
The Large Phenotypic Spectrum of Fabry Disease Requires Graduated Diagnosis and Personalized Therapy: A Meta-Analysis Can Help to Differentiate Missense Mutations

 , and
, and
Abstract
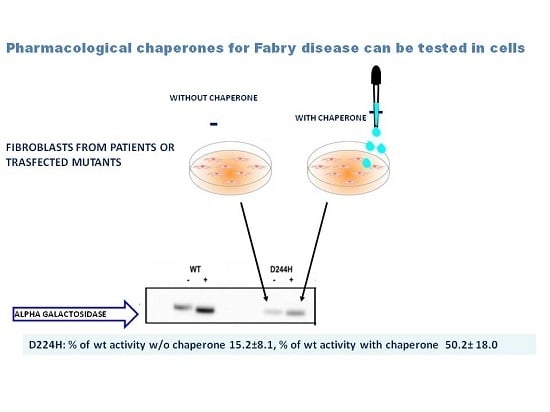
:
1. Introduction
2. Results
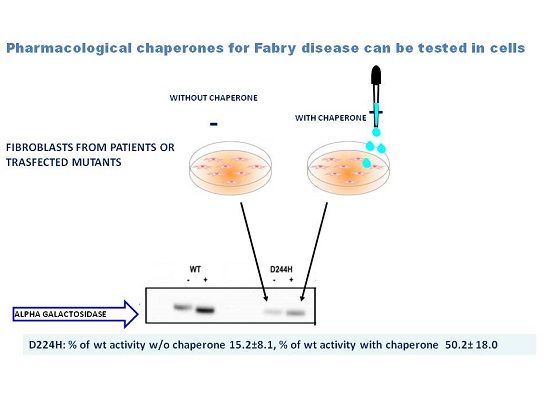
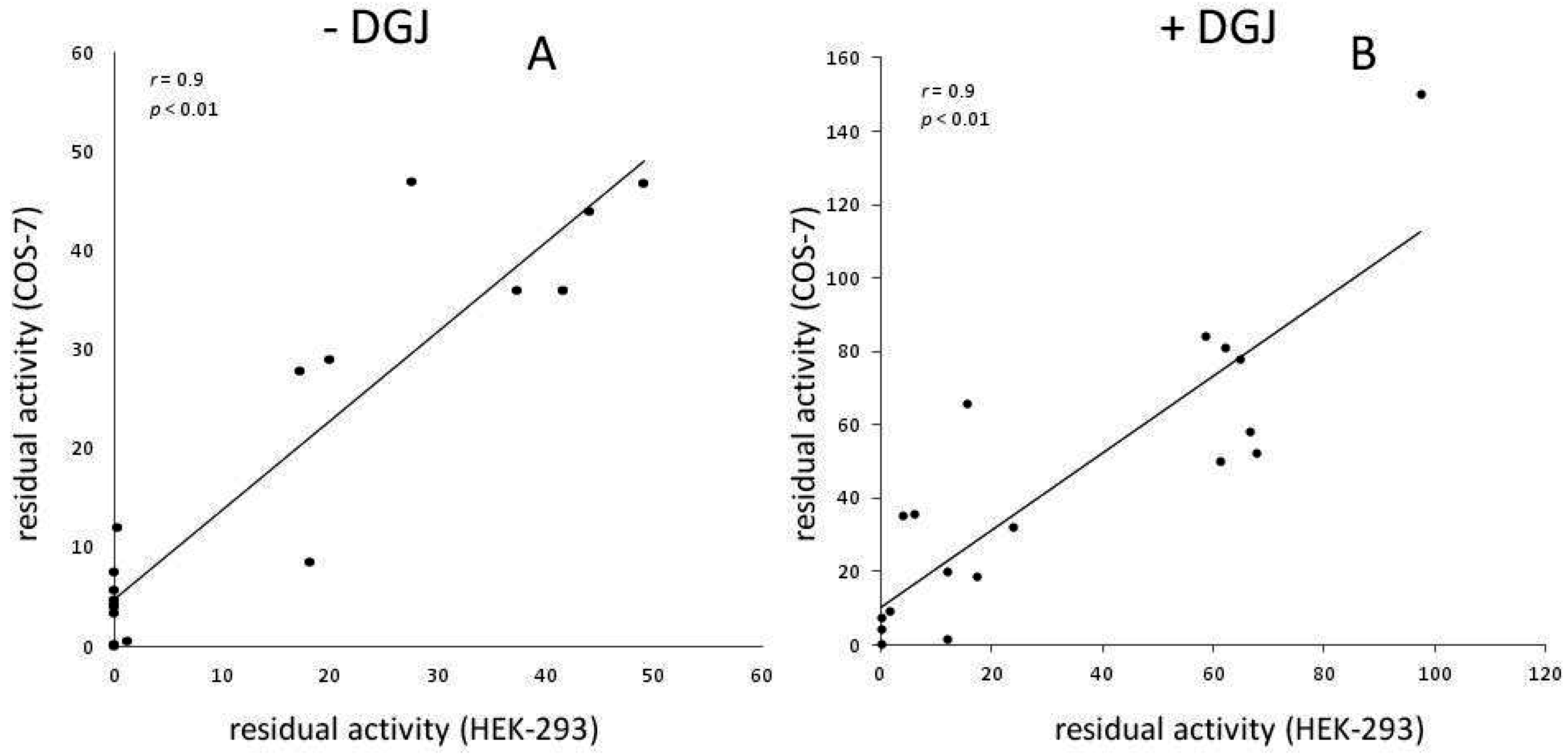
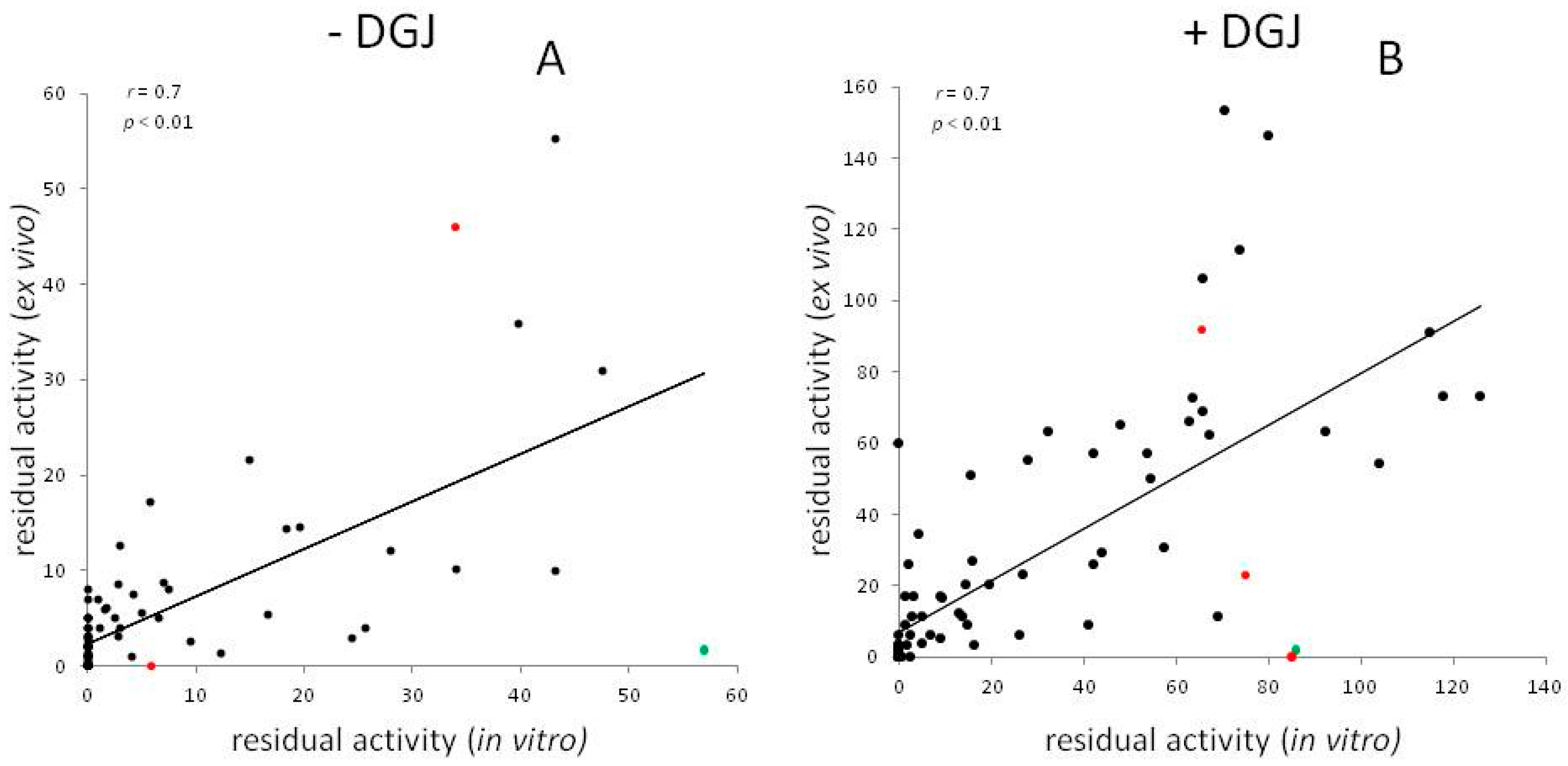
Meta-Analysis of Data Reporting Residual Activity and Responsiveness to DGJ of GLA Missense Mutations
3. Future Perspectives for Therapy
4. Methods
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sirrs, S.; Hollak, C.; Merkel, M.; Sechi, A.; Glamuzina, E.; Janssen, M.C.; Lachmann, R.; Langendonk, J.; Scarpelli, M.; Ben Omran, T.; et al. The frequencies of different inborn errors of metabolism in adult metabolic centres: Report from the ssiem adult metabolic physicians group. JIMD Rep. 2016, 27, 85–91. [Google Scholar] [PubMed]
- Lukas, J.; Giese, A.K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional characterisation of α-galactosidase a mutations as a basis for a new classification system in Fabry disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, D.P. Fabry disease. Orphanet. J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Wallin, E.F.; Clatworthy, M.R.; Pritchard, N.R. Fabry disease: Results of the first UK hemodialysis screening study. Clin. Nephrol. 2011, 75, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Noiri, E.; Ishizu, T.; Negishi, K.; Suzuki, Y.; Hamasaki, Y.; Honda, K.; Fujita, T.; Tsukimura, T.; Togawa, T.; et al. High-throughput screening identified disease-causing mutants and functional variants of α-galactosidase a gene in japanese male hemodialysis patients. J. Hum. Genet. 2012, 57, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, A.; Fazekas, F.; Grittner, U.; Dichgans, M.; Martus, P.; Holzhausen, M.; Bottcher, T.; Heuschmann, P.U.; Tatlisumak, T.; Tanislav, C.; et al. Acute cerebrovascular disease in the young: The stroke in young Fabry patients study. Stroke 2013, 44, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, B.; Takenaka, T.; Teraguchi, H.; Tei, C.; Lee, P.; McKenna, W.J.; Elliott, P.M. Prevalence of anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002, 105, 1407–1411. [Google Scholar] [CrossRef] [PubMed]
