Peptide-Lipid Interactions: Experiments and Applications

 ,
, 

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Importance of Membrane Interacting Peptides
2. Molecular Basis for Cell Selectivity: The Membrane Bilayer of Different Cells
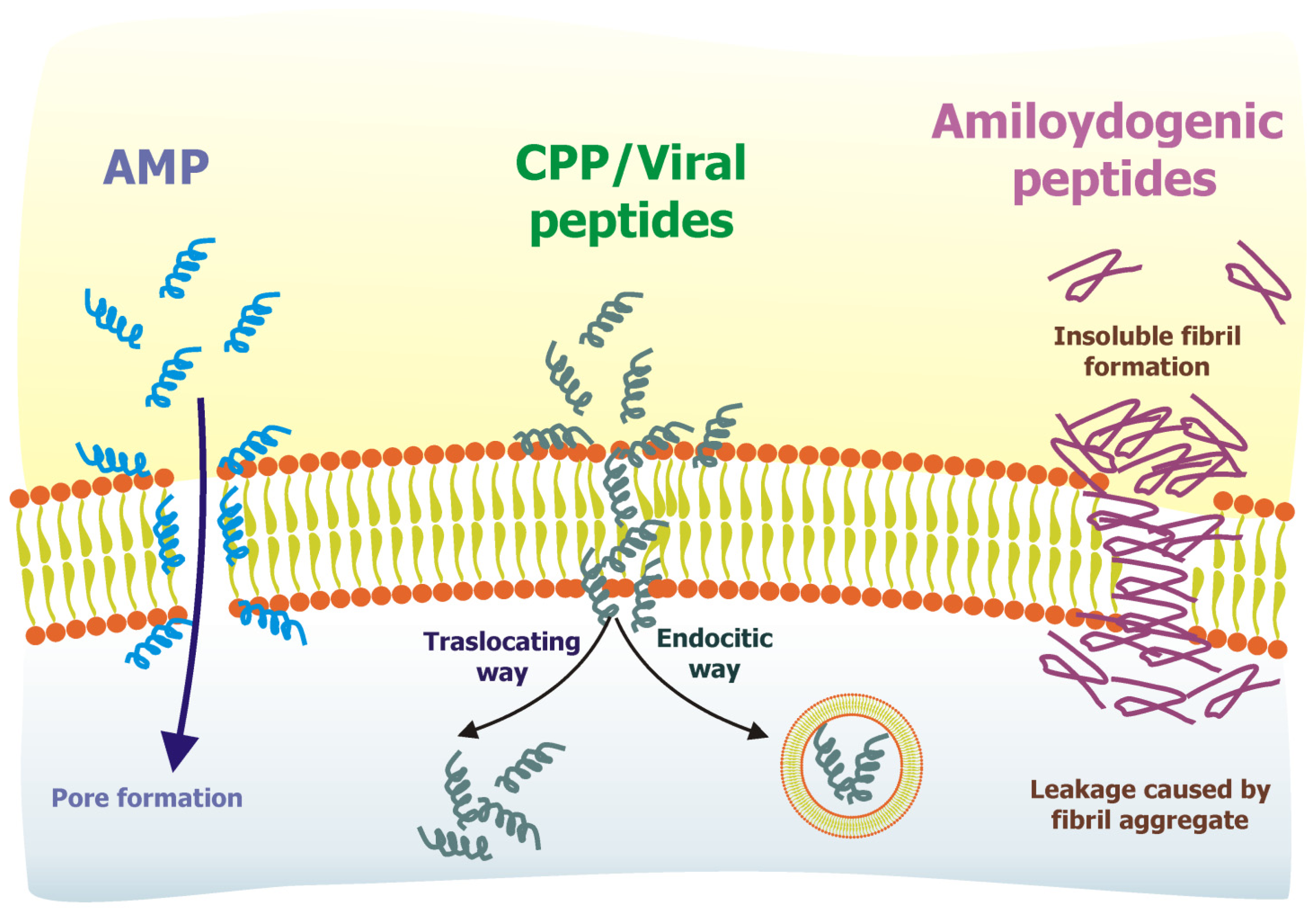
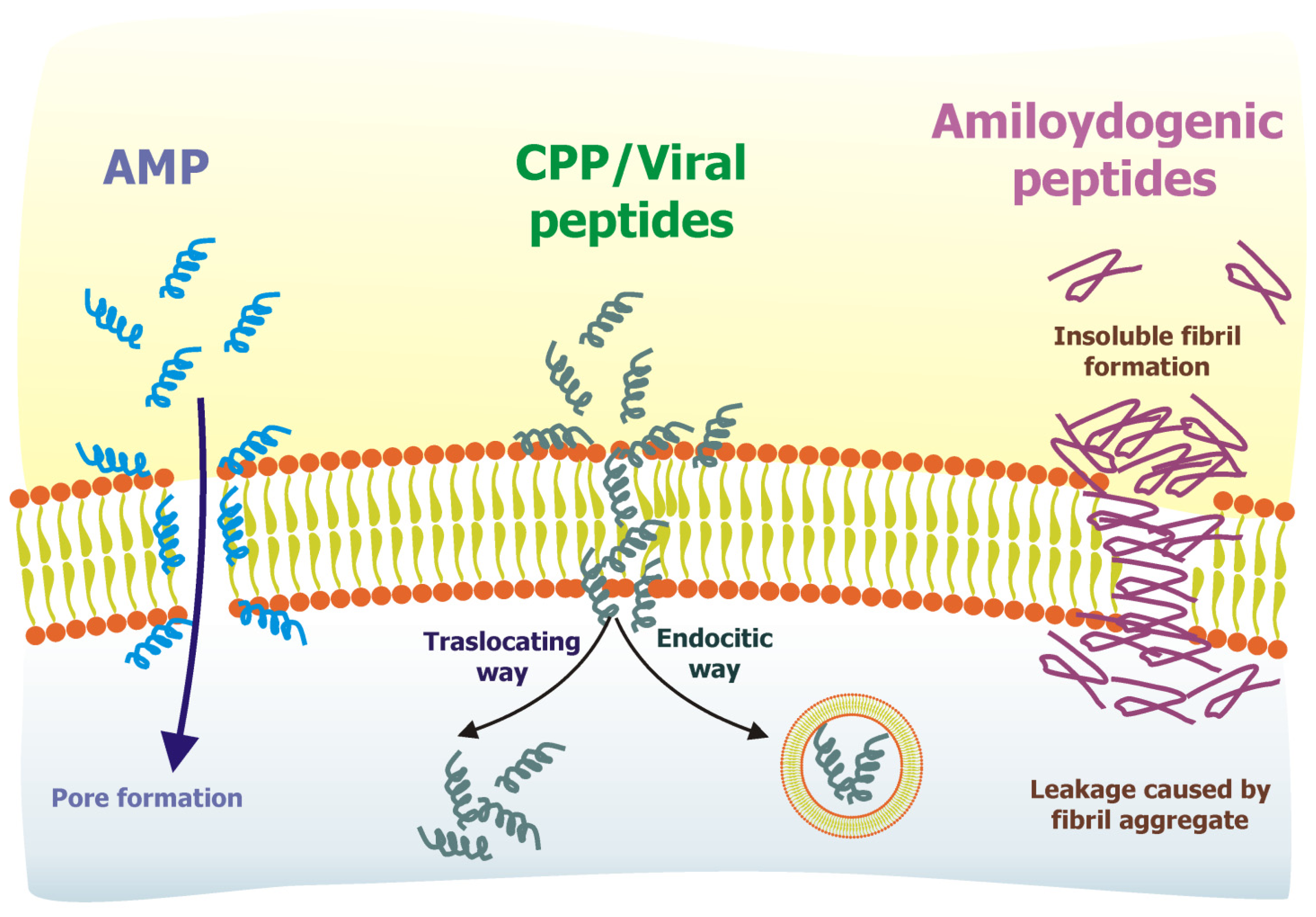
3. Examples of Membrane-Interacting Peptides: AMP, CPP, Viral Peptides, Amyloidogenic Peptides
3.1. Antimicrobial Peptides
3.2. Cell-Penetrating Peptides
3.3. Viral Peptides
3.4. Amyloidogenic Peptides
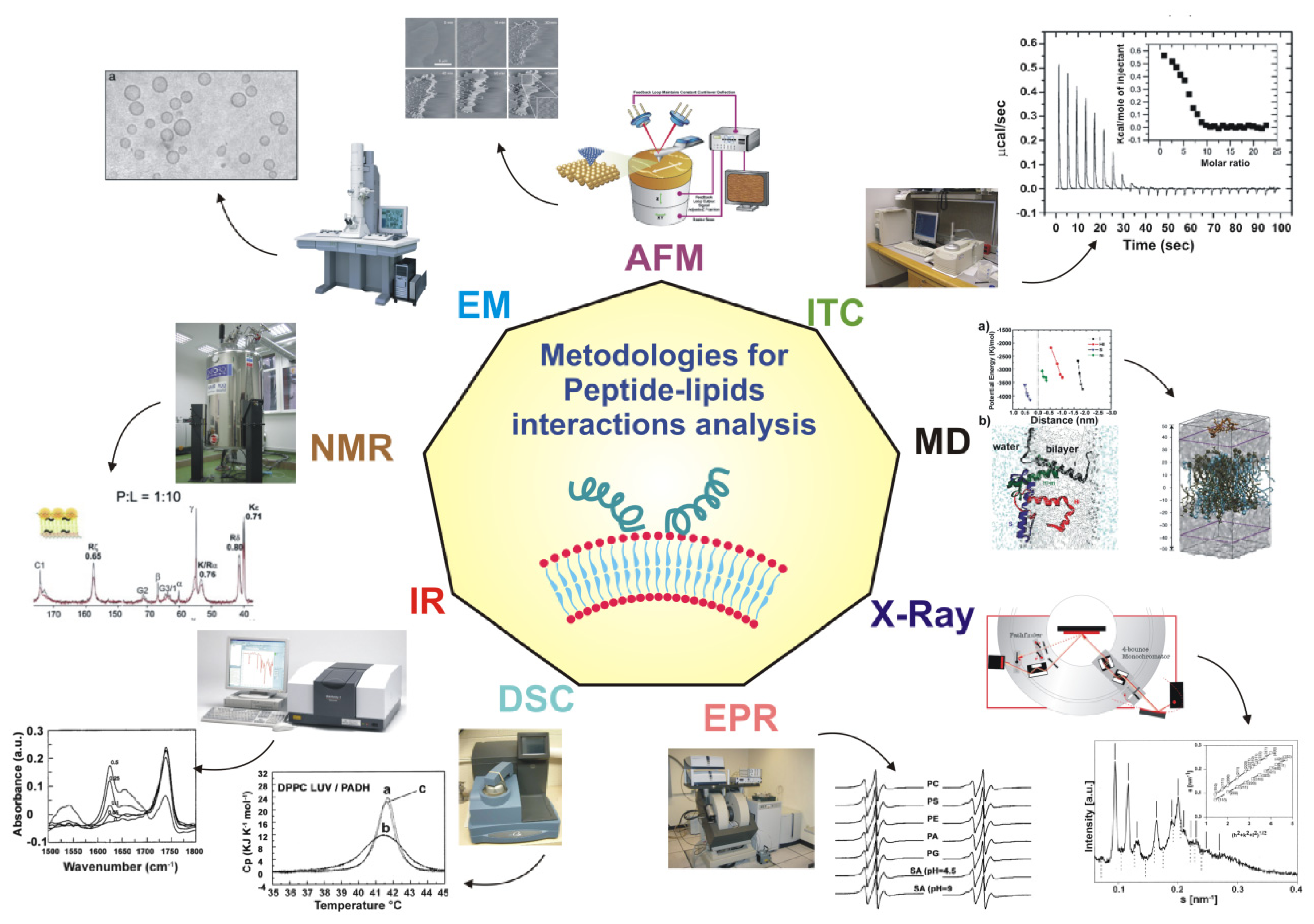
4. Experimental and Theoretical Techniques
4.1. Structural Studies: X-Ray, NMR, EPR, AFM, NR
4.2. Computer Simulations
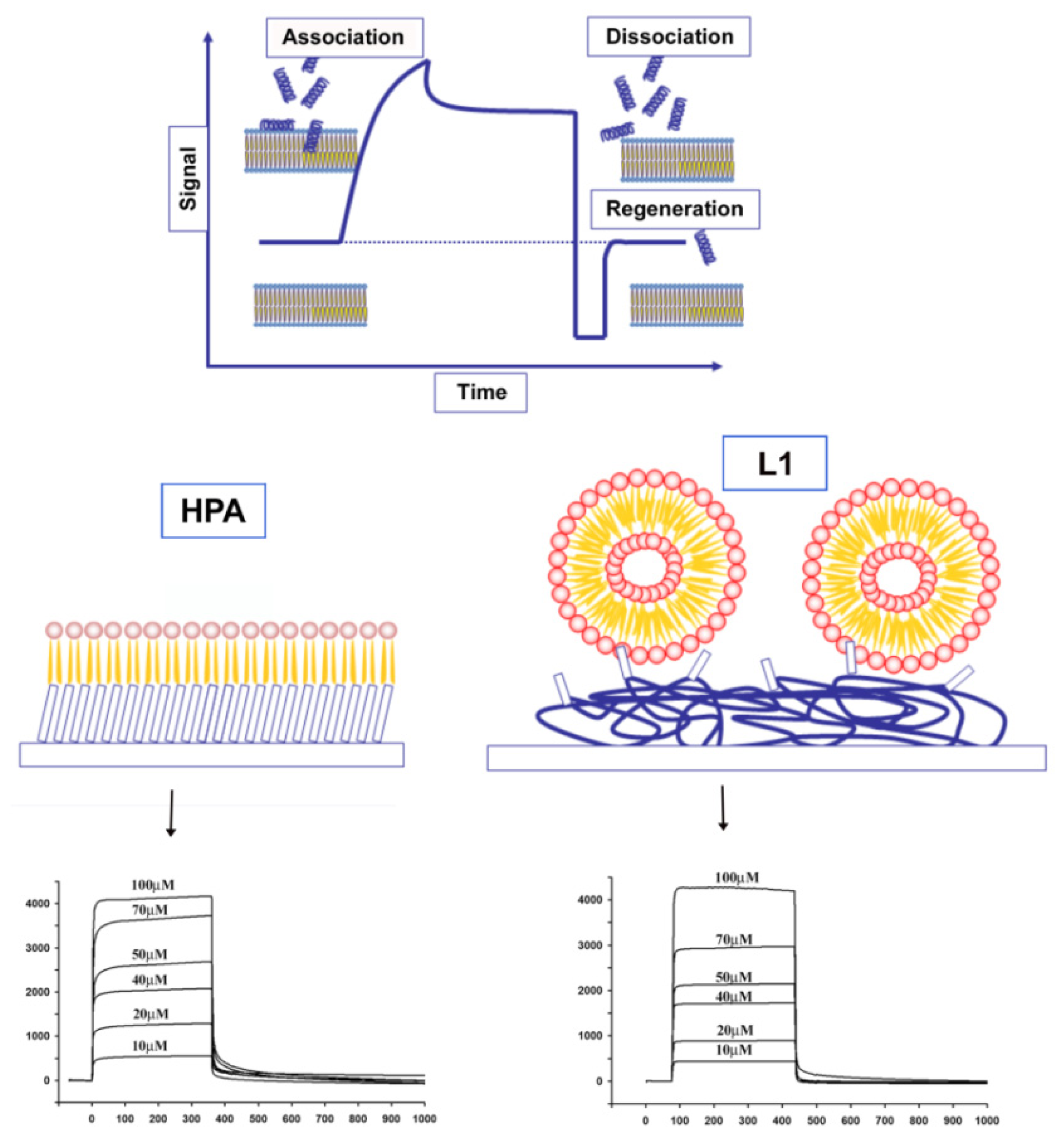
4.3. Other Experimental Approaches to Understand Peptide Location in the Bilayer: SPR, Fluorescence, Calorimetry, CD
5. Examples of Applications: Antimicrobial Compound, Drug Delivery, Antiamyloidonegic Compound
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Galdiero, S.; Vitiello, M.; Falanga, A.; Cantisani, M.; Incoronato, N.; Galdiero, M. Intracellular delivery: Exploiting viral membranotropic peptides. Curr. Drug Metab 2012, 13, 93–104. [Google Scholar]
- Jung, J.J.; Inamdar, S.M.; Tiwari, A.; Choudhury, A. Regulation of intracellular membrane trafficking and cell dynamics by syntaxin-6. Biosci. Rep 2012, 32, 383–391. [Google Scholar]
- Koch, M.; Holt, M. Coupling exo- and endocytosis: An essential role for PIP(2) at the synapse. Biochim. Biophys. Acta 2012, 1821, 1114–1132. [Google Scholar]
- Galdiero, S. Editorial: Developments in membrane fusion. Protein Pept. Lett 2009, 16, 711. [Google Scholar]
- Falanga, A.; Cantisani, M.; Pedone, C.; Galdiero, S. Membrane fusion and fission: Enveloped viruses. Protein Pept. Lett 2009, 16, 751–759. [Google Scholar]
- Sikorska, E.; Ilowska, E.; Wyrzykowski, D.; Kwiatkowska, A. Membrane structure and interactions of peptide hormones with model lipid bilayers. Biochim. Biophys. Acta 2012, 1818, 2982–2993. [Google Scholar]
- Romano, R.; Musiol, H.J.; Weyher, E.; Dufresne, M.; Moroder, L. Peptide hormone-membrane interactions: The aggregational and conformational state of lipo-gastrin derivatives and their receptor binding affinity. Biopolymers 1992, 32, 1545–1558. [Google Scholar]
- Romano, R.; Dufresne, M.; Prost, M.C.; Bali, J.P.; Bayerl, T.M.; Moroder, L. Peptide hormone-membrane interactions. Intervesicular transfer of lipophilic gastrin derivatives to artificial membranes and their bioactivities. Biochim. Biophys. Acta 1993, 1145, 235–242. [Google Scholar]
- Cabiaux, V.; Wolff, C.; Ruysschaert, J.M. Interaction with a lipid membrane: A key step in bacterial toxins virulence. Int. J. Biol. Macromol 1997, 21, 285–298. [Google Scholar]
- Lesieur, C.; Vecsey-Semjen, B.; Abrami, L.; Fivaz, M.; Gisou van der Goot, F. Membrane insertion: The strategies of toxins (review). Mol. Membr. Biol 1997, 14, 45–64. [Google Scholar]
- Galdiero, S.; Gouaux, E. High resolution crystallographic studies of alpha-hemolysin-phospholipid complexes define heptamer-lipid head group interactions: Implication for understanding protein-lipid interactions. Protein Sci 2004, 13, 1503–1511. [Google Scholar]
- Galdiero, S.; Galdiero, M.; Pedone, C. Beta-Barrel membrane bacterial proteins: Structure, function, assembly and interaction with lipids. Curr. Protein Pept. Sci 2007, 8, 63–82. [Google Scholar]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol 1996, 3, 842–848. [Google Scholar]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta 2008, 1778, 2308–2317. [Google Scholar]
- Sonnino, S.; Prinetti, A. Membrane domains and the “lipid raft” concept. Curr. Med. Chem 2013, 20, 4–21. [Google Scholar]
- Hancock, R.E.; Chapple, D.S. Peptide antibiotics. Antimicrob. Agents Chemother 1999, 43, 1317–1323. [Google Scholar]
- Abbassi, F.; Galanth, C.; Amiche, M.; Saito, K.; Piesse, C.; Zargarian, L.; Hani, K.; Nicolas, P.; Lequin, O.; Ladram, A. Solution structure and model membrane interactions of temporins-SH, antimicrobial peptides from amphibian skin. A NMR spectroscopy and differential scanning calorimetry study. Biochemistry 2008, 47, 10513–10525. [Google Scholar]
- Domingues, T.M.; Mattei, B.; Seelig, J.; Perez, K.R.; Miranda, A.; Riske, K.A. Interaction of the antimicrobial peptide gomesin with model membranes: A calorimetric study. Langmuir 2013. [Google Scholar] [CrossRef]
- Szyk, A.; Wu, Z.; Tucker, K.; Yang, D.; Lu, W.; Lubkowski, J. Crystal structures of human alpha-defensins HNP4, HD5, and HD6. Protein Sci 2006, 15, 2749–2760. [Google Scholar]
- Scudiero, O.; Galdiero, S.; Nigro, E.; del Vecchio, L.; di Noto, R.; Cantisani, M.; Colavita, I.; Galdiero, M.; Cassiman, J.J.; Daniele, A.; et al. Chimeric beta-defensin analogs, including the novel 3NI analog, display salt-resistant antimicrobial activity and lack toxicity in human epithelial cell lines. Antimicrob. Agents Chemother 2013, 57, 1701–1708. [Google Scholar]
- Scudiero, O.; Galdiero, S.; Cantisani, M.; di Noto, R.; Vitiello, M.; Galdiero, M.; Naclerio, G.; Cassiman, J.J.; Pedone, C.; Castaldo, G.; et al. Novel synthetic, salt-resistant analogs of human beta-defensins 1 and 3 endowed with enhanced antimicrobial activity. Antimicrob. Agents Chemother 2010, 54, 2312–2322. [Google Scholar]
- Frecer, V.; Ho, B.; Ding, J.L. De novo design of potent antimicrobial peptides. Antimicrob. Agents Chemother 2004, 48, 3349–4457. [Google Scholar]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, alpha-helical antimicrobial peptides. Biopolymers 2000, 55, 4–30. [Google Scholar]
