Effect of Lowering Asymmetric Dimethylarginine (ADMA) on Vascular Pathology in Atherosclerotic ApoE-Deficient Mice with Reduced Renal Mass

Abstract
:1. Introduction
2. Results
2.1. Animal Data and Laboratory Chemistry
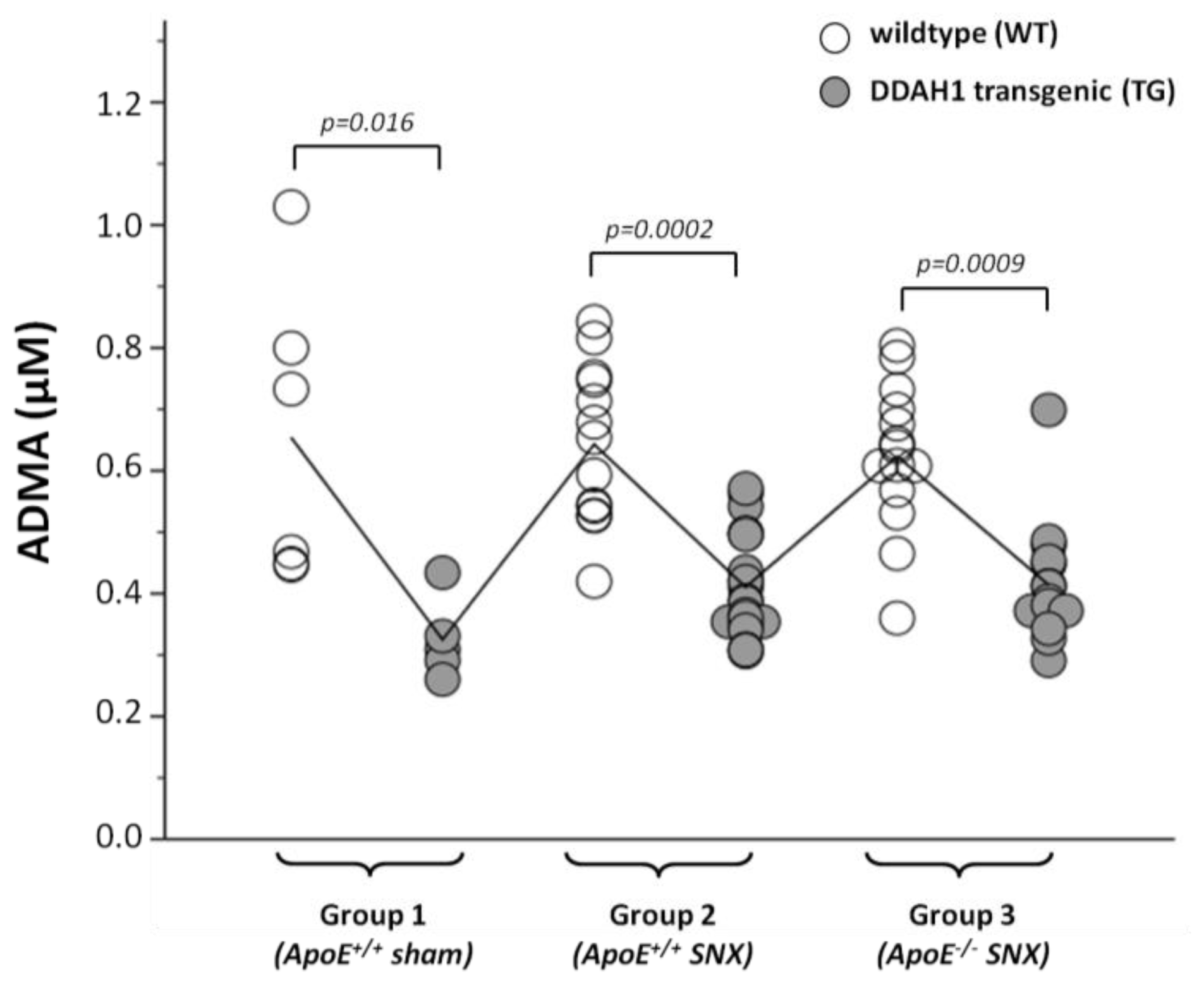
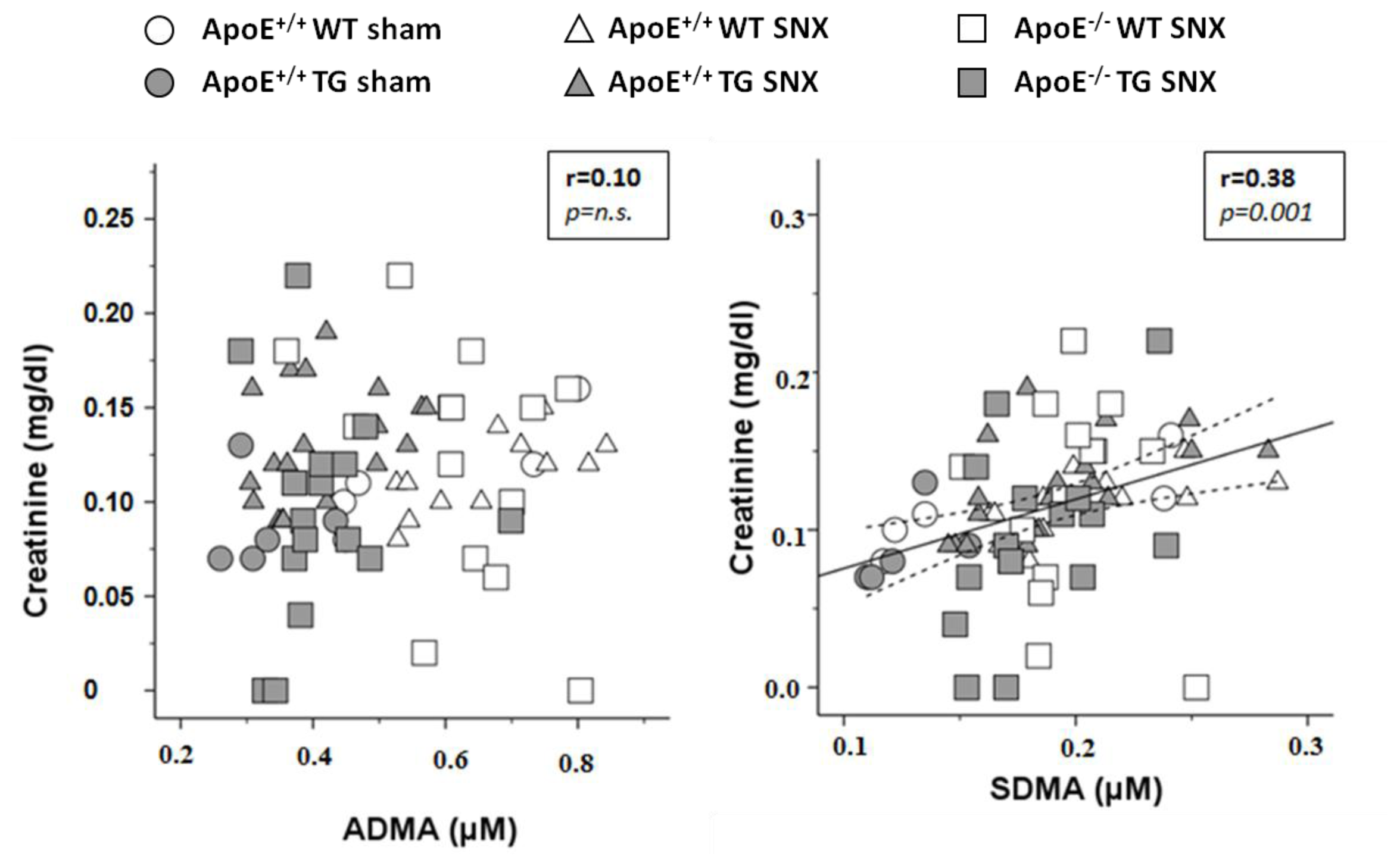
2.2. Measurement of Methylarginines and l-Arginine