- Monserrat, L.; Gimeno-Blanes, J.R.; Marin, F.; Hermida-Prieto, M.; Garcia-Honrubia, A.; Perez, I.; Fernandez, X.; de Nicolas, R.; de la Morena, G.; Paya, E.; et al. Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2007, 50, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Hwu, W.L.; Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Dobrovolny, R.; Huang, A.C.; Yeh, H.Y.; Chao, M.C.; Lin, S.J.; Kitagawa, T.; et al. Newborn screening for Fabry disease in taiwan reveals a high incidence of the later-onset GLA mutation C.936 + 919G > A (IVS4 + 919G > A). Hum. Mutat. 2009, 30, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset Fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Mellett, N.; Hein, L.K.; Brooks, D.A.; Meikle, P.J. Absence of α-galactosidase cross-correction in Fabry heterozygote cultured skin fibroblasts. Mol. Genet. Metab. 2015, 114, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; de Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B. Fabry disease: Recent advances in pathology, diagnosis, treatment and monitoring. Orphanet. J. Rare Dis. 2009, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Maruyama, H.; Nameta, M.; Yamamoto, T.; Matsuda, J.; Kulkarni, A.B.; Yoshioka, H.; Ishii, S. A symptomatic Fabry disease mouse model generated by inducing globotriaosylceramide synthesis. Biochem. J. 2013, 456, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.J.; Jeon, Y.J.; Jung, N.; Park, J.W.; Park, H.Y.; Jung, S.C. Substrate-specific gene expression profiles in different kidney cell types are associated with Fabry disease. Mol. Med. Rep. 2015, 12, 5049–5057. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.; Johnson, A.; Winchester, B. Synthesis of novel internal standards for the quantitative determination of plasma ceramide trihexoside in Fabry disease by tandem mass spectrometry. FEBS Lett. 2002, 515, 171–176. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Cyr, D.; Ntwari, A.; West, M.L.; Cox-Brinkman, J.; Bichet, D.G.; Germain, D.P.; Laframboise, R.; Melancon, S.B.; Stockley, T.; et al. Urinary globotriaosylceramide excretion correlates with the genotype in children and adults with Fabry disease. Mol. Genet. Metab. 2008, 93, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Smid, B.E.; van der Tol, L.; Biegstraaten, M.; Linthorst, G.E.; Hollak, C.E.; Poorthuis, B.J. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J. Med. Genet. 2015, 52, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Linhart, A.; Elliott, P.M. The heart in anderson-Fabry disease and other lysosomal storage disorders. Heart 2007, 93, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Moore, D.F. Neurological manifestations of Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Sunder-Plassmann, G. Renal manifestations of Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Whybra, C.; Bahner, F.; Baron, K. Measurement of disease severity and progression in Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Whybra, C.; Kampmann, C.; Krummenauer, F.; Ries, M.; Mengel, E.; Miebach, E.; Baehner, F.; Kim, K.; Bajbouj, M.; Schwarting, A.; et al. The mainz severity score index: A new instrument for quantifying the anderson-Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin. Genet. 