
Decreased Expression of SRSF2 Splicing Factor Inhibits Apoptotic Pathways in Renal Cancer

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
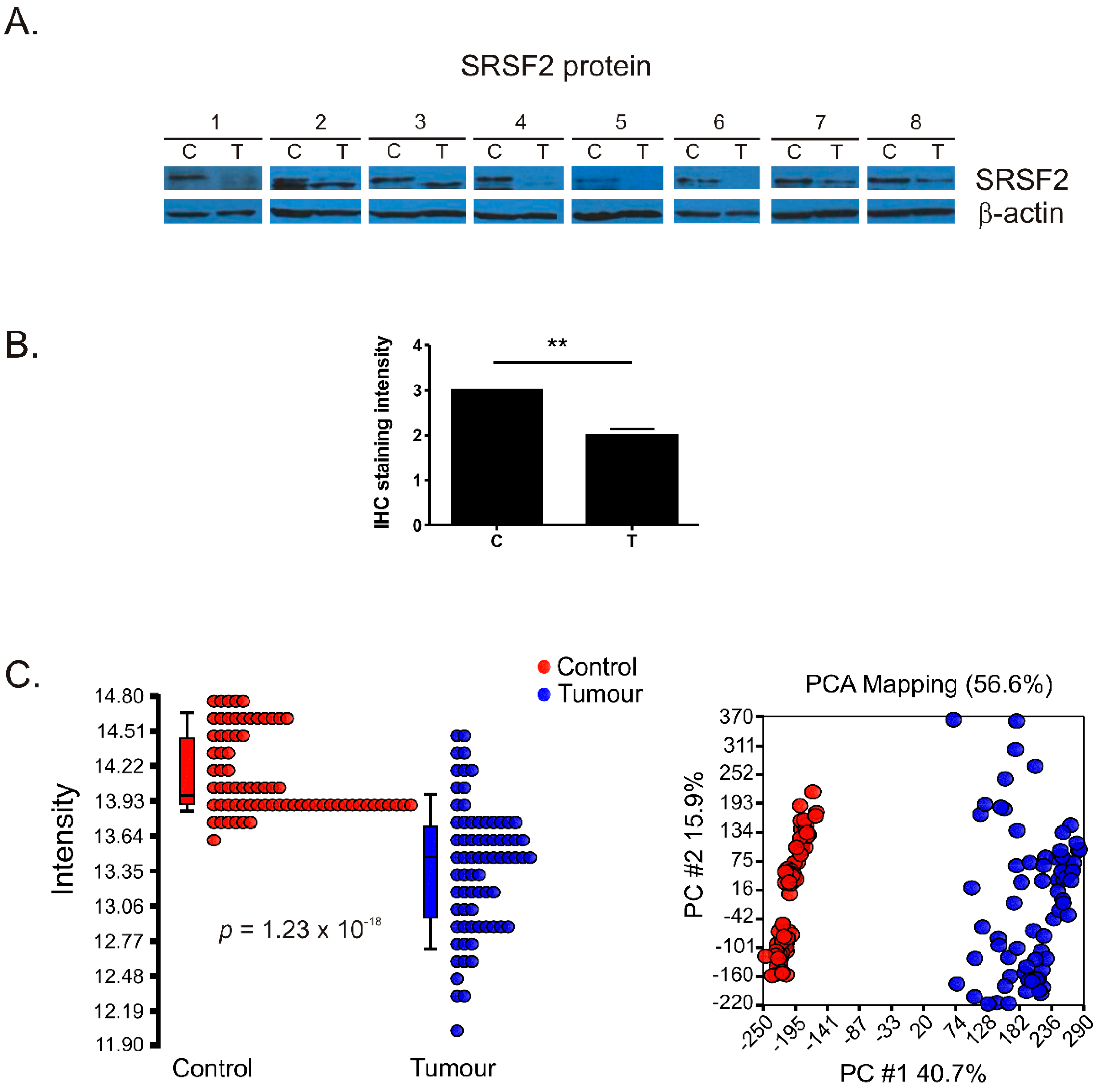
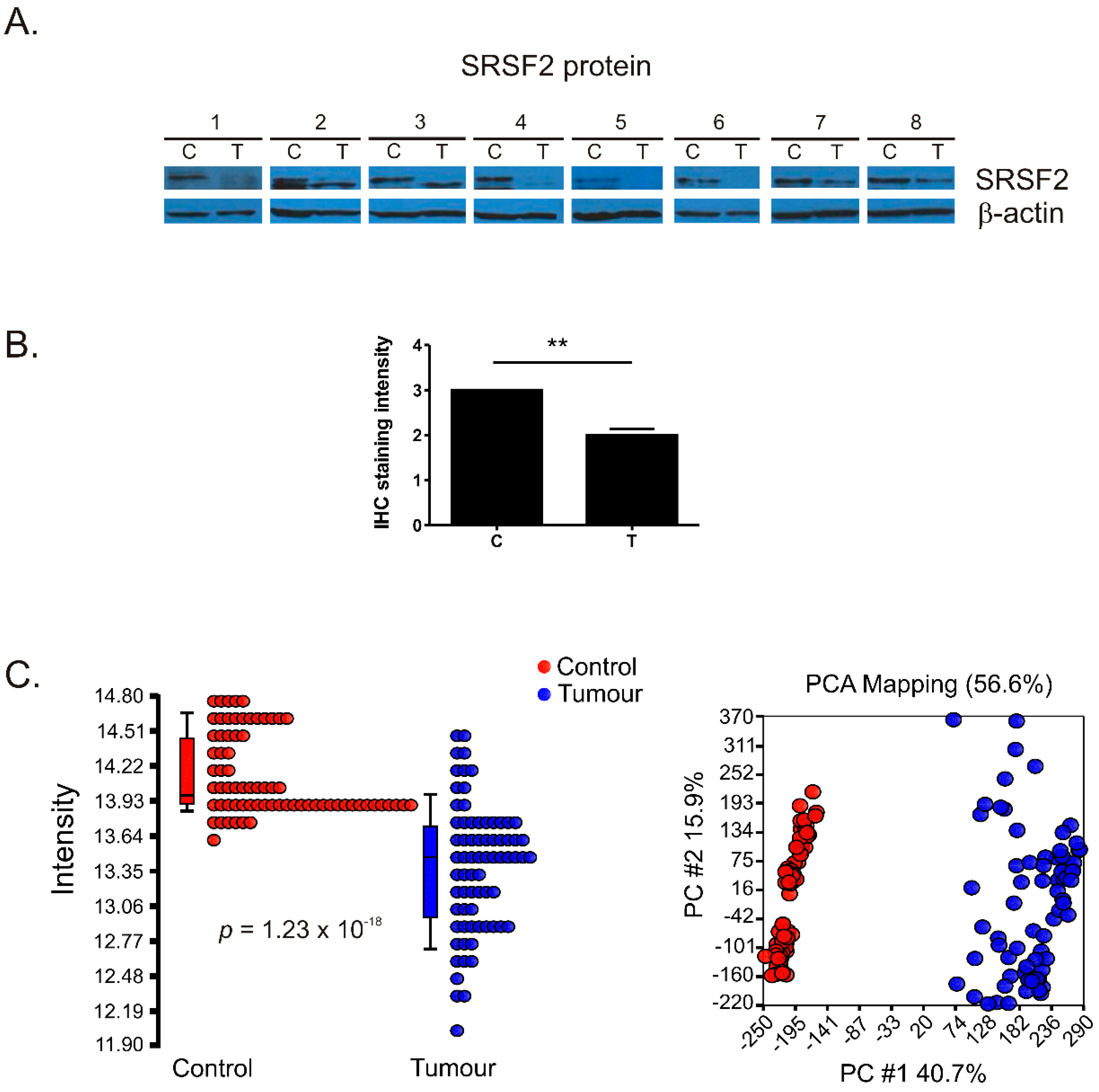
2.1. The Expression of SRSF2 Is Decreased in ccRCC
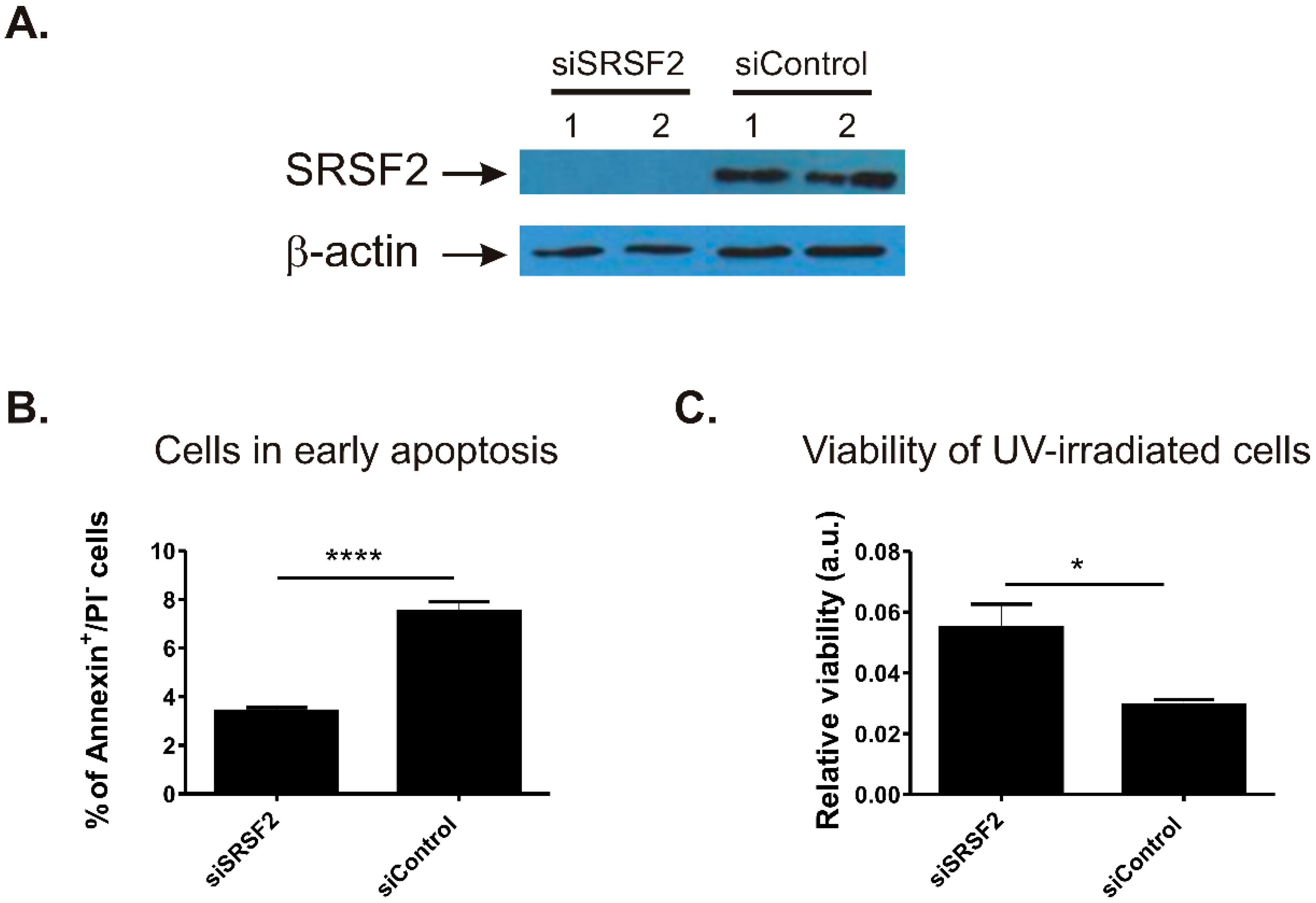
2.2. SRSF2 Silencing Protects Viability of Renal Cancer Cells
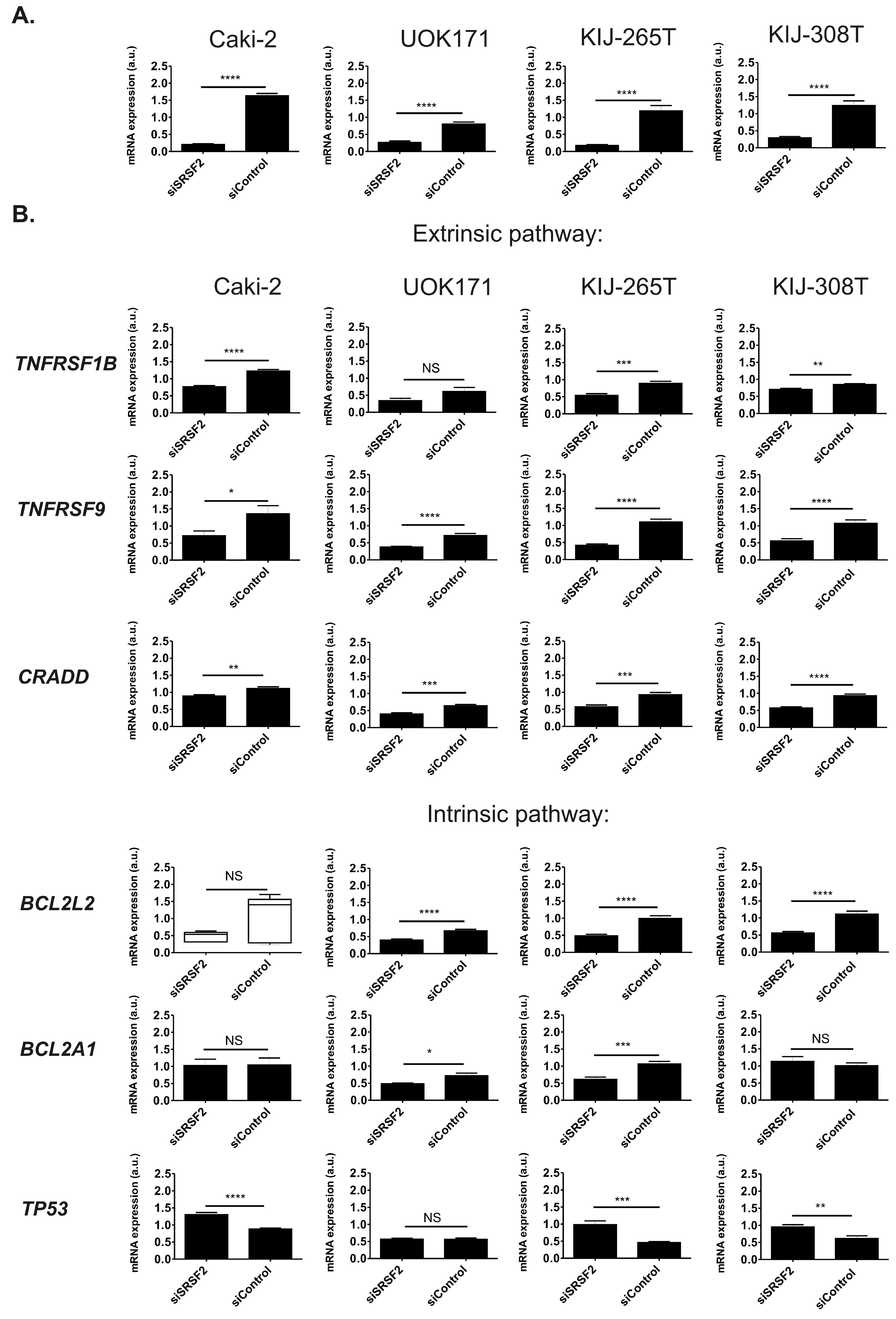
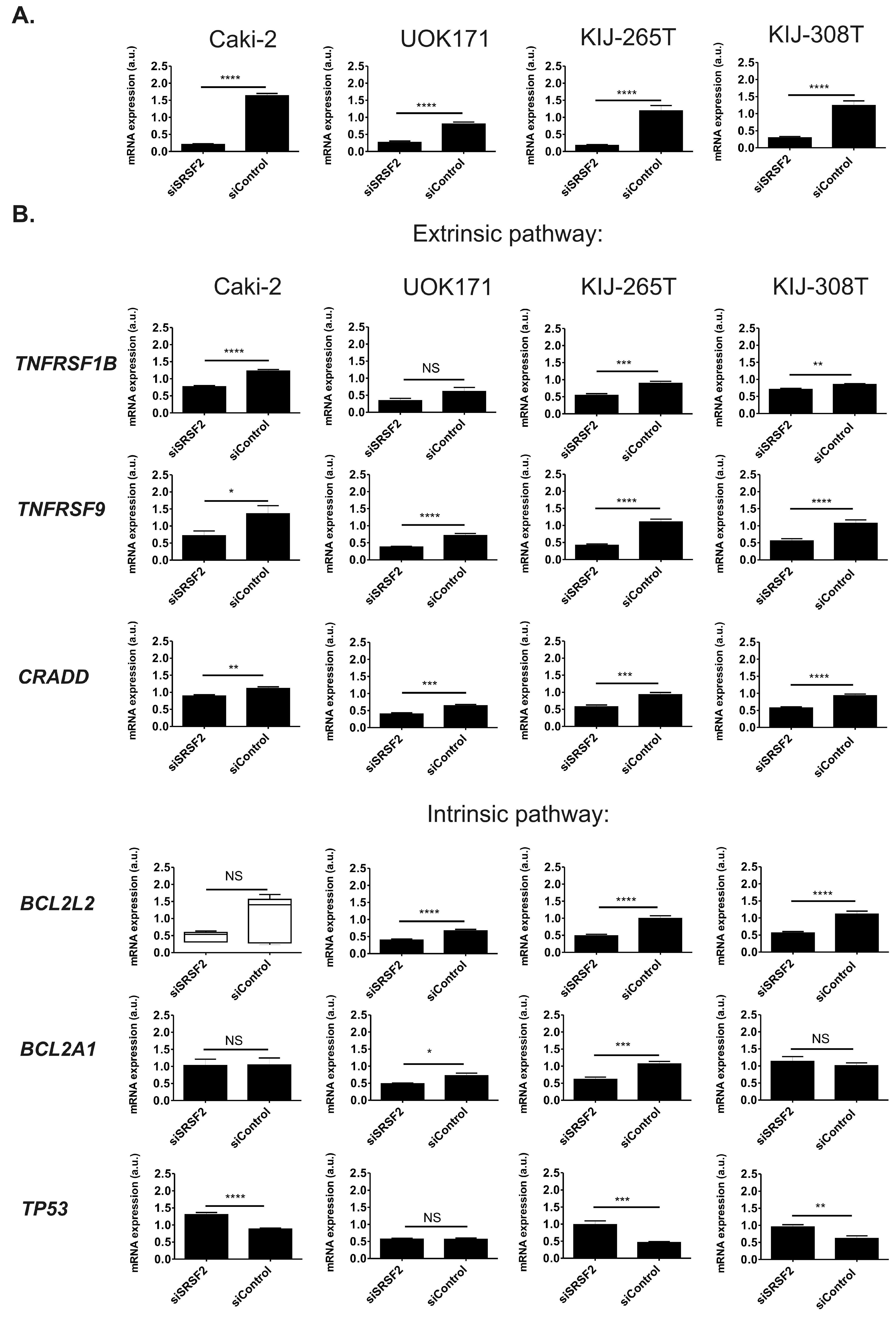
2.3. SRSF2 Silencing Induces Changes in Expression of Apoptotic Genes
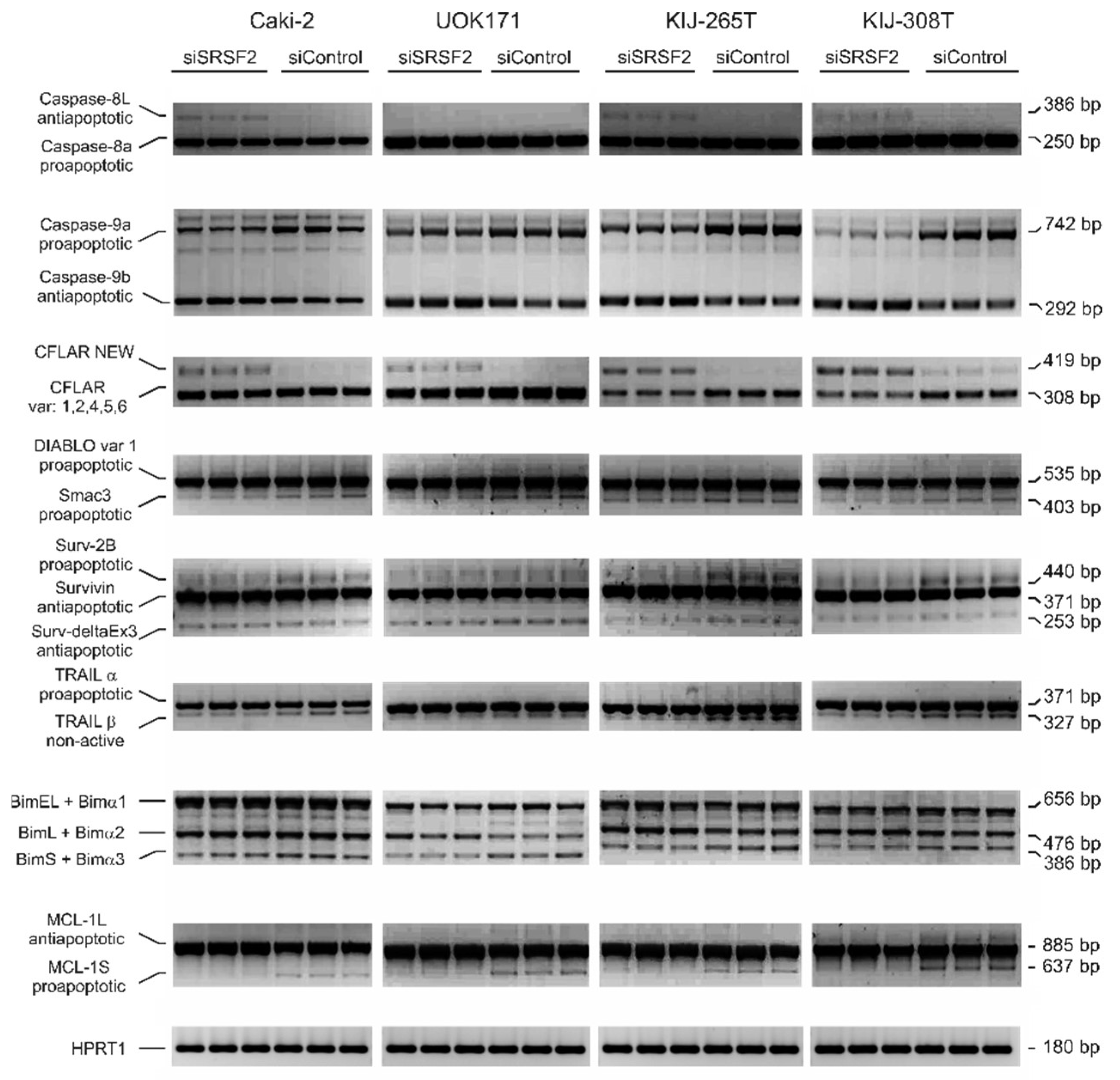
2.4. SRSF2 Silencing Affects Alternative Splicing of Apoptotic Genes
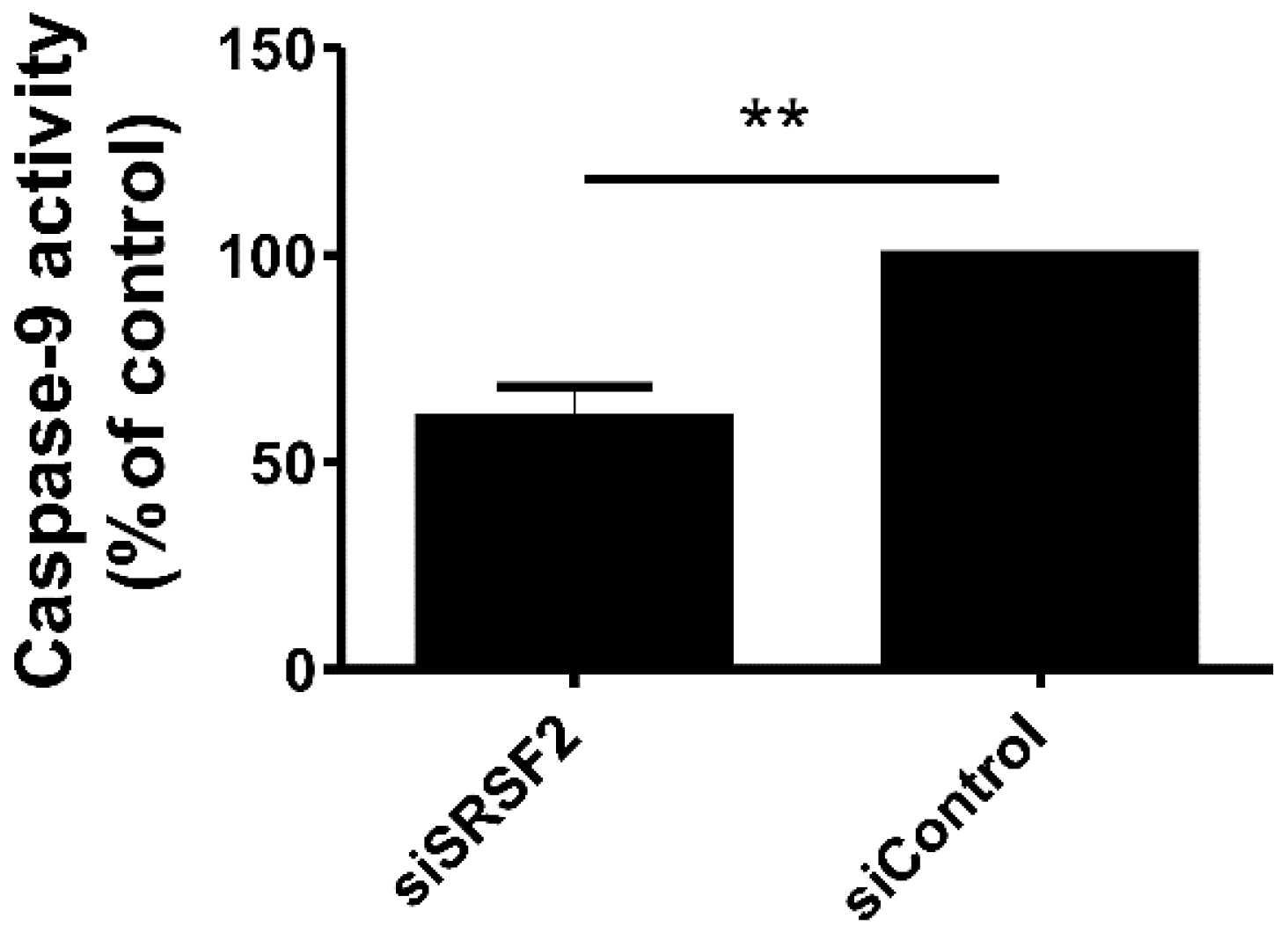
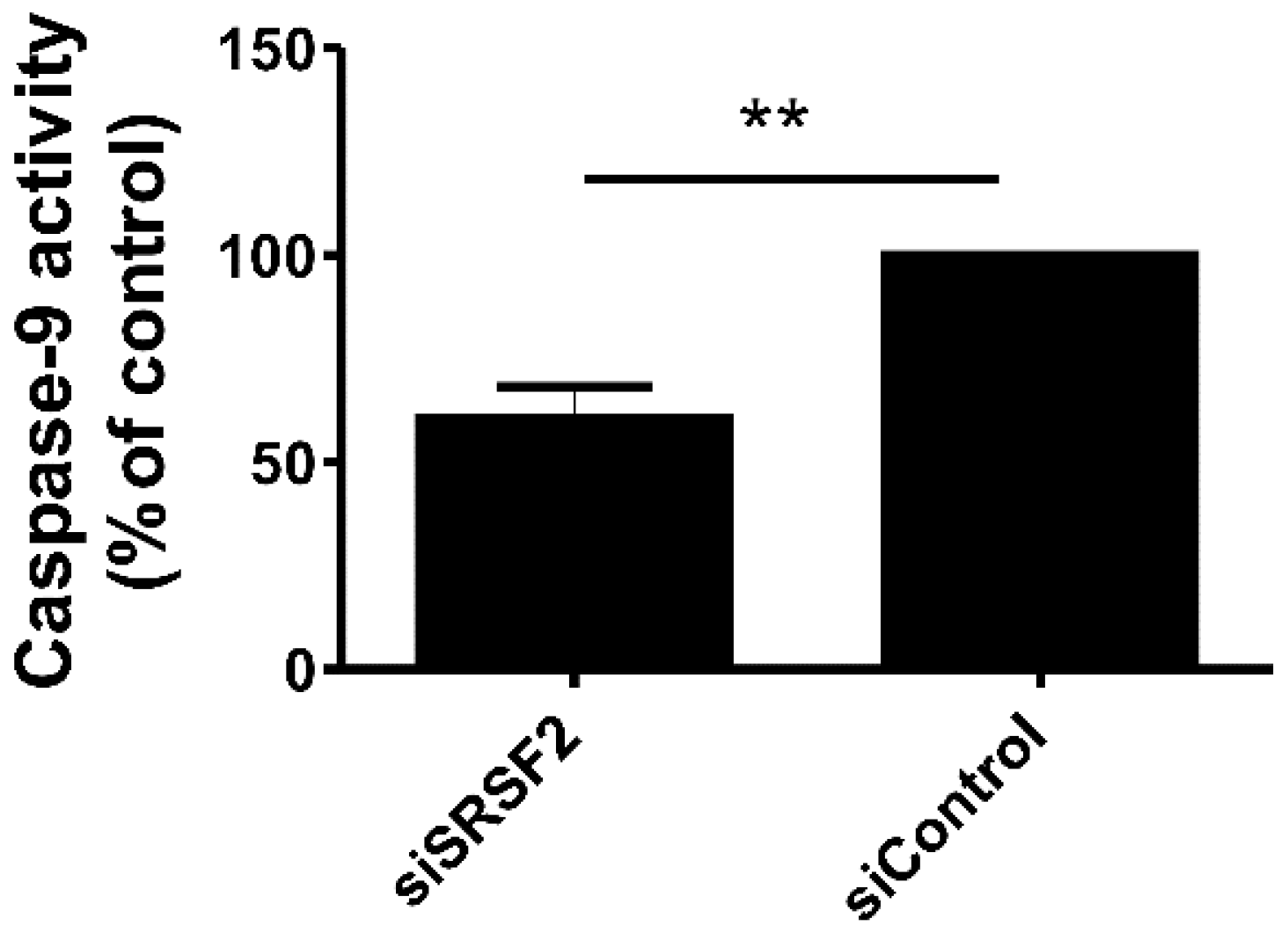