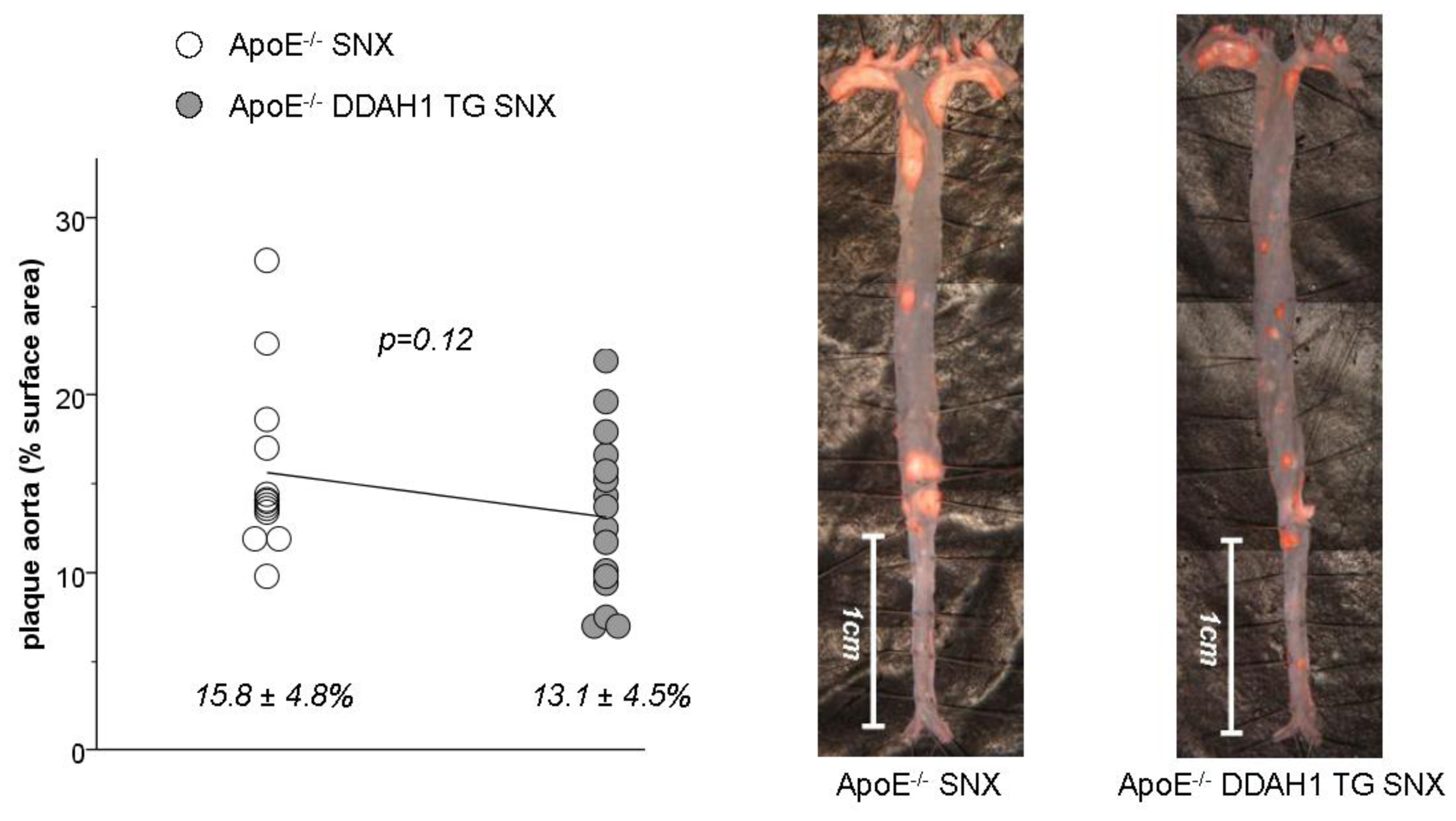
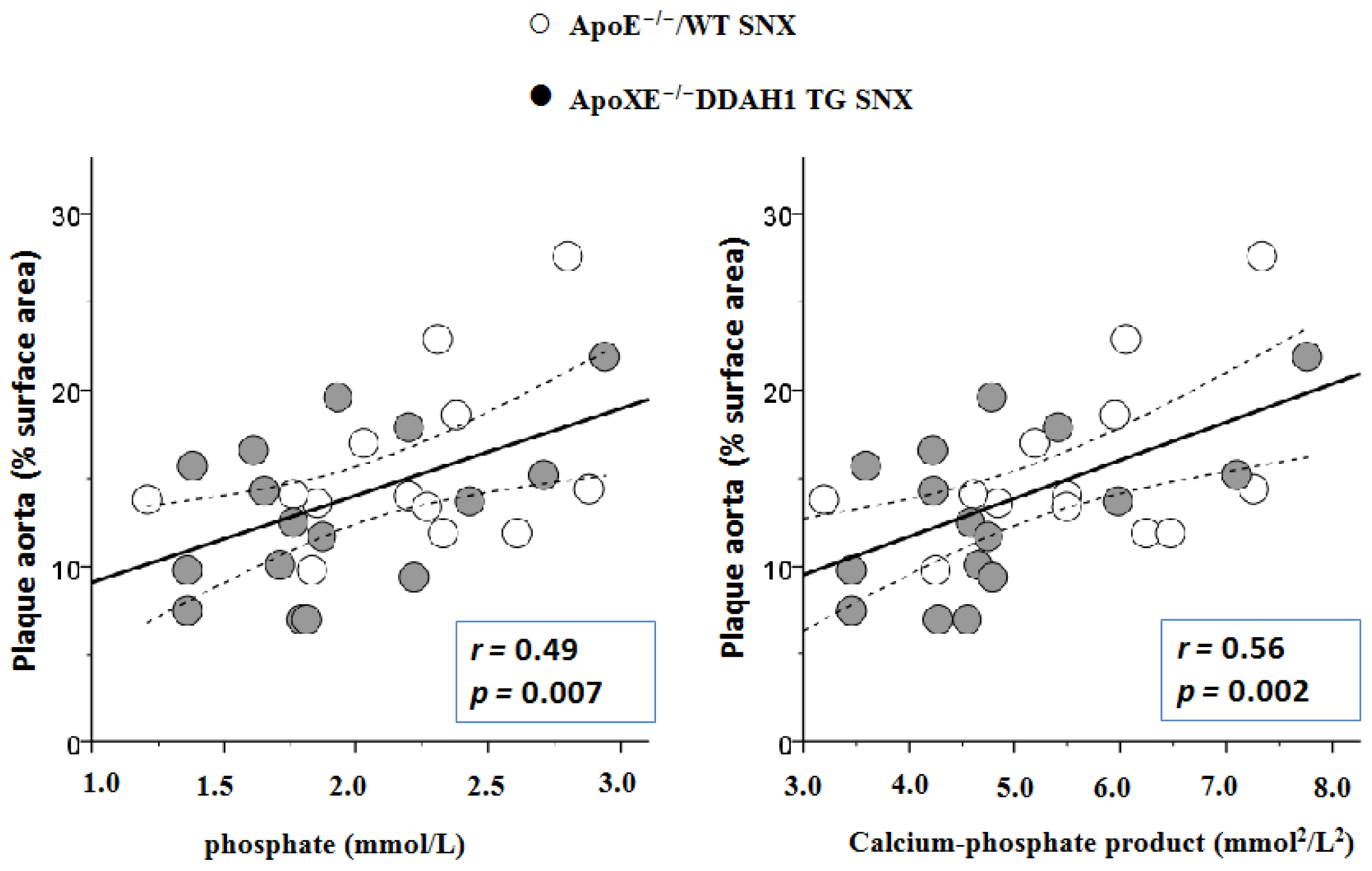
2.3. En Face Preparations of the Aorta
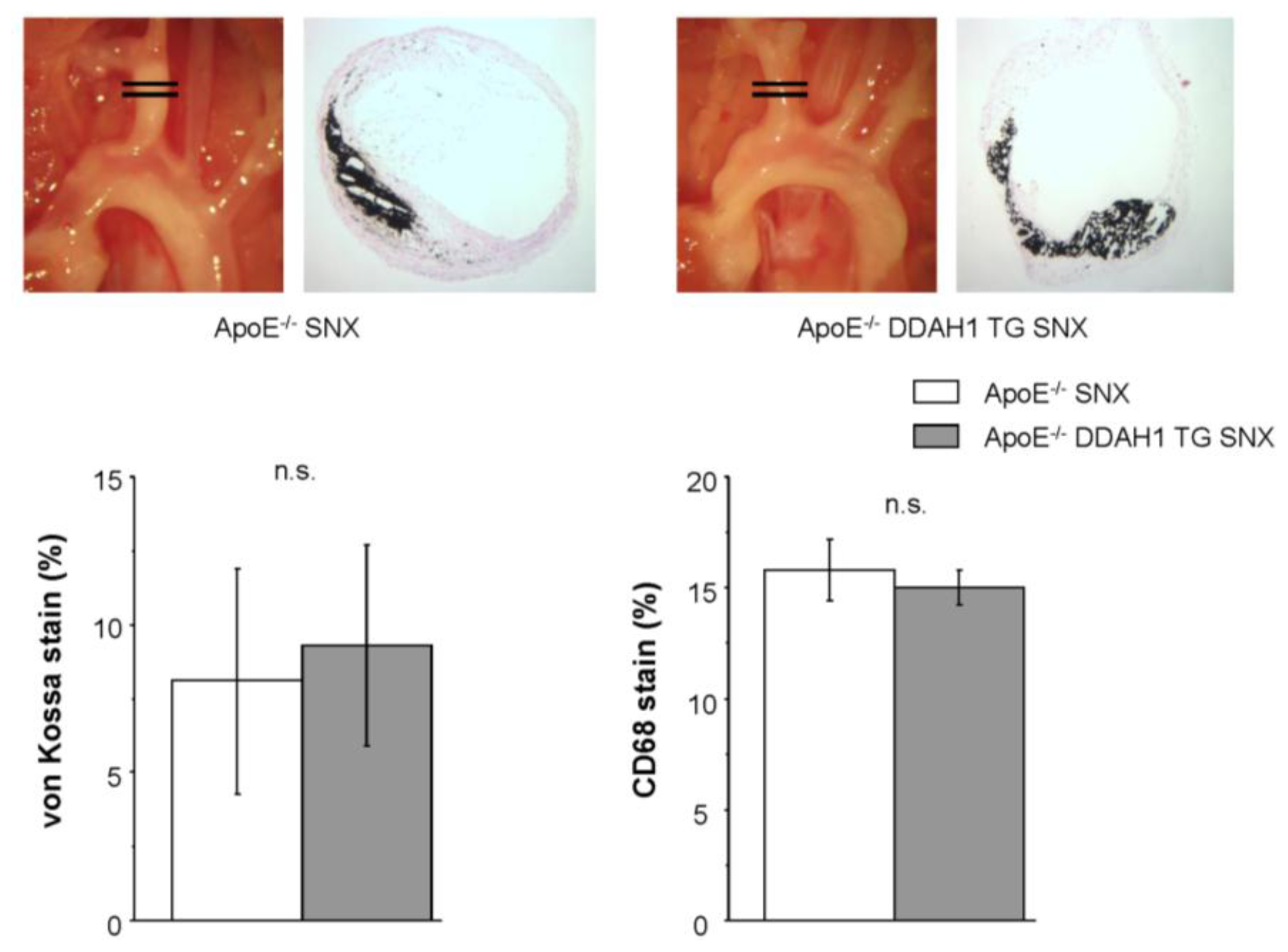
2.4. Histomorphometry and Immunohistochemistry of the Brachiocephalic Trunk
2.5. Discussion
3. Experimental Section
Animals
4. Experimental Groups
4.1. Three Groups of Mice Were Studied (Total n = 77)
4.2. Subtotal Nephrectomy (SNX)
4.3. Blood Pressure Measurements
4.4. Laboratory Tests
4.5. Measurement of Methylarginines and l-Arginine
4.6. En Face Preparations of the Aorta
4.7. Histomorphometry and Immunohistochemistry of the Brachiocephalic Trunk and Remnant Kidneys
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsJ.J.: designed the study, performed experiments and data acquisition, performed statistical analysis and wrote the manuscript; R.M.: performed measurement of methylarginines, was involved in data interpretation and writing of the manuscript; M.A.: performed experiments; N.C.: performed experiments; K.F.H.: designed and supervised the study, was involved in data interpretation and writing of the manuscript.