- Lehrer, R.I.; Ganz, T. Antimicrobial peptides in mammalian and insect host defence. Curr. Opin. Immunol 1999, 11, 23–27. [Google Scholar]
- Fidai, S.; Farmer, S.W.; Hancock, R.E. Interaction of cationic peptides with bacterial membranes. Methods Mol. Biol 1997, 78, 187–204. [Google Scholar]
- Niyonsaba, F.; Nagaoka, I.; Ogawa, H.; Okumura, K. Multifunctional antimicrobial proteins and peptides: Natural activators of immune systems. Curr. Pharm. Des 2009, 15, 2393–2413. [Google Scholar]
- Koczulla, A.R.; Bals, R. Antimicrobial peptides: Current status and therapeutic potential. Drugs 2003, 63, 389–406. [Google Scholar]
- Otvos, L., Jr. Antibacterial peptides and proteins with multiple cellular targets. J. Pept. Sci. 2005, 11, 697–706. [Google Scholar]
- Galdiero, S.; Falanga, A.; Tarallo, R.; Russo, L.; Galdiero, E.; Cantisani, M.; Morelli, G.; Galdiero, M. Peptide inhibitors against herpes simplex virus infections. J. Pept. Sci 2013, 19, 148–158. [Google Scholar]
- Hancock, R.E. Cationic peptides: Effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis 2001, 1, 156–164. [Google Scholar]
- Powers, J.P.; Hancock, R.E. The relationship between peptide structure and antibacterial activity. Peptides 2003, 24, 1681–1691. [Google Scholar]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev 2006, 19, 491–511. [Google Scholar]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol 2005, 3, 238–250. [Google Scholar]
- Lehrer, R.I. Primate defensins. Nat. Rev. Microbiol 2004, 2, 727–738. [Google Scholar]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem 1994, 269, 10444–10450. [Google Scholar]
- Said Hassane, F.; Saleh, A.F.; Abes, R.; Gait, M.J.; Lebleu, B. Cell penetrating peptides: Overview and applications to the delivery of oligonucleotides. Cell. Mol. Life Sci 2010, 67, 715–726. [Google Scholar]
- Walther, C.; Meyer, K.; Rennert, R.; Neundorf, I. Quantum dot-carrier peptide conjugates suitable for imaging and delivery applications. Bioconjug. Chem 2008, 19, 2346–2356. [Google Scholar]
- Henriques, S.T.; Costa, J.; Castanho, M.A. Translocation of beta-galactosidase mediated by the cell-penetrating peptide pep-1 into lipid vesicles and human HeLa cells is driven by membrane electrostatic potential. Biochemistry 2005, 44, 10189–10198. [Google Scholar]
- Liu, B.R.; Huang, Y.W.; Chiang, H.J.; Lee, H.J. Cell-penetrating peptide-functionalized quantum dots for intracellular delivery. J. Nanosci. Nanotechnol 2010, 10, 7897–7905. [Google Scholar]
- Xia, H.; Gao, X.; Gu, G.; Liu, Z.; Hu, Q.; Tu, Y.; Song, Q.; Yao, L.; Pang, Z.; Jiang, X.; et al. Penetratin-functionalized PEG-PLA nanoparticles for brain drug delivery. Int. J. Pharm 2012, 436, 840–850. [Google Scholar]
- Desai, P.; Patlolla, R.R.; Singh, M. Interaction of nanoparticles and cell-penetrating peptides with skin for transdermal drug delivery. Mol. Membr. Biol 2010, 27, 247–259. [Google Scholar]
- Liu, C.; Liu, F.; Feng, L.; Li, M.; Zhang, J.; Zhang, N. The targeted co-delivery of DNA and doxorubicin to tumor cells via multifunctional PEI-PEG based nanoparticles. Biomaterials 2013, 34, 2547–2564. [Google Scholar]
- Pan, L.; Liu, J.; He, Q.; Wang, L.; Shi, J. Overcoming multidrug resistance of cancer cells by direct intranuclear drug delivery using TAT-conjugated mesoporous silica nanoparticles. Biomaterials 2013, 34, 2719–2730. [Google Scholar]
- Nori, A.; Jensen, K.D.; Tijerina, M.; Kopeckova, P.; Kopecek, J. Tat-conjugated synthetic macromolecules facilitate cytoplasmic drug delivery to human ovarian carcinoma cells. Bioconjug. Chem 2003, 14, 44–50. [Google Scholar]
- Perche, F.; Torchilin, V.P. Recent trends in multifunctional liposomal nanocarriers for enhanced tumor targeting. J. Drug Deliv 2013, 2013, 705265. [Google Scholar]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med 2012, 18, 385–393. [Google Scholar]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol 2009, 157, 195–206. [Google Scholar]
- Chugh, A.; Eudes, F.; Shim, Y.S. Cell-penetrating peptides: Nanocarrier for macromolecule delivery in living cells. IUBMB Life 2010, 62, 183–193. [Google Scholar]
- Walther, C.; Ott, I.; Gust, R.; Neundorf, I. Specific labeling with potent radiolabels alters the uptake of cell-penetrating peptides. Biopolymers 2009, 92, 445–451. [Google Scholar]
- Cosset, F.-L.; Lavillette, D. 4—Cell. entry of enveloped viruses. Adv. Genet 2011, 73, 121–183. [Google Scholar]
- Joanne, P.; Nicolas, P.; el Amri, C. Antimicrobial peptides and viral fusion peptides: How different they are? Protein Pept. Lett 2009, 16, 743–750. [Google Scholar]
- Brasseur, R.; Vandenbranden, M.; Cornet, B.; Burny, A.; Ruysschaert, J.M. Orientation into the lipid bilayer of an asymmetric amphipathic helical peptide located at the N-terminus of viral fusion proteins. Biochim. Biophys. Acta 1990, 1029, 267–273. [Google Scholar]
- Tristram-Nagle, S.; Chan, R.; Kooijman, E.; Uppamoochikkal, P.; Qiang, W.; Weliky, D.P.; Nagle, J.F. HIV fusion peptide penetrates, disorders, and softens T-cell membrane mimics. J. Mol. Biol 2010, 402, 139–153. [Google Scholar]
- Arnold, E.; Luo, M.; Vriend, G.; Rossmann, M.G.; Palmenberg, A.C.; Parks, G.D.; Nicklin, M.J.; Wimmer, E. Implications of the picornavirus capsid structure for polyprotein processing. Proc. Natl. Acad. Sci. USA 1987, 84, 21–25. [Google Scholar]
- Ivanovic, T.; Agosto, M.A.; Zhang, L.; Chandran, K.; Harrison, S.C.; Nibert, M.L. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J 2008, 27, 1289–1298. [Google Scholar]