2.5. Silencing of SRSF2 Inhibits Activation of Caspase-9
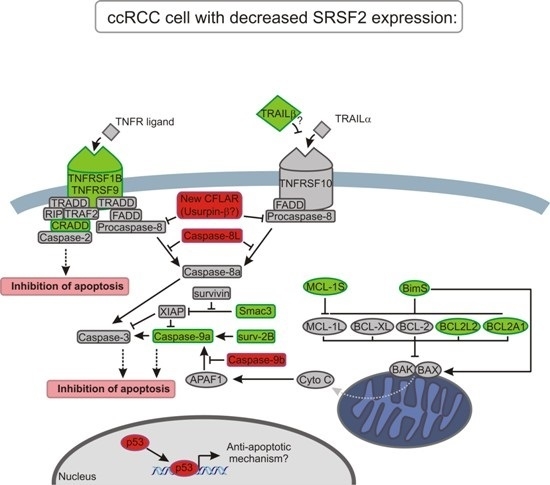
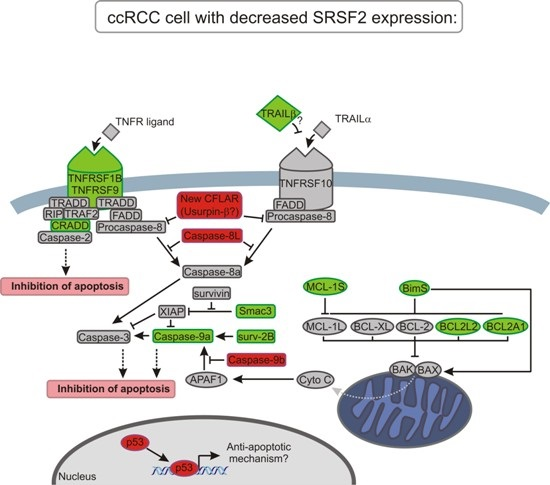
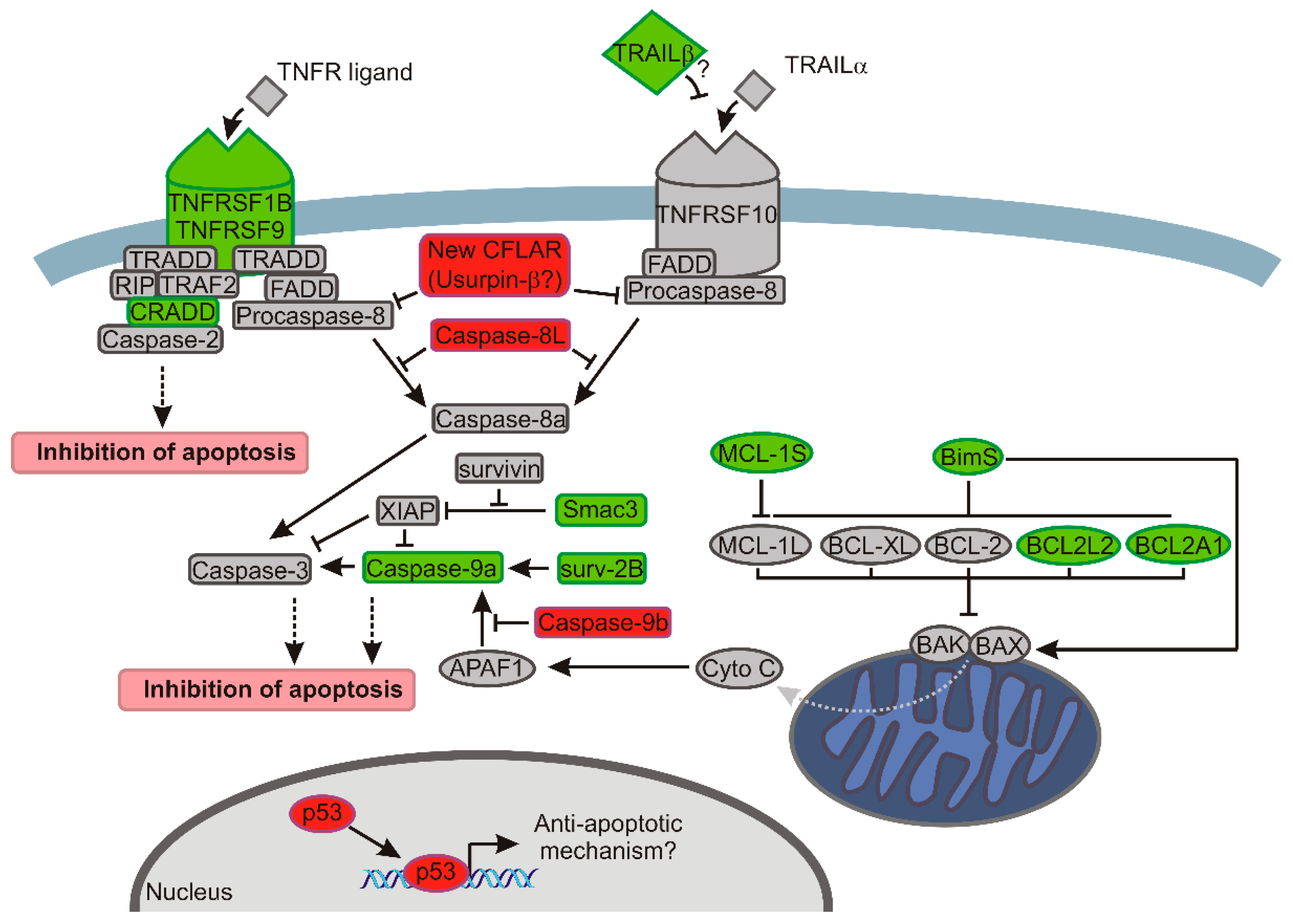
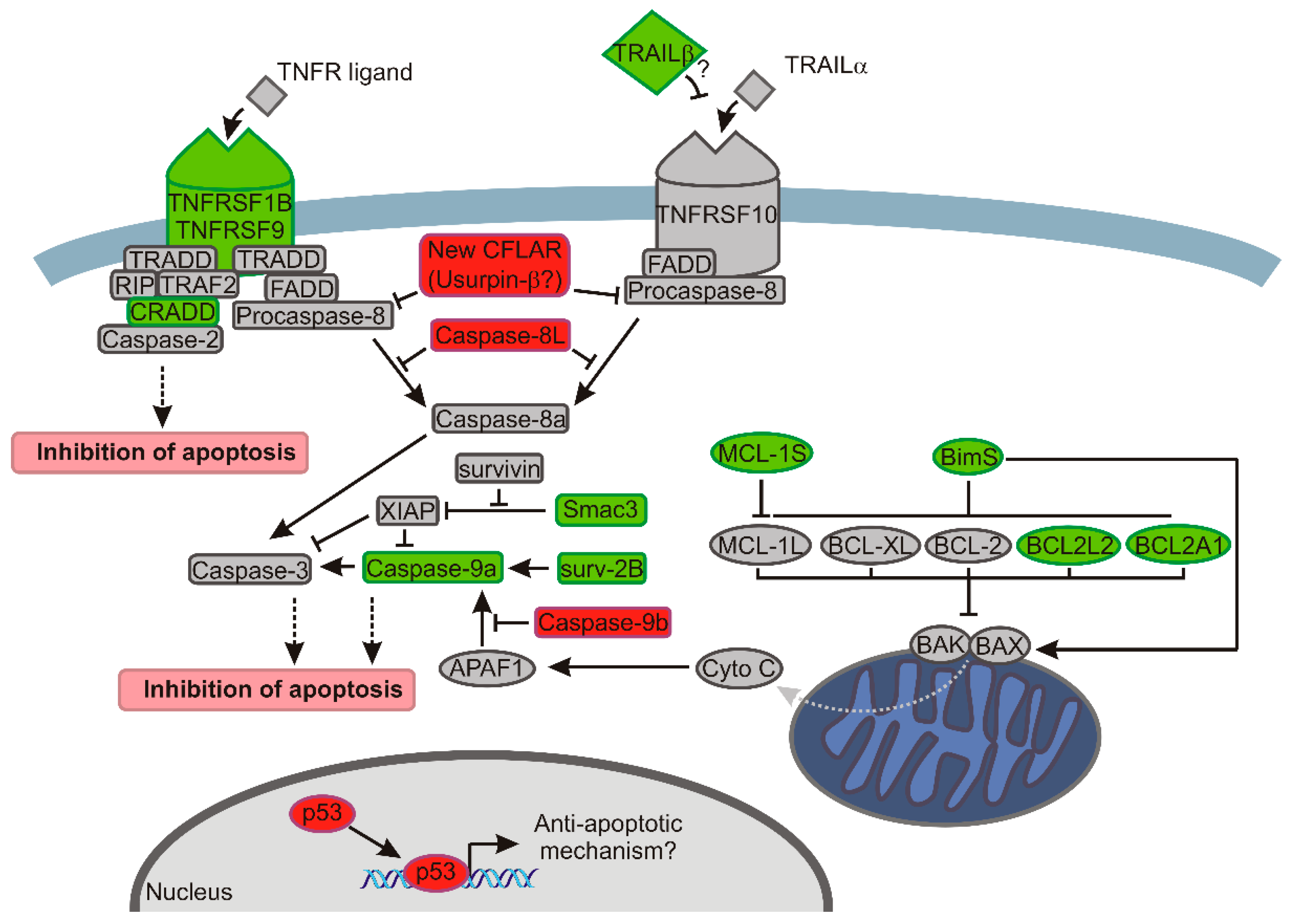
3. Discussion
4. Materials and Methods
4.1. Tissue Samples
4.2. Cell Lines
4.3. Transfections with siRNA
4.4. RNA Isolation and cDNA Synthesis
4.5. PCR and qPCR
4.6. Analysis of Apoptosis
4.7. Cell Viability Assay
4.8. Caspase-9 Activity Assay
4.9. Cloning of the New CFLAR Splice Variant
4.10. Protein Isolation and Western Blot
4.11. Analysis of TCGA Data
4.12. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Edmond, V.; Merdzhanova, G.; Gout, S.; Brambilla, E.; Gazzeri, S.; Eymin, B. A