References
- Cooke, J.P. Asymmetrical dimethylarginine: The Uber marker? Circulation 2004, 109, 1813–1818. [Google Scholar]
- Dayoub, H.; Achan, V.; Adimoolam, S.; Jacobi, J.; Stuehlinger, M.C.; Wang, B.Y.; Tsao, P.S.; Kimoto, M.; Vallance, P.; Patterson, A.J.; et al. Dimethylarginine dimethylaminohydrolase regulates nitric oxide synthesis: Genetic and physiological evidence. Circulation 2003, 108, 3042–3047. [Google Scholar]
- Hasegawa, K.; Wakino, S.; Tatematsu, S.; Yoshioka, K.; Homma, K.; Sugano, N.; Kimoto, M.; Hayashi, K.; Itoh, H. Role of asymmetric dimethylarginine in vascular injury in transgenic mice overexpressing dimethylarginie dimethylaminohydrolase 2. Circ. Res 2007, 101, e2–e10. [Google Scholar]
- Hu, X.; Atzler, D.; Xu, X.; Zhang, P.; Guo, H.; Lu, Z.; Fassett, J.; Schwedhelm, E.; Böger, R.H.; Bache, R.J.; et al. Dimethylarginine dimethylaminohydrolase-1 is the critical enzyme for degrading the cardiovascular risk factor asymmetrical dimethylarginine. Arterioscler. Thromb. Vasc. Biol. A 2011, 31, 1540–1546. [Google Scholar]
- Jacobi, J.; Maas, R.; Cardounel, A.J.; Arend, M.; Pope, A.J.; Cordasic, N.; Heusinger-Ribeiro, J.; Atzler, D.; Strobel, J.; Schwedhelm, E.; et al. Dimethylarginine dimethylaminohydrolase overexpression ameliorates atherosclerosis in apolipoprotein E-deficient mice by lowering asymmetric dimethylarginine. Am. J. Pathol 2010, 176, 2559–2570. [Google Scholar]
- Leiper, J.; Nandi, M.; Torondel, B.; Murray-Rust, J.; Malaki, M.; O’Hara, B.; Rossiter, S.; Anthony, S.; Madhani, M.; Selwood, D.; et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat. Med 2007, 13, 198–203. [Google Scholar]
- Leiper, J.; Nandi, M. The therapeutic potential of targeting endogenous inhibitors of nitric oxide synthesis. Nat. Rev. Drug Discov 2011, 10, 277–291. [Google Scholar]
- Leiper, J.M.; Santa Maria, J.; Chubb, A.; MacAllister, R.J.; Charles, I.G.; Whitley, G.S.; Vallance, P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem. J 1999, 343, 209–214. [Google Scholar]
- Fliser, D.; Kronenberg, F.; Kielstein, J.T.; Morath, C.; Bode-Boger, S.M.; Haller, H.; Ritz, E. Asymmetric dimethylarginine and progression of chronic kidney disease: The mild to moderate kidney disease study. J. Am. Soc. Nephrol 2005, 16, 2456–2461. [Google Scholar]
- Kielstein, J.T.; Boger, R.H.; Bode-Boger, S.M.; Frolich, J.C.; Haller, H.; Ritz, E.; Fliser, D. Marked increase of asymmetric dimethylarginine in patients with incipient primary chronic renal disease. J. Am. Soc. Nephrol 2002, 13, 170–176. [Google Scholar]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar]
- Zoccali, C.; Bode-Boger, S.; Mallamaci, F.; Benedetto, F.; Tripepi, G.; Malatino, L.; Cataliotti, A.; Bellanuova, I.; Fermo, I.; Frölich, J.; et al. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: A prospective study. Lancet 2001, 358, 2113–2117. [Google Scholar]
