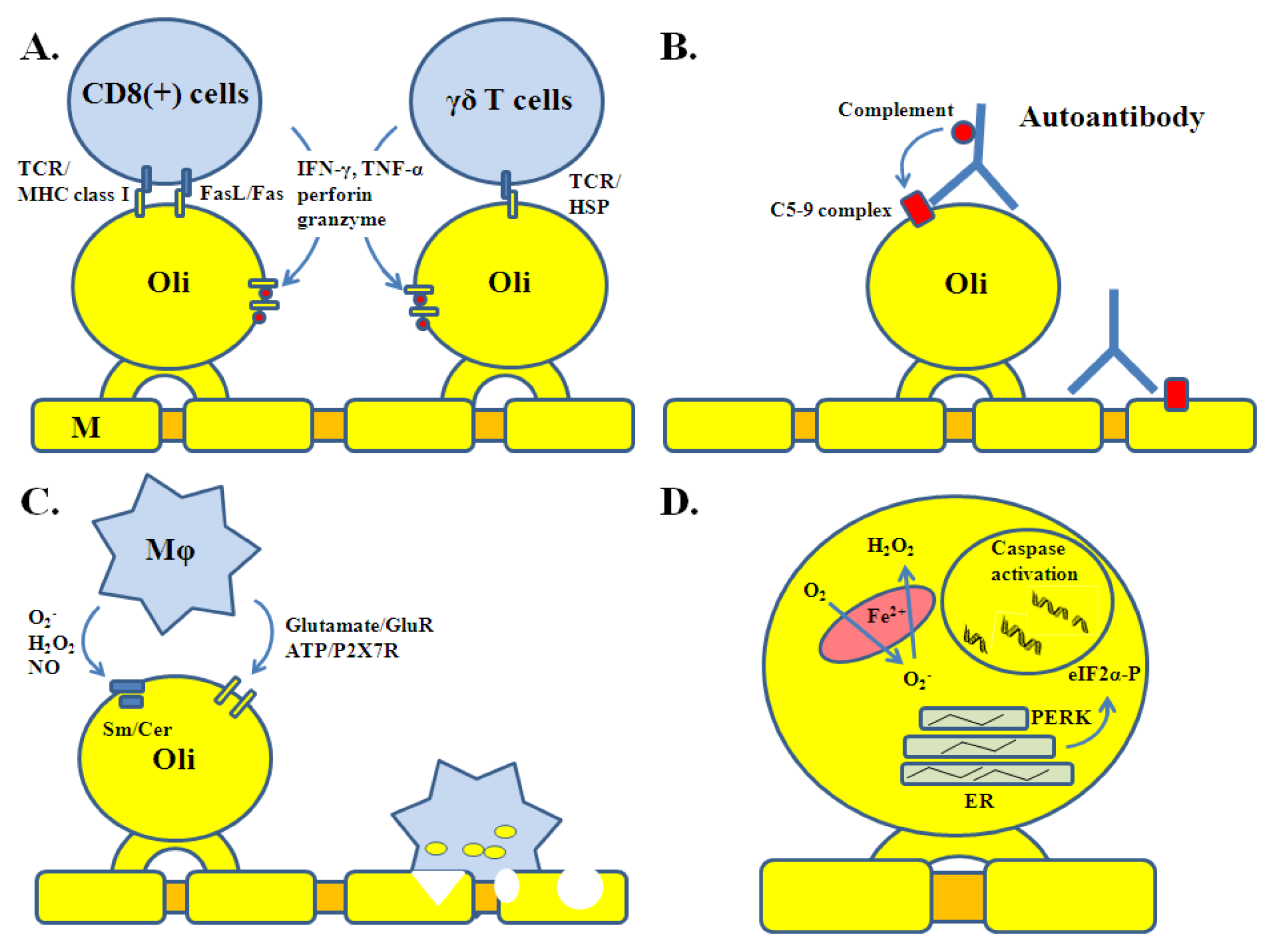
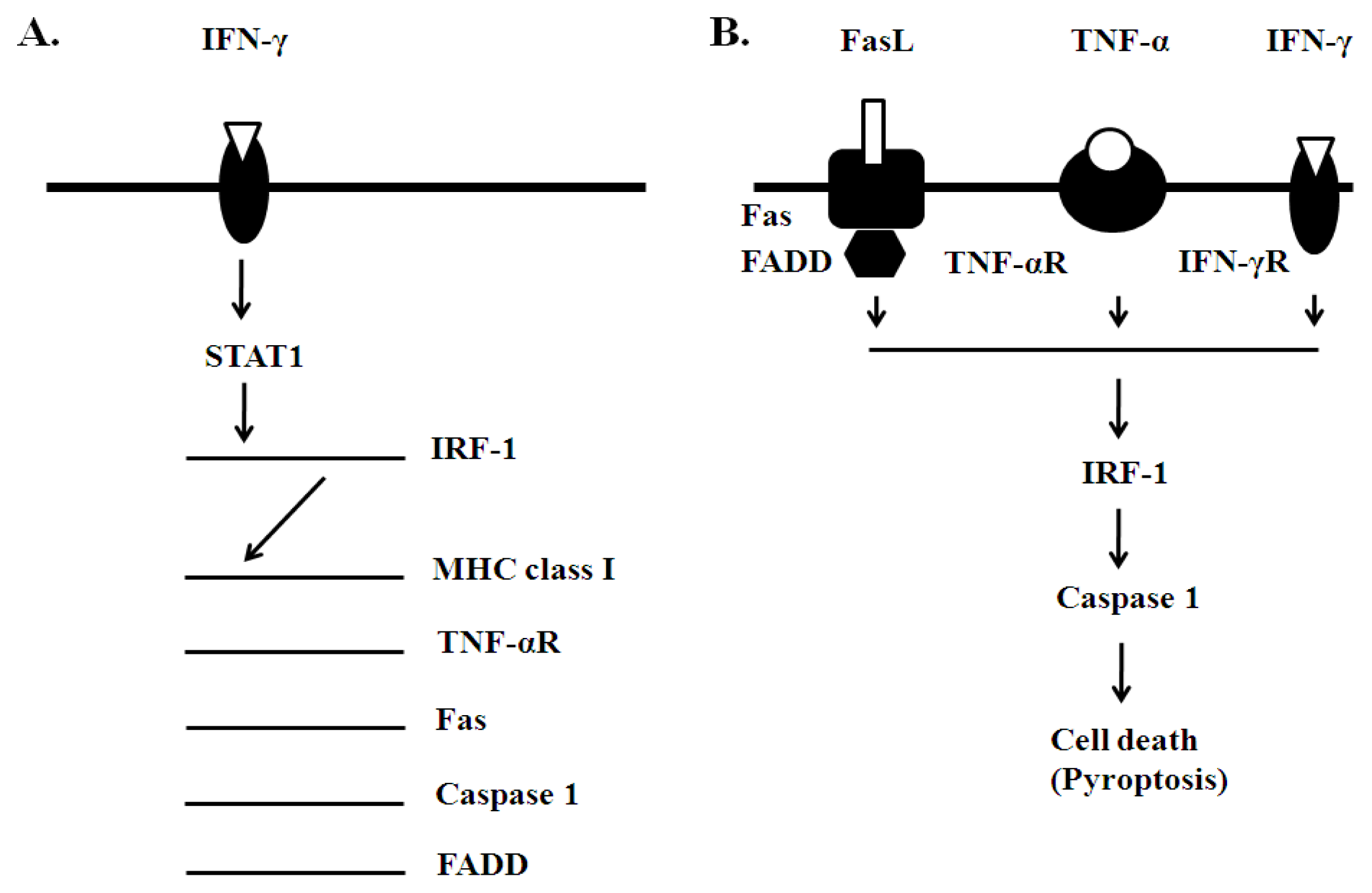
2. Oligodendrocyte Injury in MS
3. Oligodendrocyte Injury: Insights from Transgenic Models of EAE
4. Conclusion
References
- Noseworthy, J.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med 2000, 343, 938–952. [Google Scholar]
- Hafler, D.A. Multiple sclerosis. J. Clin. Invest 2004, 113, 788–794. [Google Scholar]
- Lassmann, H. Multiple sclerosis pathology: Evaluation of pathogenic concepts. Brain Pathol 2005, 15, 217–222. [Google Scholar]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheitauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol 2000, 47, 707–717. [Google Scholar]
- Swanborg, R. Experimental allergic encephalomyelitis. Methods Enzymol 1988, 162, 413–421. [Google Scholar]
- Wingerchuk, D.M. Environmental factors in multiple sclerosis: Epstein-Barr virus, vitamin D, and cigarette smoking. Mt. Sinai J. Med 2011, 78, 221–230. [Google Scholar]
- International Multiple Sclerosis Genetics Consortium (MSGC). Refining genetic associations in multiple sclerosis. Lancet Neurol. 2008, 7, 567–569.