new function of the splicing factor SRSF2 in the control of E2F1-mediated cell cycle progression in neuroendocrine lung tumors. Cell Cycle 2013, 12, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Coutinho-Mansfield, G.; Wang, D.; Pandit, S.; Fu, X.D. The splicing factor SC35 has an active role in transcriptional elongation. Nat. Struct. Mol. Biol. 2008, 15, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Zhou, Y.; Pandit, S.; Huang, J.; Li, H.; Lin, C.Y.; Xiao, R.; Burge, C.B.; Fu, X.D. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell 2013, 153, 855–868. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, M.; MacDonald, A.I.; Stevenson, A.; Graham, S.V. Human papillomavirus 16 oncoprotein expression is controlled by the cellular splicing factor SRSF2 (SC35). J. Virol. 2015, 89, 5276–5287. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X.; Liu, F. Splicing factor SC35 promotes tau expression through stabilization of its mRNA. FEBS Lett. 2011, 585, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Sun, Y.; Ding, J.H.; Lin, S.; Rose, D.W.; Rosenfeld, M.G.; Fu, X.D.; Li, X. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol. Cell. Biol. 2007, 27, 5393–5402. [Google Scholar] [CrossRef] [PubMed]
- Staehler, M.; Haseke, N.; Schoeppler, G.; Stadler, T.; Gratzke, C.; Stief, C. Modern therapeutic approaches in Metastatic Renal cell carcinoma. EAU-EBU Update Ser. 2007, 5, 26–37. [Google Scholar] [CrossRef]