- Jacobi, J.; Tsao, P.S. Asymmetrical dimethylarginine in renal disease: Limits of variation or variation limits? A systematic review. Am. J. Nephrol 2008, 28, 224–237. [Google Scholar]
- Jacobi, J.; Maas, R.; Cordasic, N.; Koch, K.; Schmieder, R.E.; Boger, R.H.; Hilgers, K.F. Role of asymmetric dimethylarginine for angiotensin II-induced target organ damage in mice. Am. J. Physiol. Heart Circ. Physiol 2008, 294, H1058–H1066. [Google Scholar]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Occurrence of a new enzyme catalyzing the direct conversion of NG,NG-dimethyl-l-arginine to l-citrulline in rats. Biochem. Biophys. Res. Commun 1987, 148, 671–677. [Google Scholar]
- Caplin, B.; Wang, Z.; Slaviero, A.; Tomlinson, J.; Dowsett, L.; Delahaye, M.; Salama, A.; Wheeler, D.C.; Leiper, J. International Consortium for Blood Pressure Genome-Wide Association Studies. Alanine-glyoxylate aminotransferase-2 metabolizes endogenous methylarginines, regulates NO, and controls blood pressure. Arterioscler. Thromb. Vasc. Biol 2012, 32, 2892–2900. [Google Scholar]
- Bugnicourt, J.M.; Da Silveira, C.; Bengrine, A.; Godefroy, O.; Baumbach, G.; Sevestre, H.; Bode-Boeger, S.M.; Kielstein, J.T.; Massy, Z.A.; Chillon, J.M. Chronic renal failure alters endothelial function in cerebral circulation in mice. Am. J. Physiol. Heart Circ. Physiol 2011, 301, H1143–H1152. [Google Scholar]
- Kajimoto, H.; Kai, H.; Aoki, H.; Yasuoka, S.; Anegawa, T.; Aoki, Y.; Ueda, S.; Okuda, S.; Imaizumi, T. Inhibition of eNOS phosphorylation mediates endothelial dysfunction in renal failure: New effect of asymmetric dimethylarginine. Kidney Int 2012, 81, 762–768. [Google Scholar]
- Mizobuchi, M.; Towler, D.; Slatopolsky, E. Vascular calcification: The killer of patients with chronic kidney disease. J. Am. Soc. Nephrol 2009, 20, 1453–1464. [Google Scholar]
- Phan, O.; Ivanovski, O.; Nikolov, I.G.; Joki, N.; Maizel, J.; Louvet, L.; Chasseraud, M.; Nguyen-Khoa, T.; Lacour, B.; Drüeke, T.B.; et al. Effect of oral calcium carbonate on aortic calcification in apolipoprotein E-deficient (apoE−/−) mice with chronic renal failure. Nephrol. Dial. Transp 2008, 23, 82–90. [Google Scholar]
- Ivanovski, O.; Nikolov, I.G.; Joki, N.; Caudrillier, A.; Phan, O.; Mentaverri, R.; Maizel, J.; Hamada, Y.; Nguyen-Khoa, T.; Fukagawa, M.; et al. The calcimimetic R-568 retards uremia-enhanced vascular calcification and atherosclerosis in apolipoprotein E deficient (apoE−/−) mice. Atherosclerosis 2009, 205, 55–62. [Google Scholar]
- Bro, S.; Bentzon, J.F.; Falk, E.; Andersen, C.B.; Olgaard, K.; Nielsen, L.B. Chronic renal failure accelerates atherogenesis in apolipoprotein E-deficient mice. J. Am. Soc. Nephrol 2003, 14, 2466–2474. [Google Scholar]
- Buzello, M.; Tornig, J.; Faulhaber, J.; Ehmke, H.; Ritz, E.; Amann, K. The apolipoprotein e knockout mouse: A model documenting accelerated atherogenesis in uremia. J. Am. Soc. Nephrol 2003, 14, 311–316. [Google Scholar]