- Arias, C.F.; Romero, P.; Alvarez, V.; Lopez, S. Trypsin activation pathway of rotavirus infectivity. J. Virol 1996, 70, 5832–5839. [Google Scholar]
- Greber, U.F.; Webster, P.; Weber, J.; Helenius, A. The role of the adenovirus protease on virus entry into cells. EMBO J 1996, 15, 1766–1777. [Google Scholar]
- Da Costa, B.; Chevalier, C.; Henry, C.; Huet, J.C.; Petit, S.; Lepault, J.; Boot, H.; Delmas, B. The capsid of infectious bursal disease virus contains several small peptides arising from the maturation process of pVP2. J. Virol 2002, 76, 2393–2402. [Google Scholar]
- Galloux, M.; Libersou, S.; Alves, I.D.; Marquant, R.; Salgado, G.F.; Rezaei, H.; Lepault, J.; Delmas, B.; Bouaziz, S.; Morellet, N. NMR structure of a viral peptide inserted in artificial membranes: A view on the early steps of the birnavirus entry process. J. Biol. Chem 2010, 285, 19409–19421. [Google Scholar]
- Harrison, S.C. Mechanism of membrane fusion by viral envelope proteins. Adv. Virus Res 2005, 64, 231–261. [Google Scholar]
- Tarallo, R.; Accardo, A.; Falanga, A.; Guarnieri, D.; Vitiello, G.; Netti, P.; D’Errico, G.; Morelli, G.; Galdiero, S. Clickable functionalization of liposomes with the gH625 peptide from Herpes simplex virus type I for intracellular drug delivery. Chemistry 2011, 17, 12659–12668. [Google Scholar]
- Falanga, A.; Vitiello, M.T.; Cantisani, M.; Tarallo, R.; Guarnieri, D.; Mignogna, E.; Netti, P.; Pedone, C.; Galdiero, M.; Galdiero, S. A peptide derived from herpes simplex virus type 1 glycoprotein H: Membrane translocation and applications to the delivery of quantum dots. Nanomedicine 2011, 7, 925–934. [Google Scholar]
- Wilson, M.R.; Yerbury, J.J.; Poon, S. Potential roles of abundant extracellular chaperones in the control of amyloid formation and toxicity. Mol. Biosyst 2008, 4, 42–52. [Google Scholar]
- Sunde, M.; Blake, C.C. From the globular to the fibrous state: Protein structure and structural conversion in amyloid formation. Q. Rev. Biophys 1998, 31, 1–39. [Google Scholar]
- Anguiano, M.; Nowak, R.J.; Lansbury, P.T., Jr. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry 2002, 41, 11338–11343. [Google Scholar]
- Ahmad, E.; Ahmad, A.; Singh, S.; Arshad, M.; Khan, A.H.; Khan, R.H. A mechanistic approach for islet amyloid polypeptide aggregation to develop anti-amyloidogenic agents for type-2 diabetes. Biochimie 2011, 93, 793–805. [Google Scholar]
- Friedman, R. Aggregation of amyloids in a cellular context: modelling and experiment. Biochem. J 2011, 438, 415–426. [Google Scholar]
- Cho, J.E.; Kim, J.R. Recent approaches targeting beta-amyloid for therapeutic intervention of Alzheimer’s disease. Recent Pat. CNS Drug Discov 2011, 6, 222–233. [Google Scholar]
- Sani, M.A.; Gehman, J.D.; Separovic, F. Lipid matrix plays a role in Abeta fibril kinetics and morphology. FEBS Lett 2011, 585, 749–754. [Google Scholar]
- Jayasinghe, S.A.; Langen, R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry 2005, 44, 12113–12119. [Google Scholar]
- Caillon, L.; Lequin, O.; Khemtemourian, L. Evaluation of membrane models and their composition for islet amyloid polypeptide-membrane aggregation. Biochim. Biophys. Acta 2013, 1828, 2091–2098. [Google Scholar]
- Tomaselli, S.; Esposito, V.; Vangone, P.; van Nuland, N.A.; Bonvin, A.M.; Guerrini, R.; Tancredi, T.; Temussi, P.A.; Picone, D. The alpha-to-beta conformational transition of Alzheimer’s Abeta-(1–42) peptide in aqueous media is reversible: A step by step conformational analysis suggests the location of beta conformation seeding. Chembiochem 2006, 7, 257–267. [Google Scholar]
- Engel, M.F.; Yigittop, H.; Elgersma, R.C.; Rijkers, D.T.; Liskamp, R.M.; de Kruijff, B.; Hoppener, J.W.; Antoinette Killian, J. Islet amyloid polypeptide inserts into phospholipid monolayers as monomer. J. Mol. Biol 2006, 356, 783–789. [Google Scholar]
- Pellistri, F.; Bucciantini, M.; Relini, A.; Nosi, D.; Gliozzi, A.; Robello, M.; Stefani, M. Nonspecific interaction of prefibrillar amyloid aggregates with glutamatergic receptors results in Ca2+ increase in primary neuronal cells. J. Biol. Chem 2008, 283, 29950–29960. [Google Scholar]
- Janson, J.; Ashley, R.H.; Harrison, D.; McIntyre, S.; Butler, P.C. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 1999, 48, 491–498. [Google Scholar]
- Wood, S.J.; Maleeff, B.; Hart, T.; Wetzel, R. Physical, morphological and functional differences between ph 5.8 and 7.4 aggregates of the Alzheimer’s amyloid peptide Abeta. J. Mol. Biol 1996, 256, 870–877. [Google Scholar]
- Klug, G.M.; Losic, D.; Subasinghe, S.S.; Aguilar, M.I.; Martin, L.L.; Small, D.H. Beta-Amyloid protein oligomers induced by metal ions and acid pH are distinct from those generated by slow spontaneous ageing at neutral pH. Eur. J. Biochem 2003, 270, 4282–4293. [Google Scholar]
- Khemtemourian, L.; Domenech, E.; Doux, J.P.; Koorengevel, M.C.; Killian, J.A. Low pH acts as inhibitor of membrane damage induced by human islet amyloid polypeptide. J. Am. Chem. Soc 2011, 133, 15598–15604. [Google Scholar]
- Jan, A.; Adolfsson, O.; Allaman, I.; Buccarello, A.L.; Magistretti, P.J.; Pfeifer, A.; Muhs, A.; Lashuel, H.A. Abeta42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Abeta42 species. J. Biol. Chem 2011, 286, 8585–8596. [Google Scholar]
- Engel, M.F.; Khemtemourian, L.; Kleijer, C.C.; Meeldijk, H.J.; Jacobs, J.; Verkleij, A.J.; de Kruijff, B.; Killian, J.A.; Hoppener, J.W. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [Google Scholar]