2004, 65, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Giannini, E.H.; Mehta, A.B.; Hilz, M.J.; Beck, M.; Bichet, D.G.; Brady, R.O.; West, M.; Germain, D.P.; Wanner, C.; Waldek, S.; et al. A validated disease severity scoring system for Fabry disease. Mol. Genet. Metab. 2010, 99, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Mignani, R.; Pieruzzi, F.; Berri, F.; Burlina, A.; Chinea, B.; Gallieni, M.; Pieroni, M.; Salviati, A.; Spada, M. Fabry stabilization index (fastex): An innovative tool for the assessment of clinical stabilization in Fabry disease. Clin. Kidney J. 2016, 9, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Teitcher, M.; Weinerman, S.; Whybra, C.; Beck, M.; Sharon, N.; Elstein, D.; Altarescu, G. Genetic polymorphisms of vitamin D receptor (VDR) in Fabry disease. Genetica 2008, 134, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Altarescu, G.; Moore, D.F.; Schiffmann, R. Effect of genetic modifiers on cerebral lesions in Fabry disease. Neurology 2005, 64, 2148–2150. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten, M.; Arngrimsson, R.; Barbey, F.; Boks, L.; Cecchi, F.; Deegan, P.B.; Feldt-Rasmussen, U.; Geberhiwot, T.; Germain, D.P.; Hendriksz, C.; et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: The european Fabry working group consensus document. Orphanet. J. Rare Dis. 2015, 10, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banikazemi, M.; Bultas, J.; Waldek, S.; Wilcox, W.R.; Whitley, C.B.; McDonald, M.; Finkel, R.; Packman, S.; Bichet, D.G.; Warnock, D.G.; et al. Agalsidase-β therapy for advanced Fabry disease: A randomized trial. Ann. Intern. Med. 2007, 146, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Bierer, G.; Balfe, D.; Wilcox, W.R.; Mosenifar, Z. Improvement in serial cardiopulmonary exercise testing following enzyme replacement therapy in Fabry disease. J. Inherit. Metab. Dis. 2006, 29, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Elliott, P.M.; Shah, J.; Zuckerman, J.; Coghlan, G.; Brookes, J.; Mehta, A.B. Effects of enzyme replacement therapy on the cardiomyopathy of anderson-Fabry disease: A randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart 2008, 94, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Rombach, S.M.; Smid, B.E.; Linthorst, G.E.; Dijkgraaf, M.G.; Hollak, C.E. Natural course of Fabry disease and the effectiveness of enzyme replacement therapy: A systematic review and meta-analysis: Effectiveness of ert in different disease stages. J. Inherit. Metab. Dis. 2014, 37, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Thurberg, B.L.; Rennke, H.; Colvin, R.B.; Dikman, S.; Gordon, R.E.; Collins, A.B.; Desnick, R.J.; O’Callaghan, M. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002, 62, 1933–1946. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Waldek, S.; Banikazemi, M.; Bushinsky, D.A.; Charrow, J.; Desnick, R.J.; Lee, P.; Loew, T.; Vedder, A.C.; Abichandani, R.; et al. Sustained, long-term renal stabilization after 54 months of agalsidase β therapy in patients with Fabry disease. J. Am. Soc. Nephrol. 2007, 18, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Tondel, C.; Bostad, L.; Larsen, K.K.; Hirth, A.; Vikse, B.E.; Houge, G.; Svarstad, E. Agalsidase benefits renal histology in young patients with Fabry disease. J. Am. Soc. Nephrol. 2013, 24, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Rombach, S.M.; Smid, B.E.; Bouwman, M.G.; Linthorst, G.E.; Dijkgraaf, M.G.; Hollak, C.E. Long term enzyme replacement therapy for Fabry disease: Effectiveness on kidney, heart and brain. Orphanet. J. Rare Dis. 2013, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Buechner, S.; Moretti, M.; Burlina, A.P.; Cei, G.; Manara, R.; Ricci, R.; Mignani, R.; Parini, R.; di Vito, R.; Giordano, G.P.; et al. Central nervous system involvement in anderson-Fabry disease: A clinical and MRI retrospective study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Jardim, L.; Vedolin, L.; Schwartz, I.V.; Burin, M.G.; Cecchin, C.; Kalakun, L.; Matte, U.; Aesse, F.; Pitta-Pinheiro, C.; Marconato, J.; et al. Cns involvement in Fabry disease: Clinical and imaging studies before and after 12 months of enzyme replacement therapy. J. Inherit. Metab. Dis. 2004, 27, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Jardim, L.B.; Aesse, F.; Vedolin, L.M.; Pitta-Pinheiro, C.; Marconato, J.; Burin, M.G.; Cecchin, C.; Netto, C.B.; Matte, U.S.; Pereira, F.; et al. White matter lesions in Fabry disease before and after enzyme replacement therapy: A 2-year follow-up. Arq. Neuropsiquiatr. 2006, 64, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Niemann, M.; Stork, S.; Breunig, F.; Beer, M.; Sommer, C.; Herrmann, S.; Ertl, G.; Wanner, C. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: Evidence for disease progression towards serious complications. J. Intern. Med. 2013, 274, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.E.; Weinreb, N.J. The attenuated/late onset lysosomal storage disorders: Therapeutic goals and indications for enzyme replacement treatment in gaucher and Fabry disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Monticelli, M.; Cubellis, M.V. Looking for protein stabilizing drugs with thermal shift assay. Drug Test. Anal. 2015, 7, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Citro, V.; Correra, A.; Cubellis, M.V. A thermodynamic assay to test pharmacological chaperones for Fabry disease. Biochim. Biophys. Acta 2014, 1840, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.R.; Khanna, R.; Schilling, A.; Flanagan, J.J.; Pellegrino, L.J.; Brignol, N.; Lun, Y.; Guillen, D.; Ranes, B.E.; Frascella, M.; et al. Co-administration with the pharmacological chaperone AT1001 increases recombinant human α-galactosidase a tissue uptake and improves substrate reduction in Fabry mice. Mol. Ther. 2012, 20, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Warnock, D.G.; Bichet, D.G.; Holida, M.; Goker-Alpan, O.; Nicholls, K.; Thomas, M.; Eyskens, F.; Shankar, S.; Adera, M.; Sitaraman, S.; et al. Oral migalastat HCL leads to greater systemic exposure and tissue levels of active α-galactosidase a in Fabry patients when co-administered with infused agalsidase. PLoS ONE 2015, 10, e0134341. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lun, Y.; Brignol, N.; Hamler, R.; Schilling, A.; Frascella, M.; Sullivan, S.; Boyd, R.E.; Chang, K.; Soska, R.; et al. Coformulation of a novel human α-galactosidase a with the pharmacological chaperone AT1001 leads to improved substrate reduction in Fabry mice. Mol. Ther. 2015, 23, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Young-Gqamana, B.; Brignol, N.; Chang, H.H.; Khanna, R.; Soska, R.; Fuller, M.; Sitaraman, S.A.; Germain, D.P.; Giugliani, R.; Hughes, D.A.; et al. Migalastat hcl reduces globotriaosylsphingosine (lyso-Gb3) in Fabry transgenic mice and in the plasma of Fabry patients. PLoS ONE 2013, 8, e57631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, D.P.; Fan, J.Q. Pharmacological chaperone therapy by active-site-specific chaperones in Fabry disease: In vitro and preclinical studies. Int. J. Clin. Pharmacol. Ther. 2009, 47, S111–S117. [Google Scholar] [PubMed]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of fabry’s disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Suzuki, Y.; Fan, J.Q. Role of ser-65 in the activity of α-galactosidase a: Characterization of a point mutation (S65T) detected in a patient with Fabry disease. Arch. Biochem. Biophys. 2000, 377, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Murray, G.J.; Kluepfel-Stahl, S.; Cooney, A.M.; Quirk, J.M.; Schiffmann, R.; Brady, R.O.; Kaneski, C.R. Screening for pharmacological chaperones in Fabry disease. Biochem. Biophys. Res. Commun. 