- Massy, Z.A.; Ivanovski, O.; Nguyen-Khoa, T.; Angulo, J.; Szumilak, D.; Mothu, N.; Phan, O.; Daudon, M.; Lacour, B.; Drüeke, T.B.; et al. Uremia accelerates both atherosclerosis and arterial calcification in apolipoprotein E knockout mice. J. Am. Soc. Nephrol 2005, 16, 109–116. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ApoE+/+ WT sham (n = 6) | ApoE+/+ DDAH1 TG sham (n = 5) | ApoE+/+ WT SNX (n = 13) | ApoE+/+ DDAH1 TG SNX (n = 23) | ApoE−/− WT SNX (n = 14) | ApoE−/− DDAH1 TG SNX (n = 16) | ANOVA p-value | DDAH1 effect p-value | SNX effect p-value | ApoE effect p-value | |
|---|---|---|---|---|---|---|---|---|---|---|
| weight (g) | 31.6 ± 1.2 | 32.3 ± 1.0 | 30.2 ± 1.9 | 31.3 ± 1.9 | 30.5 ± 2.6 | 31.8 ± 2.4 | n.s. | 0.043 | n.s. | n.s. |
| kidney weight (mg/g BW) | 16.9 ± 1.5 | 16.8 ± 1.9 | 7.5 ± 1.1 | 7.4 ± 1.9 | 9.0 ± 1.6 | 8.6 ± 1.0 | 0.0002 | n.s. | 0.0001 | n.s. |
| glomerular diameter (μm) | 169 ± 15 | 170 ± 10 | 195 ± 14 | 187 ± 17 | 234 ± 36 | 224 ± 30 | 0.0002 | n.s. | 0.001 | 0.0007 |
| glomerular collagen 4 (%) | 2.24 ± 1.70 | 3.55 ± 4.31 | 5.85 ± 3.81 | 4.78 ± 4.09 | 26.23 ± 7.68 | 21.26 ± 5.59 | 0.0001 | n.s. | 0.006 | 0.0005 |
| mean arterial BP (mmHg)* | 107 ± 7 | 111 ± 5 | 109 ± 5 | 109 ± 7 | 102 ± 12 | 108 ± 12 | n.s. | n.s. | n.s. | n.s. |
| heart rate (beats/min)* | 610 ± 35 | 596 ± 38 | 594 ± 54 | 630 ± 36 | 574 ± 53 | 610 ± 44 | n.s. | n.s. | n.s. | n.s. |
| creatinine (mg/dL) | 0.11 ± 0.03 | 0.09 ± 0.03 | 0.12 ± 0.02 | 0.13 ± 0.03 | 0.12 ± 0.06 | 0.10 ± 0.06 | n.s. | n.s. | 0.010 | n.s. |
| urea (mg/dL) | 73.8 ± 11.6 | 68.1 ± 18.4 | 87.3 ± 18.0 | 92.1 ± 15.3 | 104.7 ± 33.5 | 102.9 ± 17.7 | 0.002 | n.s. | 0.0004 | 0.001 |
| cholesterol (mg/dL) | 78 ± 7 | 76 ± 15 | 76 ± 13 | 80 ± 7 | 480 ± 142 | 447 ± 166 | 0.0002 | n.s. | 0.011 | 0.0001 |
| triglycerides (mg/dL) | 108 ± 21 | 81 ± 28 | 74 ± 42 | 90 ± 39 | 138 ± 84 | 136 ± 72 | 0.013 | n.s. | n.s. | 0.0003 |
| total protein (g/L) | 46.7 ± 7.4 | 43.5 ± 7.0 | 49.0 ± 19.9 | 48.4 ± 3.3 | 40.0 ± 14.2 | 45.0 ± 6.2 | n.s. | n.s. | n.s. | n.s. |
| albumin (g/L) | 30.7 ± 3.9 | 29.5 ± 3.9 | 32.3 ± 9.4 | 32.3 ± 2.3 | 23.6 ± 8.0 | 27.5 ± 3.7 | 0.004 | n.s. | n.s. | 0.0002 |
| phosphate (mmol/L) | 1.92 ± 0.28 | 2.27 ± 0.22 | 2.02 ± 0.54 | 1.88 ± 0.27 | 2.11 ± 0.53 | 1.92 ± 0.47 | n.s. | n.s. | n.s. | n.s. |
| ADMA ELISA (μmol/L) | 0.68 ± 0.33 | 0.27 ± 0.15 | 0.66 ± 0.08 | 0.48 ± 0.15 | 0.75 ± 0.20 | 0.59 ± 0.12 | 0.0008 | 0.0002 | n.s. | n.s. |
| ADMA (μmol/L) | 0.65 ± 0.24 | 0.32 ± 0.07 | 0.64 ± 0.13 | 0.41 ± 0.08 | 0.62 ± 0.12 | 0.41 ± 0.09 | 0.0003 | 0.0006 | n.s. | n.s. |
| SDMA (μmol/L) | 0.20 ± 0.08 | 0.13 ± 0.02 | 0.20 ± 0.04 | 0.19 ± 0.03 | 0.20 ± 0.02 | 0.18 ± 0.03 | 0.007 | 0.049 | 0.026 | n.s. |