- Willer, C.J.; Dyment, D.A.; Risch, N.J.; Sadovnick, A.D.; Ebers, G.C. Canadian Collaborative Study Group. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 12877–12882. [Google Scholar]
- Raine, C.S. Oligodendrocytes and Central Nervous System Myelin. In Textbook of Neuropathology, 2nd ed; Davis, R.L., Robertson, D.M., Eds.; Williams and Wilkins: Baltimore, MD, USA, 1991; pp. 115–141. [Google Scholar]
- Hartline, D.K.; Colman, D.R. Rapid conduction and the evolution of giant axons and myelinated fibers. Curr. Biol 2007, 17, R29–R35. [Google Scholar]
- McTigue, D.M.; Tripathi, R.B. The life, death and replacement of oligodendrocytes in the adult CNS. J. Neurochem 2008, 107, 1–19. [Google Scholar]
- Brady, S.T.; Witt, A.S.; Kirkpatrick, L.L.; de Waegh, S.M.; Readhead, C.; Tu, P.-H.; Lee, V.M.-Y. Formation of compact myelin is required for maturation of the axonal cytoskeleton. J. Neurosci 1999, 19, 7278–7288. [Google Scholar]
- Smith, K.; McDonald, W. The pathophysiology of multiple sclerosis: The mechanisms underlying the production of symptoms and the natural history of the disease. Phil. Trans. R. Soc. Lond. B 1999, 354, 1649–1673. [Google Scholar]
- Trapp, B.D.; Peterson, J.; Ransohoff, M.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med 1998, 338, 278–285. [Google Scholar]
- Waxman, S.G. Acquired channelopathies in nerve injury and MS. Neurology 2001, 56, 1621–1627. [Google Scholar]
- Barnett, M.H.; Prineas, J.W. Relapsing and remitting multiple sclerosis: Pathology of the newly forming lesion. Ann. Neurol 2004, 55, 458–468. [Google Scholar]
- Rodriguez, M.; Scheithauer, B.W.; Forbes, G.; Kelly, P.J. Oligodendrocyte injury is an early event in multiple scleosis. Mayo Clin. Proc 1993, 68, 627–636. [Google Scholar]
- Hisahara, S.; Araki, T.; Sugiyama, F.; Yagami, K.; Suzuki, M.; Abe, K.; Yamamura, K.; Miyazaki, J.; Momoi, T.; Satura, T.; et al. Targeted expression of baculovirus p35 caspase inhibitor in oligodendrocytes protects mice against autoimmune-mediated demyelination. EMBO J 2000, 19, 341–348. [Google Scholar]
- Hovelmeyer, N.; Hao, Z.; Kranidioti, K.; Kassiotis, G.; Buch, T.; Frommer, F.; von Hoch, L.; Kramer, D.; Minichiello, L.; Kollias, G.; et al. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J. Immunol 2005, 175, 5875–5884. [Google Scholar]
- Balabanov, R.; Strand, K.; Goswami, R.; McMahon, E.; Begolka, W.; Miller, S.D.; Popko, B. Interferon-γ-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J. Neurosci 2007, 27, 2013–2024. [Google Scholar]
- McGuire, C.; Volckaert, T.; Wolke, U.; Sze, M.; de Rycke, R.; Waisman, A.; Prinz, M.; Beyaert, R.; Rasparakis, M.; van Loo, G. Oligodendrocyte-specific FADD deletion protects mice from autoimmune-mediated demyelination. J. Immunol 2010, 185, 7646–7653. [Google Scholar]
- Ren, Z.; Wang, Y.; Tao, D.; Liebenson, D.; Liggett, T.; Goswami, R.; Stefoski, D.; Balabanov, R. Overexpression of the dominant negative form of interferon regulatory factor 1 in oligodendrocytes protects against experimental autoimmune encephalomyelitis. J. Neurosci 2011, 83, 8329–8341. [Google Scholar]
- Mi, S.; Hu, B.; Hahm, K.; Luo, Y.; Hui, E.S.K.; Yuan, Q.; Wong, W.; Wang, L.; Su, H.; Chu, T.; et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat. Med 2005, 13, 1228–1233. [Google Scholar]