- Galdiero, S.; Capasso, D.; Vitiello, M.; D’Isanto, M.; Pedone, C.; Galdiero, M. Role of surface-exposed loops of Haemophilus influenzae protein P2 in the mitogen-activated protein kinase cascade. Infect. Immun 2003, 71, 2798–2809. [Google Scholar]
- Oreopoulos, J.; Epand, R.F.; Epand, R.M.; Yip, C.M. Peptide-Induced domain formation in supported lipid bilayers: Direct evidence by combined atomic force and polarized total internal reflection fluorescence microscopy. Biophys. J 2010, 98, 815–823. [Google Scholar]
- El Kirat, K.; Morandat, S.; Dufrene, Y.F. Nanoscale analysis of supported lipid bilayers using atomic force microscopy. Biochim. Biophys. Acta 2010, 1798, 750–765. [Google Scholar]
- Garcia-Saez, A.J.; Chiantia, S.; Salgado, J.; Schwille, P. Pore formation by a bax-derived peptide: Effect on the line tension of the membrane probed by AFM. Biophys. J 2007, 93, 103–112. [Google Scholar]
- Volinsky, R.; Kolusheva, S.; Berman, A.; Jelinek, R. Investigations of antimicrobial peptides in planar film systems. Biochim. Biophys. Acta 2006, 1758, 1393–1407. [Google Scholar]
- Lohner, K.; Prenner, E.J. Differential scanning calorimetry and X-ray diffraction studies of the specificity of the interaction of antimicrobial peptides with membrane-mimetic systems. Biochim. Biophys. Acta 1999, 1462, 141–156. [Google Scholar]
- Pott, T.; Maillet, J.C.; Abad, C.; Campos, A.; Dufourcq, J.; Dufourc, E.J. The lipid charge density at the bilayer surface modulates the effects of melittin on membranes. Chem. Phys. Lipids 2001, 109, 209–223. [Google Scholar]
- Faucon, J.F.; Bonmatin, J.M.; Dufourcq, J.; Dufourc, E.J. Acyl chain length dependence in the stability of melittin-phosphatidylcholine complexes. A light scattering and 31P-NMR study. Biochim. Biophys. Acta 1995, 1234, 235–243. [Google Scholar]
- Castano, S.; Desbat, B. Structure and orientation study of fusion peptide FP23 of gp41 from HIV-1 alone or inserted into various lipid membrane models (mono-, bi- and multibi-layers) by FT-IR spectroscopies and Brewster angle microscopy. Biochim. Biophys. Acta 2005, 1715, 81–95. [Google Scholar]
- Sani, M.A.; Loudet, C.; Grobner, G.; Dufourc, E.J. Pro-apoptotic bax-alpha1 synthesis and evidence for beta-sheet to alpha-helix conformational change as triggered by negatively charged lipid membranes. J. Pept. Sci 2007, 13, 100–106. [Google Scholar]
- Jean-Francois, F.; Khemtemourian, L.; Odaert, B.; Castano, S.; Grelard, A.; Manigand, C.; Bathany, K.; Metz-Boutigue, M.H.; Dufourc, E.J. Variability in secondary structure of the antimicrobial peptide Cateslytin in powder, solution, DPC micelles and at the air-water interface. Eur. Biophys. J 2007, 36, 1019–1027. [Google Scholar]
- Galdiero, S.; Falanga, A.; Vitiello, M.; Browne, H.; Pedone, C.; Galdiero, M. Fusogenic domains in herpes simplex virus type 1 glycoprotein H. J. Biol. Chem 2005, 280, 28632–28643. [Google Scholar]
- Galdiero, S.; Falanga, A.; Vitiello, M.; D’Isanto, M.; Collins, C.; Orrei, V.; Browne, H.; Pedone, C.; Galdiero, M. Evidence for a role of the membrane-proximal region of herpes simplex virus Type 1 glycoprotein H in membrane fusion and virus inhibition. Chembiochem 2007, 8, 885–895. [Google Scholar]
- Galdiero, S.; Vitiello, M.; D’Isanto, M.; Falanga, A.; Cantisani, M.; Browne, H.; Pedone, C.; Galdiero, M. The identification and characterization of fusogenic domains in herpes virus glycoprotein B molecules. Chembiochem 2008, 9, 758–767. [Google Scholar]
- Galdiero, S.; Falanga, A.; Vitiello, M.; Raiola, L.; Fattorusso, R.; Browne, H.; Pedone, C.; Isernia, C.; Galdiero, M. Analysis of a membrane interacting region of herpes simplex virus type 1 glycoprotein H. J. Biol. Chem 2008, 283, 29993–30009. [Google Scholar]
- Galdiero, S.; Falanga, A.; Vitiello, M.; Raiola, L.; Russo, L.; Pedone, C.; Isernia, C.; Galdiero, M. The presence of a single N-terminal histidine residue enhances the fusogenic properties of a membranotropic peptide derived from herpes simplex virus type 1 glycoprotein H. J. Biol. Chem 2010, 285, 17123–17136. [Google Scholar]
- Vitiello, G.; Falanga, A.; Galdiero, M.; Marsh, D.; Galdiero, S.; D’Errico, G. Lipid composition modulates the interaction of peptides deriving from herpes simplex virus type I glycoproteins B and H with biomembranes. Biochim. Biophys. Acta 2011, 1808, 2517–2526. [Google Scholar]
- D’Errico, G.; Ercole, C.; Lista, M.; Pizzo, E.; Falanga, A.; Galdiero, S.; Spadaccini, R.; Picone, D. Enforcing the positive charge of N-termini enhances membrane interaction and antitumor activity of bovine seminal ribonuclease. Biochim. Biophys. Acta 2011, 1808, 3007–3015. [Google Scholar]
- Falanga, A.; Tarallo, R.; Vitiello, G.; Vitiello, M.; Perillo, E.; Cantisani, M.; D’Errico, G.; Galdiero, M.; Galdiero, S. Biophysical characterization and membrane interaction of the two fusion loops of glycoprotein B from herpes simplex type I virus. PLoS One 2012, 7, e32186. [Google Scholar]
- Castano, S.; Desbat, B.; Laguerre, M.; Dufourcq, J. Structure, orientation and affinity for interfaces and lipids of ideally amphipathic lytic LiKj(i = 2j) peptides. Biochim. Biophys. Acta 1999, 1416, 176–194. [Google Scholar]
- Frey, S.; Tamm, L.K. Orientation of melittin in phospholipid bilayers. A polarized attenuated total reflection infrared study. Biophys. J 1991, 60, 922–930. [Google Scholar]
- Bechinger, B.; Resende, J.M.; Aisenbrey, C. The structural and topological analysis of membrane-associated polypeptides by oriented solid-state NMR spectroscopy: Established concepts and novel developments. Biophys. Chem 2011, 153, 115–125. [Google Scholar]