- Gupta, K.; Miller, J.D.; Li, J.Z.; Russell, M.W.; Charbonneau, C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): A literature review. Cancer Treat. Rev. 2008, 34, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Lughezzani, G.; Perrotte, P.; Karakiewicz, P.I. Treatment of metastatic renal cell carcinoma. Nat. Rev. Urol. 2010, 7, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar]
- Buczek, M.; Escudier, B.; Bartnik, E.; Szczylik, C.; Czarnecka, A. Resistance to tyrosine kinase inhibitors in clear cell renal cell carcinoma: from the patient's bed to molecular mechanisms. Biochim. Biophys. Acta Rev. Cancer 2014, 1845, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Zantl, N.; Weirich, G.; Zall, H.; Seiffert, B.M.; Fischer, S.F.; Kirschnek, S.; Hartmann, C.; Fritsch, R.M.; Gillissen, B.; Daniel, P.T.; et al. Frequent loss of expression of the pro-apoptotic protein Bim in renal cell carcinoma: Evidence for contribution to apoptosis resistance. Oncogene 2007, 26, 7038–7048. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Merdzhanova, G.; Edmond, V.; de Seranno, S.; van den Broeck, A.; Corcos, L.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death Differ. 2008, 15, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Database. Available online: http://cancergenome.nih.gov (accessed on 20 February 2015).