- Butzkueven, H.; Zhang, J.-G.; Soilu-Hanninen, M.; Hochrein, H.; Chionh, F.; Shipham, K.A.; Emery, B.; Turnley, A.M.; Petratos, S.; Ernst, M.; et al. LIF receptor signaling limits immune mediated demyelination by enhancing oligodendrocyte survival. Nat. Med 2002, 8, 613–619. [Google Scholar]
- Linker, R.; Maüer, M.; Gaupp, S.; Martini, R.; Holtmann, L.; Giess, R.; Rieckmann, P.; Lassmann, H.; Toyka, K.; Sendtner, M.; et al. CNTF is a major protective factor in demyelinating CNS disease: A neuroprotective cytokine as modulator in neuroinflammation. Nat. Med 2002, 8, 620–624. [Google Scholar]
- Steinmann, L. Multiple sclerosis: A coordinated immunological attack against myelin in the central nervous system. Cell 1996, 185, 299–302. [Google Scholar]
- Franklin, R.J.M. Why does remyelination fail in multiple sclerosis? Nat. Rev. Neurosci 2002, 3, 705–714. [Google Scholar]
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schröder, R.; Deckert, M.; Schmidt, S.; et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med 2000, 192, 393–404. [Google Scholar]
- Neumann, H.; Medana, I.M.; Bauer, J.; Lassmann, H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci 2002, 25, 313–319. [Google Scholar]
- Höftberger, R.; Aboul-Enein, F.; Brueck, W.; Lucchinetti, C.; Rodriguez, M.; Schmidbauer, M.; Jellinger, K.; Lassmann, H. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain Pathol 2004, 14, 43–50. [Google Scholar]
- Dowling, P.; Shang, G.; Raval, S.; Menonna, J.; Cook, S.; Husar, W. Involvement of the CD95 (APO-1/Fas) receptor/ligand system in multiple sclerosis brain. J. Exp. Med 1996, 184, 1513–1518. [Google Scholar]
- Selmaj, K.; Raine, C. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann. Neurol 1988, 23, 339–346. [Google Scholar]
- Vartanian, T.; Li, Y.; Zhao, M.; Stefansson, K. Interferon-γ induced oligodendrocyte cell death: Implications for the pathogenesis of multiple sclerosis. Mol. Med 1995, 1, 732–743. [Google Scholar]
- Pouly, S.; Belcher, B.; Blain, M.; Antel, J.P. Interferon-gamma modulates human oligodendrocyte susceptibility to Fas-mediated injury. J. Neuropathol. Exp. Neurol 2000, 59, 280–286. [Google Scholar]
- Aquino, D.A.; Selmaj, K. Heat-shock proteins and gamma-delta T cell responses in the central nervous system. Chem. Immunol 1992, 53, 86–101. [Google Scholar]
- Van Noort, J.M.; van Sechel, A.C.; Bajramovic, J.J.; el Ouagmiri, M.; Polman, C.H.; Lassmann, H.; Ravid, R. The small heat-shock protein alpha B-crystallin as candidate autoantigen in multiple sclerosis. Nature 1995, 375, 798–801. [Google Scholar]
- Genain, C.P.; Cannella, B.; Hauser, S.L.; Raine, C.S. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med 1999, 5, 170–175. [Google Scholar]
- Dubois-Dalcq, M.; Niedieck, B.; Buyse, M. Action of anti-cerebroside sera on myelinated nervous tissue cultures. Pathol. Eur 1970, 5, 331–347. [Google Scholar]
- Schenck, M.; Carpinteiro, A.; Grassme, H.; Lang, F.; Gulbins, E. Ceramide: Physiological and pathophysiological aspects. Arch. Biochem. Biophys 2007, 462, 171–175. [Google Scholar]
- Thorburne, S.K.; Juurlink, B.H. Low glutathione and high iron govern the susceptibility of oligodendrocyte precursors to oxidative stress. J. Neurochem 1996, 67, 1014–1022. [Google Scholar]
- Lin, W.; Kemper, A.; Dupree, J.L.; Harding, H.P.; Ron, D.; Popko, B. Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain 2006, 129, 1306–1318. [Google Scholar]