- Resende, J.M.; Moraes, C.M.; Munhoz, V.H.; Aisenbrey, C.; Verly, R.M.; Bertani, P.; Cesar, A.; Pilo-Veloso, D.; Bechinger, B. Membrane structure and conformational changes of the antibiotic heterodimeric peptide distinctin by solid-state NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 16639–16644. [Google Scholar]
- Salnikov, E.; Rosay, M.; Pawsey, S.; Ouari, O.; Tordo, P.; Bechinger, B. Solid-state NMR spectroscopy of oriented membrane polypeptides at 100 K with signal enhancement by dynamic nuclear polarization. J. Am. Chem. Soc 2010, 132, 5940–5941. [Google Scholar]
- Galdiero, S.; Russo, L.; Falanga, A.; Cantisani, M.; Vitiello, M.; Fattorusso, R.; Malgieri, G.; Galdiero, M.; Isernia, C. Structure and orientation of the gH625–644 membrane interacting region of herpes simplex virus type 1 in a membrane mimetic system. Biochemistry 2012, 51, 3121–3128. [Google Scholar]
- La Rocca, P.; Biggin, P.C.; Tieleman, D.P.; Sansom, M.S. Simulation studies of the interaction of antimicrobial peptides and lipid bilayers. Biochim. Biophys. Acta 1999, 1462, 185–200. [Google Scholar]
- Biggin, P.C.; Sansom, M.S. Interactions of alpha-helices with lipid bilayers: A review of simulation studies. Biophys. Chem 1999, 76, 161–183. [Google Scholar]
- Karle, I.L.; Perozzo, M.A.; Mishra, V.K.; Balaram, P. Crystal structure of the channel-forming polypeptide antiamoebin in a membrane-mimetic environment. Proc. Natl. Acad. Sci. USA 1998, 95, 5501–5504. [Google Scholar]
- Merlino, A.; Vitiello, G.; Grimaldi, M.; Sica, F.; Busi, E.; Basosi, R.; D’Ursi, A.M.; Fragneto, G.; Paduano, L.; D’Errico, G. Destabilization of lipid membranes by a peptide derived from glycoprotein gp36 of feline immunodeficiency virus: A combined molecular dynamics/experimental study. J. Phys. Chem. B 2012, 116, 401–412. [Google Scholar]
- Vitiello, G.; Fragneto, G.; Petruk, A.A.; Falanga, A.; Galdiero, S.; D’Ursi, A.M.; Merlino, A.; D’Errico, G. Cholesterol 1 modulates the fusogenic activity of a membranotropic domain of the FIV glycoprotein gp36. Soft Matter 2013. [Google Scholar] [CrossRef]
- Rankenberg, J.M.; Vostrikov, V.V.; Greathouse, D.V.; Grant, C.V.; Opella, S.J.; Koeppe, R.E., 2nd. Properties of membrane-incorporated WALP peptides that are anchored on only one end. Biochemistry 2012, 51, 10066–10074. [Google Scholar]
- Park, S.H.; de Angelis, A.A.; Nevzorov, A.A.; Wu, C.H.; Opella, S.J. Three-dimensional structure of the transmembrane domain of Vpu from HIV-1 in aligned phospholipid bicelles. Biophys. J 2006, 91, 3032–3042. [Google Scholar]
- Strandberg, E.; Zerweck, J.; Wadhwani, P.; Ulrich, A.S. Synergistic insertion of antimicrobial magainin-family peptides in membranes depends on the lipid spontaneous curvature. Biophys. J 2013, 104, L9–L11. [Google Scholar]
- Song, C.; Weichbrodt, C.; Salnikov, E.S.; Dynowski, M.; Forsberg, B.O.; Bechinger, B.; Steinem, C.; de Groot, B.L.; Zachariae, U.; Zeth, K. Crystal structure and functional mechanism of a human antimicrobial membrane channel. Proc. Natl. Acad. Sci. USA 2013, 110, 4586–4591. [Google Scholar]
- D’Ursi, A.M.; Armenante, M.R.; Guerrini, R.; Salvadori, S.; Sorrentino, G.; Picone, D. Solution structure of amyloid beta-peptide (25–35) in different media. J. Med. Chem 2004, 47, 4231–4238. [Google Scholar]
- Warschawski, D.E.; Arnold, A.A.; Beaugrand, M.; Gravel, A.; Chartrand, E.; Marcotte, I. Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biochim. Biophys. Acta 2011, 1808, 1957–1974. [Google Scholar]
- Kallick, D.A.; Tessmer, M.R.; Watts, C.R.; Li, C.Y. The use of dodecylphosphocholine micelles in solution NMR. J. Magn. Reson. B 1995, 109, 60–65. [Google Scholar]
- Keifer, P.A.; Peterkofsky, A.; Wang, G. Effects of detergent alkyl chain length and chemical structure on the properties of a micelle-bound bacterial membrane targeting peptide. Anal. Biochem 2004, 331, 33–39. [Google Scholar]
- Prosser, R.S.; Evanics, F.; Kitevski, J.L.; Al-Abdul-Wahid, M.S. Current applications of bicelles in NMR studies of membrane-associated amphiphiles and proteins. Biochemistry 2006, 45, 8453–8465. [Google Scholar]
- Tamm, L.K.; Abildgaard, F.; Arora, A.; Blad, H.; Bushweller, J.H. Structure, dynamics and function of the outer membrane protein A (OmpA) and influenza hemagglutinin fusion domain in detergent micelles by solution NMR. FEBS Lett 2003, 555, 139–143. [Google Scholar]
- Shenkarev, Z.O.; Balashova, T.A.; Efremov, R.G.; Yakimenko, Z.A.; Ovchinnikova, T.V.; Raap, J.; Arseniev, A.S. Spatial structure of zervamicin IIB bound to DPC micelles: Implications for voltage-gating. Biophys. J 2002, 82, 762–771. [Google Scholar]
- Gao, X.; Wong, T.C. Studies of the binding and structure of adrenocorticotropin peptides in membrane mimics by NMR spectroscopy and pulsed-field gradient diffusion. Biophys. J 1998, 74, 1871–1888. [Google Scholar]
- Appelt, C.; Wessolowski, A.; Soderhall, J.A.; Dathe, M.; Schmieder, P. Structure of the antimicrobial, cationic hexapeptide cyclo(RRWWRF) and its analogues in solution and bound to detergent micelles. Chembiochem 2005, 6, 1654–1662. [Google Scholar]
- Tjandra, N.; Bax, A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 1997, 278, 1111–1114. [Google Scholar]
- Brown, L.R.; Bosch, C.; Wuthrich, K. Location and orientation relative to the micelle surface for glucagon in mixed micelles with dodecylphosphocholine: EPR and NMR studies. Biochim. Biophys. Acta 1981, 642, 296–312. [Google Scholar]