2007, 359, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Chang, H.H.; Kawasaki, K.; Yasuda, K.; Wu, H.L.; Garman, S.C.; Fan, J.Q. Mutant α-galactosidase a enzymes identified in Fabry disease patients with residual enzyme activity: Biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem. J. 2007, 406, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Kluepfel-Stahl, S.; Cooney, A.M.; Kaneski, C.R.; Quirk, J.M.; Schiffmann, R.; Brady, R.O.; Murray, G.J. Prediction of response of mutated α-galactosidase a to a pharmacological chaperone. Pharmacogenet. Genom. 2008, 18, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, G.H.; Kim, S.S.; Ko, J.M.; Lee, J.J.; Yoo, H.W. Effects of a chemical chaperone on genetic mutations in α-galactosidase a in Korean patients with Fabry disease. Exp. Mol. Med. 2009, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.R.; Flanagan, J.J.; Schilling, A.; Chang, H.H.; Agarwal, L.; Katz, E.; Wu, X.; Pine, C.; Wustman, B.; Desnick, R.J.; et al. The pharmacological chaperone 1-deoxygalactonojirimycin increases α-galactosidase a levels in Fabry patient cell lines. J. Inherit. Metab. Dis. 2009, 32, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Filoni, C.; Caciotti, A.; Carraresi, L.; Cavicchi, C.; Parini, R.; Antuzzi, D.; Zampetti, A.; Feriozzi, S.; Poisetti, P.; Garman, S.C.; et al. Functional studies of new GLA gene mutations leading to conformational Fabry disease. Biochim. Biophys. Acta 2010, 1802, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Katz, E.; Della Valle, M.C.; Mascioli, K.; Flanagan, J.J.; Castelli, J.P.; Schiffmann, R.; Boudes, P.; Lockhart, D.J.; Valenzano, K.J.; et al. A pharmacogenetic approach to identify mutant forms of α-galactosidase a that respond to a pharmacological chaperone for Fabry disease. Hum. Mutat. 2011, 32, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Citro, V.; de Crescenzo, A.; Orlando, P.; Cammisa, M.; Correra, A.; Cubellis, M.V. Therapy of Fabry disease with pharmacological chaperones: From in silico predictions to in vitro tests. Orphanet. J. Rare Dis. 2011, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Waldek, S.; Germain, D.P.; Nicholls, K.; Bichet, D.G.; Simosky, J.K.; Bragat, A.C.; Castelli, J.P.; Benjamin, E.R.; Boudes, P.F. A phase 2 study of migalastat hydrochloride in females with Fabry disease: Selection of population, safety and pharmacodynamic effects. Mol. Genet. Metab. 2013, 109, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Scalia, S.; Eichler, S.; Pockrandt, A.M.; Dehn, N.; Cozma, C.; Giese, A.K.; Rolfs, A. Functional and clinical consequences of novel α-galactosidase a mutations in Fabry disease. Hum. Mutat. 