- McDonald, J.W.; Althomsons, S.P.; Hyrc, K.L.; Choi, D.W.; Goldberg, M.P. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainite receptor-mediated cytotoxicity. Nat. Med 1998, 4, 291–297. [Google Scholar]
- Matute, C.; Torre, I.; Pérez-Gerdá, F.; Pérez-Samartín, A.; Alberdi, E.; Etxerbaria, E.; Arranz, A.M.; Ravid, R.; Rodriguez-Antigüedad, A.; Sánchez-Gómez, M.; et al. P2×7 receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorate experimental autoimmune encephalomyelitis. J. Neurosci 2007, 27, 9525–9533. [Google Scholar]
- Singh, I.; Pahan, K.; Khan, M.; Singh, A.K. Cytokine-mediated induction of ceramide production is redox-sensitive. Implications to proinflammatory cytokine mediated apoptosis in demyelinating diseases. J. Biol. Chem 1998, 273, 20354–20362. [Google Scholar]
- Boullerne, A.I.; Rodriguez, J.J.; Touil, T.; Brochet, B.; Schmidt, S.; Abrous, N.D.; Le Moal, M.; Pua, J.R.; Jensen, M.A.; Mayo, W.; et al. Anti-S-nitrosocysteine antibodies are a predictive marker for demyelination in experimental autoimmune encephalomyelitis: Implications for multiple sclerosis. J. Neurosci 2002, 22, 123–132. [Google Scholar]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. A quantitative analysis of oligodendrocytes in multiple sclerosis lesions. A study of 113 cases. Brain 1999, 122, 2279–2295. [Google Scholar]
- Keirstead, K.S.; Blakemore, W.F. Identification of post-mitotic oligodendrocytes incapable of remyelination within demyelinated adult spinal cord. J. Neuropathol. Exp. Neurol 1997, 56, 1191–1201. [Google Scholar]
- Wolswijk, G. Oligodendrocyte precursor cells in the demyelinated multiple sclerosis spinal cord. Brain 2002, 125, 338–349. [Google Scholar]
- Baerwald, K.D.; Popko, B. Developing and mature oligodendrocytes respond differently to the immune cytokine interferon-gamma. J. Neurosci. Res 1998, 52, 230–239. [Google Scholar]
- Bernardo, A.; Greco, A.; Levi, G.; Minghetti, L. Differential lipid peroxidation, Mn superoxide, and bcl-2 expression contribute to the maturation-dependent vulnerability of oligodendrocytes to oxidative stress. J. Neuropathol. Exp. Neurol 2003, 62, 509–519. [Google Scholar]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Brück, W.; Lucchinetti, C.; Lassmann, H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006, 129, 3165–3172. [Google Scholar]
- Wolswijk, G. Oligodendrocyte survival, loss and birth in lesions of chronic-stage multiple sclerosis. Brain 2000, 123, 105–115. [Google Scholar]
- Ludwin, S.K.; Maitland, M. Long-term remyelination fails to reconstitute normal thickness of central myelin sheaths. J. Neurol. Sci 1984, 64, 193–198. [Google Scholar]
- Bramow, S.; Frischer, J.M.; Lassmann, H.; Koch-Henriksen, N.; Lucchinetti, C.F.; Sørensen, P.S.; Laursen, H. Demyelination versus remyelination in progressive multiple sclerosis. Brain 2010, 133, 2983–2998. [Google Scholar]
- Rodriguez, M.; Scheithauer, B. Ultrasctucture od multiple sclerosis. Ultratruct. Pathol 1994, 18, 3–13. [Google Scholar]
- Aboul-Enein, F.; Rauschka, H.; Kornek, B.; Stadelmann, C.; Stefferl, A.; Brück, W.; Lucchinetti, C.; Schmidbauer, M.; Jellinger, K.; Lassmann, H. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J. Neuropathol. Exp. Neurol 2003, 62, 25–33. [Google Scholar]