- Prosser, R.S.; Luchette, P.A.; Westerman, P.W. Using O2 to probe membrane immersion depth by 19F NMR. Proc. Natl. Acad. Sci. USA 2000, 97, 9967–9971. [Google Scholar]
- Hilty, C.; Wider, G.; Fernandez, C.; Wuthrich, K. Membrane protein-lipid interactions in mixed micelles studied by NMR spectroscopy with the use of paramagnetic reagents. Chembiochem 2004, 5, 467–473. [Google Scholar]
- Laws, D.D.; Bitter, H.M.; Jerschow, A. Solid-state NMR spectroscopic methods in chemistry. Angew. Chem. Int. Ed. Engl 2002, 41, 3096–3129. [Google Scholar]
- Marsh, D. Orientation and peptide-lipid interactions of alamethicin incorporated in phospholipid membranes: Polarized infrared and spin-label EPR spectroscopy. Biochemistry 2009, 48, 729–737. [Google Scholar]
- D’Errico, G.; D’Ursi, A.M.; Marsh, D. Interaction of a peptide derived from glycoprotein gp36 of feline immunodeficiency virus and its lipoylated analogue with phospholipid membranes. Biochemistry 2008, 47, 5317–5327. [Google Scholar]
- Inbaraj, J.J.; Cardon, T.B.; Laryukhin, M.; Grosser, S.M.; Lorigan, G.A. Determining the topology of integral membrane peptides using EPR spectroscopy. J. Am. Chem. Soc 2006, 128, 9549–9554. [Google Scholar]
- Spadaccini, R.; D’Errico, G.; D’Alessio, V.; Notomista, E.; Bianchi, A.; Merola, M.; Picone, D. Structural characterization of the transmembrane proximal region of the hepatitis C virus E1 glycoprotein. Biochim. Biophys. Acta 2010, 1798, 344–353. [Google Scholar]
- Inbaraj, J.J.; Laryukhin, M.; Lorigan, G.A. Determining the helical tilt angle of a transmembrane helix in mechanically aligned lipid bilayers using EPR spectroscopy. J. Am. Chem. Soc 2007, 129, 7710–7711. [Google Scholar]
- Mayo, D.J.; Inbaraj, J.J.; Subbaraman, N.; Grosser, S.M.; Chan, C.A.; Lorigan, G.A. Comparing the structural topology of integral and peripheral membrane proteins utilizing electron paramagnetic resonance spectroscopy. J. Am. Chem. Soc 2008, 130, 9656–9657. [Google Scholar]
- Galdiero, S.; Falanga, A.; Vitiello, G.; Vitiello, M.; Pedone, C.; D’Errico, G.; Galdiero, M. Role of membranotropic sequences from herpes simplex virus type I glycoproteins B and H in the fusion process. Biochim. Biophys. Acta 2010, 1798, 579–591. [Google Scholar]
- Esposito, C.; Tedeschi, A.; Scrima, M.; D’Errico, G.; Ottaviani, M.F.; Rovero, P.; D’Ursi, A.M. Exploring interaction of beta-amyloid segment (25–35) with membrane models through paramagnetic probes. J. Pept. Sci. 2006, 12, 766–774. [Google Scholar]
- Im, W.; Feig, M.; Brooks, C.L., 3rd. An implicit membrane generalized born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 2003, 85, 2900–2918. [Google Scholar]
- Im, W.; Lee, M.S.; Brooks, C.L., III. Generalized born model with a simple smoothing function. J. Comput. Chem. 2003, 24, 1691–1702. [Google Scholar]
- Ulmschneider, M.B.; Sansom, M.S.; di Nola, A. Properties of integral membrane protein structures: Derivation of an implicit membrane potential. Proteins 2005, 59, 252–265. [Google Scholar]
- Ulmschneider, M.B.; Ulmschneider, J.P.; Sansom, M.S.; di Nola, A. A generalized born implicit-membrane representation compared to experimental insertion free energies. Biophys. J 2007, 92, 2338–2349. [Google Scholar]
- La Rocca, P.; Shai, Y.; Sansom, M.S. Peptide-bilayer interactions: Simulations of dermaseptin B, an antimicrobial peptide. Biophys. Chem 1999, 76, 145–159. [Google Scholar]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial peptides in action. J. Am. Chem. Soc 2006, 128, 12156–12161. [Google Scholar]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [Google Scholar]
- Yesylevskyy, S.; Marrink, S.J.; Mark, A.E. Alternative mechanisms for the interaction of the cell-penetrating peptides penetratin and the TAT peptide with lipid bilayers. Biophys. J 2009, 97, 40–49. [Google Scholar]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar]
- Bond, P.J.; Sansom, M.S. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc 2006, 128, 2697–2704. [Google Scholar]
- Bond, P.J.; Parton, D.L.; Clark, J.F.; Sansom, M.S. Coarse-grained simulations of the membrane-active antimicrobial Peptide maculatin 1.1. Biophys. J 2008, 95, 3802–3815. [Google Scholar]
- Bond, P.J.; Holyoake, J.; Ivetac, A.; Khalid, S.; Sansom, M.S. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol 2007, 157, 593–605. [Google Scholar]
- Cox, K.; Sansom, M.S. One membrane protein, two structures and six environments: A comparative molecular dynamics simulation study of the bacterial outer membrane protein PagP. Mol. Membr. Biol 2009, 26, 205–214. [Google Scholar]
- Khalid, S.; Bond, P.J.; Holyoake, J.; Hawtin, R.W.; Sansom, M.S. DNA and lipid bilayers: Self-assembly and insertion. J. R. Soc. Interface 2008, 5, S241–S250. [Google Scholar]
- Risselada, H.J.; Marrink, S.J. Curvature effects on lipid packing and dynamics in liposomes revealed by coarse grained molecular dynamics simulations. Phys. Chem. Chem. Phys 2009, 11, 2056–2067. [Google Scholar]
- Gkeka, P.; Sarkisov, L. Spontaneous formation of a barrel-stave pore in a coarse-grained model of the synthetic LS3 peptide and a DPPC lipid bilayer. J. Phys. Chem. B 2009, 113, 6–8. [Google Scholar]
- Ladokhin, A.S.; Jayasinghe, S.; White, S.H. How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Anal. Biochem 2000, 285, 235–245. [Google Scholar]
- Christiaens, B.; Symoens, S.; Verheyden, S.; Engelborghs, Y.; Joliot, A.; Prochiantz, A.; Vandekerckhove, J.; Rosseneu, M.; Vanloo, B. Tryptophan fluorescence study of the interaction of penetratin peptides with model membranes. Eur. J. Biochem 2002, 269, 2918–2926. [Google Scholar]