- Rasper, D.M.; Vaillancourt, J.P.; Hadano, S.; Houtzager, V.M.; Seiden, I.; Keen, S.L.; Tawa, P.; Xanthoudakis, S.; Nasir, J.; Martindale, D.; et al. Cell death attenuation by ’Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998, 5, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Jin, Y.; Arend, L.J. Smac3, a novel Smac/DIABLO splicing variant, attenuates the stability and apoptosis-inhibiting activity of X-linked inhibitor of apoptosis protein. J. Biol. Chem. 2003, 278, 52660–52672. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Cheng, Q.; Black, J.D.; Li, F. Forced expression of survivin-2B abrogates mitotic cells and induces mitochondria-dependent apoptosis by blockade of tubulin polymerization and modulation of Bcl-2, Bax, and survivin. J. Biol. Chem. 2007, 282, 27204–27214. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Leo, C.P.; Hsu, S.Y.; Hsueh, A.J. MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J. Biol. Chem. 2000, 275, 25255–25261. [Google Scholar] [CrossRef] [PubMed]
- O'Connor, L.; Strasser, A.; O'Reilly, L.A.; Hausmann, G.; Adams, J.M.; Cory, S.; Huang, D.C. Bim: A novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998, 17, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Paschen, S.A.; Heger, K.; Wilfling, F.; Frankenberg, T.; Bauerschmitt, H.; Seiffert, B.M.; Kirschnek, S.; Wagner, H.; Häcker, G. BimS-induced apoptosis requires mitochondrial localization but not interaction with anti-apoptotic Bcl-2 proteins. J. Cell Biol. 2007, 177, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Happo, L.; Strasser, A.; Cory, S. BH3-only proteins in apoptosis at a glance. J. Cell Sci. 2012, 125, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Djerbi, M.; Darreh-Shori, T.; Zhivotovsky, B.; Grandien, A. Characterization of the human FLICE-inhibitory protein locus and comparison of the anti-apoptotic activity of four different flip isoforms. Scand. J. Immunol. 2001, 54, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, Y.; Tian, B.; Jamaluddin, M.; Mitra, A.; Yang, J.; Rowicka, M.; Brasier, A.R.; Kudlicki, A. Modulation of gene expression regulated by the transcription factor NF-κB/RelA. J. Biol. Chem. 2014, 289, 11927–11944. [Google Scholar] [CrossRef] [PubMed]
- Pileczki, V.; Cojocneanu-Petric, R.; Maralani, M.; Neagoe, I.B.; Sandulescu, R. MicroRNAs as regulators of apoptosis mechanisms in cancer. Clujul. Med. 2016, 89, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Butkytė, S.; Čiupas, L.; Jakubauskienė, E.; Vilys, L.; Mocevicius, P.; Kanopka, A.; Vilkaitis, G. Splicing-dependent expression of microRNAs of mirtron origin in human digestive and excretory system cancer cells. Clin. Epigenet. 2016, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Rajabi, H.; Kufe, D. miR-1226 targets expression of the mucin 1 oncoprotein and induces cell death. Int. J. Oncol. 2010, 37, 61–69. [Google Scholar] [PubMed]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis—The p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Garner, E.; Martinon, F.; Tschopp, J.; Beard, P.; Raj, K. Cells with defective p53-p21-pRb pathway are susceptible to apoptosis induced by p84N5 via caspase-6. Cancer Res. 2007, 67, 7631–7637. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Figg, N.; Stoneman, V.; Braganza, D.; Bennett, M.R. Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ. Res. 2005, 96, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.R.; Thakur, V.S.; Gupta, K.; Jackson, M.W.; Harada, H.; Agarwal, M.K.; Shin, D.M.; Wald, D.N.; Agarwal, M.L. Restoration of p53 functions protects cells from concanavalin A-induced apoptosis. Mol. Cancer Ther. 2010, 9, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.S.; Joo, H.J.; Oh, D.K.; Kang, J.H.; Kim, Y.S.; Lee, K.B.; Kim, S.J. Cyclooxygenase-2 and p53 expression as prognostic indicators in conventional renal cell carcinoma. Yonsei Med. J. 2005, 46, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Erdem, H.; Oktay, M.; Yildirim, U.; Uzunlar, A.K.; Kayikci, M.A. Expression of AEG-1 and p53 and their clinicopathological significance in malignant lesions of renal cell carcinomas: a microarray study. Pol. J. Pathol. 2013, 64, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Haitel, A.; Wiener, H.G.; Baethge, U.; Marberger, M.; Susani, M. mdm2 expression as a prognostic indicator in clear cell renal cell carcinoma: comparison with p53 overexpression and clinicopathological parameters. Clin. Cancer Res. 2000, 6, 1840–1844. [Google Scholar] [PubMed]
- Kankaya, D.; Kiremitci, S.; Tulunay, O.; Baltaci, S. Gelsolin, NF-κB, and p53 expression in clear cell renal cell carcinoma: Impact on outcome. Pathol. Res. Pract. 2015, 211, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Tolstov, Y.; Arslan, A.; Roth, W.; Grüllich, C.; Pahernik, S.; Hohenfellner, M.; Duensing, S. Harnessing the p53-PUMA axis to overcome DNA damage resistance in renal cell carcinoma. Neoplasia 2014, 16, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Bilim, V.; Yuuki, K.; Itoi, T.; Muto, A.; Kato, T.; Nagaoka, A.; Motoyama, T.; Tomita, Y. Double inhibition of XIAP and Bcl-2 axis is beneficial for retrieving sensitivity of renal cell cancer to apoptosis. Br. J. Cancer 2008, 98, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.S.; Fialkov, J.M.; Scott, D.L.; Azuhata, T.; Williams, R.D.; Wall, N.R.; Altieri, D.C.; Sandler, A.D. Induction and regulation of tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand-mediated apoptosis in renal cell carcinoma. Cancer Res. 2002, 62, 3093–3099. [Google Scholar] [PubMed]
- Oya, M.; Ohtsubo, M.; Takayanagi, A.; Tachibana, M.; Shimizu, N.; Murai, M. Constitutive activation of nuclear factor-κB prevents TRAIL-induced apoptosis in renal cancer cells. Oncogene 2001, 20, 3888–3896. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Kandasamy, K.; Kraft, A.S. ABT-737 induces expression of the death receptor 5 and sensitizes human cancer cells to TRAIL-induced apoptosis. J. Biol. Chem. 2008, 283, 25003–25013. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.M.; Min, K.J.; Seo, B.R.; Nam, J.O.; Choi, K.S.; Yoo, Y.H.; Kwon, T.K. Cafestol overcomes ABT-737 resistance in Mcl-1-overexpressed renal carcinoma Caki cells through downregulation of Mcl-1 expression and upregulation of Bim expression. Cell Death Dis. 2014, 5, e1514. [Google Scholar] [CrossRef] [PubMed]