- Bo, L.; Vedeler, C.A.; Nyland, H.I.; Trapp, B.D.; Mork, S.J. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J. Neuropathol. Exp. Neurol 2003, 62, 723–732. [Google Scholar]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 2004, 14, 164–174. [Google Scholar]
- Magliozzi, R.; Howell, O.W.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Serafini, B.; Aloisi, F.; Reynolds, R.A. A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol 2010, 68, 477–493. [Google Scholar]
- Steinman, L.; Zamvil, S.S. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann. Neurol 2006, 60, 12–21. [Google Scholar]
- Gao, X.; Kemper, A.; Popko, B. Advanced transgenic and gene-targeting approaches. Neurochem. Res. 1999, 24, 1181–1188. [Google Scholar]
- Wang, Y.; Ren, Z.; Tao, D.; Tilwalli, S.; Goswami, R.; Balabanov, R. STAT1/IRF-1 signaling pathway mediates the injurious effect of interferon-gamma on oligodendrocyte progenitor cells. Glia 2010, 58, 195–208. [Google Scholar]
- Balabanov, R.; Stand, K.; Kemper, A.; Lee, J.Y.; Popko, B. Suppressor of cytokine signaling 1 expression protects oligodendrocytes from the deleterious effects of interferon-gamma. J. Neurosci 2006, 26, 5143–5152. [Google Scholar]
- Agresti, C.; Bernardo, A.; Del Russo, N.; Marziali, G.; Battistini, A.; Aloisi, F.; Levi, G.; Coccia, E.M. Synergistic stimulation of MHC class I and IRF-1 gene expression by IFN-gamma and TNF-alpha in oligodendrocytes. Eur. J. Neurosci 1998, 10, 2975–8293. [Google Scholar]
- Stang, M.T.; Armstrong, M.J.; Watson, G.A.; Sung, K.Y.; Liu, Y.; Ren, B.; Yim, J.H. Interferon regulatory factor-1-induced apoptosis mediated by a ligand-independent fas-associated death domain pathway in breast cancer cells. Oncogene 2007, 26, 6420–6430. [Google Scholar]
- Conte, E.; Manzella, L.; Zeuner, A.; Cocchiaro, G.; Conticello, C.; Zammataro, L.; Messina, C.G.; de Maria, R.; Messina, A. Involvement of interferon regulatory factor-1 in monocyte CD95 expression and CD95-mediated apoptosis. Cell Death Differ 2003, 10, 615–617. [Google Scholar]
- Tompkins, S.; Padilla, M.; Dal Canto, J.; Ting, J.; Van Kaer, L.; Miller, S. De novo Central Nervous System processing of myelin antigen is required for the initiation of Experimental Autoimmune Encephalomyelitis. J. Immunol 2002, 168, 4173–4183. [Google Scholar]
- Kassmann, C.M.; Lappe-Siefke, C.; Baes, M.; Brügger, B.; Mildner, A.; Werner, H.B.; Natt, O.; Michaelis, T.; Prinz, M.; Frahm, J.; et al. Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat. Genet 2007, 39, 969–976. [Google Scholar]
- Bergsbaken, T.; Fink, L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol 2009, 7, 99–109. [Google Scholar]



© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Patel, J.; Balabanov, R. Molecular Mechanisms of Oligodendrocyte Injury in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2012, 13, 10647-10659. https://doi.org/10.3390/ijms130810647
Patel J, Balabanov R. Molecular Mechanisms of Oligodendrocyte Injury in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences. 2012; 13(8):10647-10659. https://doi.org/10.3390/ijms130810647
Chicago/Turabian StylePatel, Jilpa, and Roumen Balabanov. 2012. "Molecular Mechanisms of Oligodendrocyte Injury in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis" International Journal of Molecular Sciences 13, no. 8: 10647-10659. https://doi.org/10.3390/ijms130810647