- De Kroon, A.I.; Soekarjo, M.W.; de Gier, J.; de Kruijff, B. The role of charge and hydrophobicity in peptide-lipid interaction: A comparative study based on tryptophan fluorescence measurements combined with the use of aqueous and hydrophobic quenchers. Biochemistry 1990, 29, 8229–8240. [Google Scholar]
- Wimley, W.C.; White, S.H. Determining the membrane topology of peptides by fluorescence quenching. Biochemistry 2000, 39, 161–170. [Google Scholar]
- Esbjorner, E.K.; Oglecka, K.; Lincoln, P.; Graslund, A.; Norden, B. Membrane binding of pH-sensitive influenza fusion peptides. Positioning, configuration, and induced leakage in a lipid vesicle model. Biochemistry 2007, 46, 13490–13504. [Google Scholar]
- Ortiz, A.; Cajal, Y.; Haro, I.; Reig, F.; Alsina, M.A. Fluorescence study on the interaction of a multiple antigenic peptide from hepatitis A virus with lipid vesicles. Biopolymers 2000, 53, 455–466. [Google Scholar]
- Moreno, M.R.; Perez-Berna, A.J.; Guillen, J.; Villalain, J. Biophysical characterization and membrane interaction of the most membranotropic region of the HIV-1 gp41 endodomain. Biochim. Biophys. Acta 2008, 1778, 1298–1307. [Google Scholar]
- Reshetnyak, Y.K.; Segala, M.; Andreev, O.A.; Engelman, D.M. A monomeric membrane peptide that lives in three worlds: In solution, attached to, and inserted across lipid bilayers. Biophys. J 2007, 93, 2363–2372. [Google Scholar]
- Bittova, L.; Stahelin, R.V.; Cho, W. Roles of ionic residues of the C1 domain in protein kinase C-alpha activation and the origin of phosphatidylserine specificity. J. Biol. Chem 2001, 276, 4218–4226. [Google Scholar]
- Stahelin, R.V.; Burian, A.; Bruzik, K.S.; Murray, D.; Cho, W. Membrane binding mechanisms of the PX domains of NADPH oxidase p40phox and p47phox. J. Biol. Chem 2003, 278, 14469–14479. [Google Scholar]
- Thomas, C.J.; Surolia, N.; Surolia, A. Surface plasmon resonance studies resolve the enigmatic endotoxin neutralizing activity of polymyxin B. J. Biol. Chem 1999, 274, 29624–29627. [Google Scholar]
- Mozsolits, H.; Wirth, H.J.; Werkmeister, J.; Aguilar, M.I. Analysis of antimicrobial peptide interactions with hybrid bilayer membrane systems using surface plasmon resonance. Biochim. Biophys. Acta 2001, 1512, 64–76. [Google Scholar]
- Papo, N.; Shai, Y. Can we predict biological activity of antimicrobial peptides from their interactions with model phospholipid membranes? Peptides 2003, 24, 1693–1703. [Google Scholar]
- Papo, N.; Shai, Y. Exploring peptide membrane interaction using surface plasmon resonance: Differentiation between pore formation versus membrane disruption by lytic peptides. Biochemistry 2003, 42, 458–466. [Google Scholar]
- Kremer, J.J.; Murphy, R.M. Kinetics of adsorption of beta-amyloid peptide Abeta(1–40) to lipid bilayers. J. Biochem. Biophys. Methods 2003, 57, 159–169. [Google Scholar]
- Kalb, E.; Frey, S.; Tamm, L.K. Formation of supported planar bilayers by fusion of vesicles to supported phospholipid monolayers. Biochim. Biophys. Acta 1992, 1103, 307–316. [Google Scholar]
- Myszka, D.G. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Curr. Opin. Biotechnol 1997, 8, 50–57. [Google Scholar]
- Karlsson, R.; Falt, A. Experimental design for kinetic analysis of protein-protein interactions with surface plasmon resonance biosensors. J. Immunol. Methods 1997, 200, 121–133. [Google Scholar]
- Schuck, P. Reliable determination of binding affinity and kinetics using surface plasmon resonance biosensors. Curr. Opin. Biotechnol 1997, 8, 498–502. [Google Scholar]
- Eid, M.; Rippa, S.; Castano, S.; Desbat, B.; Chopineau, J.; Rossi, C.; Beven, L. Exploring the membrane mechanism of the bioactive peptaibol ampullosporin a using lipid monolayers and supported biomimetic membranes. J. Biophys 2010, 2010, 179641. [Google Scholar]
- Joanne, P.; Galanth, C.; Goasdoué, N.; Nicolas, P.; Sagan, S.; Lavielle, S.; Chassaing, G.; el Amri, C.; Alves, I.D. Lipid reorganization induced by membrane-active peptides probed using differential scanning calorimetry. Biochim. Biophys. Acta (BBA) 2009, 1788, 1772–1781. [Google Scholar]
- Prenner, E.J.; Lewis, R.N.A.H.; Kondejewski, L.H.; Hodges, R.S.; McElhaney, R.N. Differential scanning calorimetric study of the effect of the antimicrobial peptide gramicidin S on the thermotropic phase behavior of phosphatidylcholine, phosphatidylethanolamine and phosphatidylglycerol lipid bilayer membranes. Biochim. Biophys. Acta 1999, 1417, 211–223. [Google Scholar]
- Woody, R.W. Circular dichroism. Methods Enzymol 1995, 246, 34–71. [Google Scholar]
- Wallace, B.A.; Lees, J.G.; Orry, A.J.; Lobley, A.; Janes, R.W. Analyses of circular dichroism spectra of membrane proteins. Protein Sci 2003, 12, 875–884. [Google Scholar]
- Wu, Y.; Huang, H.W.; Olah, G.A. Method of oriented circular dichroism. Biophys. J 1990, 57, 797–806. [Google Scholar]
- Nielsen, S.B.; Otzen, D.E. Impact of the antimicrobial peptide Novicidin on membrane structure and integrity. J. Colloid Interface Sci 2010, 345, 248–256. [Google Scholar]
- Cheng, J.T.; Hale, J.D.; Elliot, M.; Hancock, R.E.; Straus, S.K. Effect of membrane composition on antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys. J 2009, 96, 552–565. [Google Scholar]
- Alves, I.D.; Jiao, C.Y.; Aubry, S.; Aussedat, B.; Burlina, F.; Chassaing, G.; Sagan, S. Cell biology meets biophysics to unveil the different mechanisms of penetratin internalization in cells. Biochim. Biophys. Acta 2010, 1798, 2231–2239. [Google Scholar]