- Shultz, J.C.; Goehe, R.W.; Murudkar, C.S.; Wijesinghe, D.S.; Mayton, E.K.; Massiello, A.; Hawkins, A.J.; Mukerjee, P.; Pinkerman, R.L.; Park, M.A.; et al. SRSF1 regulates the alternative splicing of caspase 9 via a novel intronic splicing enhancer affecting the chemotherapeutic sensitivity of non-small cell lung cancer cells. Mol. Cancer Res. 2011, 9, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Vu, N.T.; Park, M.A.; Shultz, J.C.; Goehe, R.W.; Hoeferlin, L.A.; Shultz, M.D.; Smith, S.A.; Lynch, K.W.; Chalfant, C.E. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNPL. J. Biol. Chem. 2013, 288, 8575–8584. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Deng, H.; Li, Y.; Zhang, C.; Liu, X.; Liu, Q.; Zhang, D.; Wang, L.; Pu, Y.; Zhang, H.; et al. The DNA methylation-regulated miR-193a-3p dictates the multi-chemoresistance of bladder cancer via repression of SRSF2/PLAU/HIC2 expression. Cell Death Dis. 2014, 5, e1402. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; He, Y.; Zhang, H.; Fei, Q.; Niu, D.; Wang, D.; Ding, X.; Xu, H.; Chen, X.; Zhu, J. DNA methylation-regulated miR-193a-3p dictates resistance of hepatocellular carcinoma to 5-fluorouracil via repression of SRSF2 expression. J. Biol. Chem. 2012, 287, 5639–5649. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.; Krieg, T.; Wenzel, M.; Schmitt, M.; Ramp, U.; Fang, B.; Gabbert, H.E.; Gerharz, C.D.; Mahotka, C. TRAIL-β and TRAIL-γ: Two novel splice variants of the human TNF-related apoptosis-inducing ligand (TRAIL) without apoptotic potential. Br. J. Cancer 2003, 88, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Himeji, D.; Horiuchi, T.; Tsukamoto, H.; Hayashi, K.; Watanabe, T.; Harada, M. Characterization of caspase-8L: A novel isoform of caspase-8 that behaves as an inhibitor of the caspase cascade. Blood 2002, 99, 4070–4078. [Google Scholar] [CrossRef] [PubMed]
- Seol, D.W.; Billiar, T.R. A caspase-9 variant missing the catalytic site is an endogenous inhibitor of apoptosis. J. Biol. Chem. 1999, 274, 2072–2076. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Ahmad, M.; Guo, Y.; Zhan, Y.; Lazebnik, Y.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of an endogenous dominant-negative short isoform of caspase-9 that can regulate apoptosis. Cancer Res. 1999, 59, 999–1002. [Google Scholar] [PubMed]
- Nam, S.Y.; Sabapathy, K. p53 promotes cellular survival in a context-dependent manner by directly inducing the expression of haeme-oxygenase-1. Oncogene 2011, 30, 4476–4486. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, B.; Bensalah, K.; Canfield, S.; Dabestani, S.; Hofmann, F.; Hora, M.; Kuczyk, M.A.; Lam, T.; Marconi, L.; Merseburger, A.S.; et al. EAU Guidelines on Renal Cell Carcinoma: 2014 Update. Eur. Urol. 2015, 67, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Tun, H.W.; Marlow, L.A.; von Roemeling, C.A.; Cooper, S.J.; Kreinest, P.; Wu, K.; Luxon, B.A.; Sinha, M.; Anastasiadis, P.Z.; Copland, J.A. Pathway signature and cellular differentiation in clear cell renal cell carcinoma. PLoS ONE 2010, 5, e10696. [Google Scholar] [CrossRef] [PubMed]
- Boguslawska, J.; Piekielko-Witkowska, A.; Wojcicka, A.; Kedzierska, H.; Poplawski, P.; Nauman, A. Regulatory feedback loop between T3 and microRNAs in renal cancer. Mol. Cell. Endocrinol. 2014, 384, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Boguslawska, J.; Kedzierska, H.; Poplawski, P.; Rybicka, B.; Tanski, Z.; Piekielko-Witkowska, A. Expression of genes involved in cellular adhesion and extracellular matrix remodeling correlates with poor survival of patients with renal cancer. J. Urol. 2016, 195, 1892–1902. [Google Scholar] [CrossRef] [PubMed]
- Genbank. Available online: http://www.ncbi.nlm.nih.gov/genbank (accessed on 13 January 2014).
- Piekielko-Witkowska, A.; Wiszomirska, H.; Wojcicka, A.; Poplawski, P.; Boguslawska, J.; Tanski, Z.; Nauman, A. Disturbed expression of splicing factors in renal cancer affects alternative splicing of apoptosis regulators, oncogenes, and tumor suppressors. PLoS ONE 2010, 5, e13690. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Eisenhart, C. The assumptions underlying the analysis of variance. Biometrics 1947, 3, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Tamhane, A.C.; Dunlop, D.D. Statistics and Data Analysis from Elementary to Intermediate, 1st ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2000. [Google Scholar]
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118608_A_7_5.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118608_A_9_5.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118608_A_8_5.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118609_A_8_6.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118609_A_9_3.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118609_A_9_2.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118609_A_9_5.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118609_A_8_2.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118609_A_8_3.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118609_A_7_2.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118609_A_8_7.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118609_A_7_3.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118609_A_9_7.jpg (accessed on 29 August 2016).
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/images/49905/118609_A_7_5.jpg (accessed on 29 August 2016).
- Master, A.; Wójcicka, A.; Piekiełko-Witkowska, A.; Bogusławska, J.; Popławski, P.; Tański, Z.; Darras, V.M.; Williams, G.R.; Nauman, A. Untranslated regions of thyroid hormone receptor β 1 mRNA are impaired in human clear cell renal cell carcinoma. Biochim. Biophys. Acta 2010, 1802, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- The Champion ChiP Transcription Factor Search Portal. Available online: http://www.sabiosciences.com/chipqpcrsearch.php (accessed on 8 December 2015).
- Massiello, A.; Chalfant, C.E. SRp30a (ASF/SF2) regulates the alternative splicing of Caspase-9 pre-mRNA and is required for ceramide-responsiveness. J. Lipid Res. 2006, 47, 892–897. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kędzierska, H.; Popławski, P.; Hoser, G.; Rybicka, B.; Rodzik, K.; Sokół, E.; Bogusławska, J.; Tański, Z.; Fogtman, A.; Koblowska, M.; et al. Decreased Expression of SRSF2 Splicing Factor Inhibits Apoptotic Pathways in Renal Cancer. Int. J. Mol. Sci. 2016, 17, 1598. https://doi.org/10.3390/ijms17101598
Kędzierska H, Popławski P, Hoser G, Rybicka B, Rodzik K, Sokół E, Bogusławska J, Tański Z, Fogtman A, Koblowska M, et al. Decreased Expression of SRSF2 Splicing Factor Inhibits Apoptotic Pathways in Renal Cancer. International Journal of Molecular Sciences. 2016; 17(10):1598. https://doi.org/10.3390/ijms17101598
Chicago/Turabian StyleKędzierska, Hanna, Piotr Popławski, Grażyna Hoser, Beata Rybicka, Katarzyna Rodzik, Elżbieta Sokół, Joanna Bogusławska, Zbigniew Tański, Anna Fogtman, Marta Koblowska, and et al. 2016. "Decreased Expression of SRSF2 Splicing Factor Inhibits Apoptotic Pathways in Renal Cancer" International Journal of Molecular Sciences 17, no. 10: 1598. https://doi.org/10.3390/ijms17101598
APA StyleKędzierska, H., Popławski, P., Hoser, G., Rybicka, B., Rodzik, K., Sokół, E., Bogusławska, J., Tański, Z., Fogtman, A., Koblowska, M., & Piekiełko-Witkowska, A. (2016). Decreased Expression of SRSF2 Splicing Factor Inhibits Apoptotic Pathways in Renal Cancer. International Journal of Molecular Sciences, 17(10), 1598. https://doi.org/10.3390/ijms17101598