| l-arginine (μmol/L) | 87.9 ± 19.4 | 92.0 ± 30.2 | 70.7 ± 35.9 | 75.4 ± 28.9 | 68.5 ± 30.4 | 83.0 ± 24.4 | n.s. | n.s. | n.s. | n.s. |
| l-arginine/ADMA ratio | 141 ± 28 | 278 ± 44 | 107 ± 42 | 181 ± 50 | 111 ± 45 | 202 ± 48 | 0.0005 | 0.0003 | 0.027 | n.s. |
| ApoE+/+ WT sham (n = 6) | ApoE+/+ DDAH1 TG sham (n = 5) | ApoE+/+ WT SNX (n = 13) | ApoE+/+ DDAH1 TG SNX (n = 23) | ApoE−/− WT SNX (n = 14) | ApoE−/− DDAH1 TG SNX (n = 16) | ANOVA p-value | DDAH effect p-value | SNX effect p-value | ApoE effect p-value | |
|---|---|---|---|---|---|---|---|---|---|---|
| pH | 7.37 ± 0.06 | 7.29 ± 0.02 | 7.33 ± 0.04 | 7.37 ± 0.06 | 7.31 ± 0.07 | 7.35 ± 0.04 | 0.011 | n.s. | n.s. | n.s. |
| pO2 (mmHg) | 121 ± 3 | 126 ± 11 | 136 ± 17 | 130 ± 24 | 131 ± 17 | 113 ± 25 | n.s. | n.s. | n.s. | n.s. |
| pCO2 (mmHg) | 37 ± 4 | 41 ± 4 | 37 ± 4 | 38 ± 8 | 41 ± 6 | 38 ± 11 | n.s. | n.s. | n.s. | n.s. |
| BE (mmol/L) | −3.7 ± 1.6 | −6.5 ± 2.4 | −6.0 ± 3.0 | −4.3 ± 4.3 | −5.0 ± 4.7 | −5.8 ± 3.7 | n.s. | n.s. | n.s. | n.s. |
| bicarbonate (mmol/L) | 20.9 ± 1.2 | 19.1 ± 2.0 | 19.1 ± 2.7 | 20.2 ± 4.4 | 20.0 ± 4.2 | 19.3 ± 3.2 | n.s. | n.s. | n.s. | n.s. |
| hemoglobin (g/dL) | 14.1 ± 0.5 | 13.8 ± 1.3 | 13.1 ± 1.2 | 12.7 ± 1.1 | 12.1 ± 1.6 | 12.8 ± 1.3 | n.s. | n.s. | 0.017 | n.s. |
| hematocrit (%) | 43.1 ± 1.5 | 42.3 ± 3.8 | 40.7 ± 3.4 | 39.2 ± 3.3 | 35.4 ± 7.7 | 39.5 ± 4.1 | 0.049 | n.s. | 0.038 | 0.030 |
| sodium (mmol/L) | 145.2 ± 0.8 | 146.9 ± 1.6 | 147.2 ± 2.5 | 143.3 ± 7.4 | 145.4 ± 4.1 | 144.1 ± 5.2 | n.s. | n.s. | n.s. | n.s. |
| potassium (mmol/L) | 3.5 ± 0.3 | 3.6 ± 0.2 | 3.5 ± 0.5 | 3.9 ± 0.3 | 3.9 ± 0.5 | 3.7 ± 0.4 | n.s. | n.s. | n.s. | n.s. |
| chloride (mmol/L) | 115.7 ± 1.2 | 116.2 ± 0.8 | 118.9 ± 5.0 | 116.4 ± 4.5 | 117.0 ± 2.2 | 118.6 ± 3.2 | n.s. | n.s. | n.s. | n.s. |
| ion calcium (mmol/L) | 1.29 ± 0.02 | 1.29 ± 0.04 | 1.31 ± 0.07 | 1.26 ± 0.05 | 1.27 ± 0.05 | 1.26 ± 0.06 | n.s. | n.s. | n.s. | n.s. |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jacobi, J.; Maas, R.; Arend, M.; Cordasic, N.; Hilgers, K.F. Effect of Lowering Asymmetric Dimethylarginine (ADMA) on Vascular Pathology in Atherosclerotic ApoE-Deficient Mice with Reduced Renal Mass. Int. J. Mol. Sci. 2014, 15, 5522-5535. https://doi.org/10.3390/ijms15045522
Jacobi J, Maas R, Arend M, Cordasic N, Hilgers KF. Effect of Lowering Asymmetric Dimethylarginine (ADMA) on Vascular Pathology in Atherosclerotic ApoE-Deficient Mice with Reduced Renal Mass. International Journal of Molecular Sciences. 2014; 15(4):5522-5535. https://doi.org/10.3390/ijms15045522
Chicago/Turabian StyleJacobi, Johannes, Renke Maas, Michaela Arend, Nada Cordasic, and Karl F. Hilgers. 2014. "Effect of Lowering Asymmetric Dimethylarginine (ADMA) on Vascular Pathology in Atherosclerotic ApoE-Deficient Mice with Reduced Renal Mass" International Journal of Molecular Sciences 15, no. 4: 5522-5535. https://doi.org/10.3390/ijms15045522