- Kamei, N.; Nielsen, E.J.B.; Khafagy, E.-S.; Takeda-Morishita, M. Noninvasive insulin delivery: The great potential of cell-penetrating peptides. Ther. Deliv 2013, 4, 315–326. [Google Scholar]
- MacEwan, S.R.; Chilkoti, A. Harnessing the power of cell-penetrating peptides: Activatable carriers for targeting systemic delivery of cancer therapeutics and imaging agents. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 2013, 5, 31–48. [Google Scholar]
- Fominaya, J.; Wels, W. Target cell-specific DNA transfer mediated by a chimeric multidomain protein. Novel non-viral gene delivery system. J. Biol. Chem 1996, 271, 10560–10568. [Google Scholar]
- Deshayes, S.; Morris, M.; Heitz, F.; Divita, G. Delivery of proteins and nucleic acids using a non-covalent peptide-based strategy. Adv. Drug Deliv. Rev 2008, 60, 537–547. [Google Scholar]
- Deshayes, S.; Gerbal-Chaloin, S.; Morris, M.C.; Aldrian-Herrada, G.; Charnet, P.; Divita, G.; Heitz, F. On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids. Biochim. Biophys. Acta 2004, 1667, 141–147. [Google Scholar]
- Funhoff, A.M.; van Nostrum, C.F.; Lok, M.C.; Fretz, M.M.; Crommelin, D.J.; Hennink, W.E. Poly(3-guanidinopropyl methacrylate): A novel cationic polymer for gene delivery. Bioconjug. Chem 2004, 15, 1212–1220. [Google Scholar]
- Jiang, X.; Lok, M.C.; Hennink, W.E. Degradable-brushed pHEMA-pDMAEMA synthesized via ATRP and click chemistry for gene delivery. Bioconjug. Chem 2007, 18, 2077–2084. [Google Scholar]
- Mastrobattista, E.; Koning, G.A.; van Bloois, L.; Filipe, A.C.; Jiskoot, W.; Storm, G. Functional characterization of an endosome-disruptive peptide and its application in cytosolic delivery of immunoliposome-entrapped proteins. J. Biol. Chem 2002, 277, 27135–27143. [Google Scholar]
- Fretz, M.M.; Mastrobattista, E.; Koning, G.A.; Jiskoot, W.; Storm, G. Strategies for cytosolic delivery of liposomal macromolecules. Int. J. Pharm 2005, 298, 305–309. [Google Scholar]
- Simeoni, F.; Morris, M.C.; Heitz, F.; Divita, G. Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res 2003, 31, 2717–2724. [Google Scholar]
- Carberry, T.P.; Tarallo, R.; Falanga, A.; Finamore, E.; Galdiero, M.; Weck, M.; Galdiero, S. Dendrimer functionalization with a membrane-interacting domain of herpes simplex virus type 1: Towards intracellular delivery. Chemistry 2012, 18, 13678–13685. [Google Scholar]
- Guarnieri, D.; Falanga, A.; Muscetti, O.; Tarallo, R.; Fusco, S.; Galdiero, M.; Galdiero, S.; Netti, P.A. Shuttle-mediated nanoparticle delivery to the blood-brain barrier. Small 2013, 9, 853–862. [Google Scholar]
- Tarallo, R.; Carberry, T.P.; Falanga, A.; Vitiello, M.; Galdiero, S.; Galdiero, M.; Weck, M. Dendrimers functionalized with membrane-interacting peptides for viral inhibition. Int. J. Nanomed 2013, 8, 521–534. [Google Scholar]
- Falanga, A.; Tarallo, R.; Galdiero, E.; Cantisani, M.; Galdiero, M.; Galdiero, S. Review of a viral peptide nanosystem for intracellular delivery. J. Nanophotonics 2013, 7, 071599. [Google Scholar]
- Smaldone, G.; Falanga, A.; Capasso, D.; Guarnieri, D.; Correale, S.; Galdiero, M.; Netti, P.A.; Zollo, M.; Galdiero, S.; di Gaetano1, S.; et al. gH625 is a viral derived peptide for effective delivery of Intrinsically disordered proteins. Int. J. Nanomed 2013, 8, 2555–2565. [Google Scholar]
- Hard, T.; Lendel, C. Inhibition of amyloid formation. J. Mol. Biol 2012, 421, 441–465. [Google Scholar]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol 2013, 125, 795–813. [Google Scholar]
- Dorgeret, B.; Khemtemourian, L.; Correia, I.; Soulier, J.L.; Lequin, O.; Ongeri, S. Sugar-based peptidomimetics inhibit amyloid beta-peptide aggregation. Eur. J. Med. Chem 2011, 46, 5959–5969. [Google Scholar]
- Castelletto, V.; Cheng, G.; Hamley, I.W. Amyloid peptides incorporating a core sequence from the amyloid beta peptide and gamma amino acids: Relating bioactivity to self-assembly. Chem. Commun. (Camb) 2011, 47, 12470–12472. [Google Scholar]
- Kumar, S.; Miranker, A.D. A foldamer approach to targeting membrane bound helical states of islet amyloid polypeptide. Chem. Commun. (Camb. ) 2013, 49, 4749–4751. [Google Scholar]
- Tjernberg, L.O.; Naslund, J.; Lindqvist, F.; Johansson, J.; Karlstrom, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem 1996, 271, 8545–8548. [Google Scholar]
- Hughes, E.; Burke, R.M.; Doig, A.J. Inhibition of toxicity in the beta-amyloid peptide fragment beta-(25–35) using N-methylated derivatives: A general strategy to prevent amyloid formation. J. Biol. Chem 2000, 275, 25109–25115. [Google Scholar]
- Soto, C.; Sigurdsson, E.M.; Morelli, L.; Kumar, R.A.; Castano, E.M.; Frangione, B. Beta-Sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer’s therapy. Nat. Med 1998, 4, 822–826. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Galdiero, S.; Falanga, A.; Cantisani, M.; Vitiello, M.; Morelli, G.; Galdiero, M. Peptide-Lipid Interactions: Experiments and Applications. Int. J. Mol. Sci. 2013, 14, 18758-18789. https://doi.org/10.3390/ijms140918758
Galdiero S, Falanga A, Cantisani M, Vitiello M, Morelli G, Galdiero M. Peptide-Lipid Interactions: Experiments and Applications. International Journal of Molecular Sciences. 2013; 14(9):18758-18789. https://doi.org/10.3390/ijms140918758
Chicago/Turabian StyleGaldiero, Stefania, Annarita Falanga, Marco Cantisani, Mariateresa Vitiello, Giancarlo Morelli, and Massimiliano Galdiero. 2013. "Peptide-Lipid Interactions: Experiments and Applications" International Journal of Molecular Sciences 14, no. 9: 18758-18789. https://doi.org/10.3390/ijms140918758