2016, 37, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.K.; Mudd, P.N., Jr.; Bragat, A.; Adera, M.; Boudes, P. Pharmacokinetics and safety of migalastat HCL and effects on agalsidase activity in healthy volunteers. Clin. Pharmacol. Drug Dev. 2013, 2, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Togawa, T.; Tsukimura, T.; Kodama, T.; Tanaka, T.; Kawashima, I.; Saito, S.; Ohno, K.; Fukushige, T.; Kanekura, T.; Satomura, A.; et al. Fabry disease: Biochemical, pathological and structural studies of the α-galactosidase a with E66Q amino acid substitution. Mol. Genet. Metab. 2012, 105, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.R.; Della Valle, M.C.; Wu, X.; Katz, E.; Pruthi, F.; Bond, S.; Bronfin, B.; Williams, H.; Yu, J.; Bichet, D.G.; et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cubellis, M.V.; Baaden, M.; Andreotti, G. Taming molecular flexibility to tackle rare diseases. Biochimie 2015, 113, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Ohno, K.; Sese, J.; Sugawara, K.; Sakuraba, H. Prediction of the clinical phenotype of Fabry disease based on protein sequential and structural information. J. Hum. Genet. 2010, 55, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Riera, C.; Lois, S.; Dominguez, C.; Fernandez-Cadenas, I.; Montaner, J.; Rodriguez-Sureda, V.; de la Cruz, X. Molecular damage in Fabry disease: Characterization and prediction of α-galactosidase a pathological mutations. Proteins 2015, 83, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Sakuraba, H.; Suzuki, Y. Point mutations in the upstream region of the α-galactosidase a gene exon 6 in an atypical variant of Fabry disease. Hum. Genet. 1992, 89, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Havndrup, O.; Christiansen, M.; Stoevring, B.; Jensen, M.; Hoffman-Bang, J.; Andersen, P.S.; Hasholt, L.; Norremolle, A.; Feldt-Rasmussen, U.; Kober, L.; et al. Fabry disease mimicking hypertrophic cardiomyopathy: Genetic screening needed for establishing the diagnosis in women. Eur. J. Heart Fail. 2010, 12, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Branton, M.H.; Schiffmann, R.; Sabnis, S.G.; Murray, G.J.; Quirk, J.M.; Altarescu, G.; Goldfarb, L.; Brady, R.O.; Balow, J.E.; Austin Iii, H.A.; et al. Natural history of Fabry renal disease: Influence of α-galactosidase a activity and genetic mutations on clinical course. Medicine (Baltimore) 2002, 81, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Resnick-Silverman, L.A.; Niehaus, D.J.; Astrin, K.H.; Desnick, R.J. Nature and frequency of mutations in the α-galactosidase a gene that cause Fabry disease. Am. J. Hum. Genet. 1993, 53, 1186–1197. [Google Scholar] [PubMed]
- Shabbeer, J.; Yasuda, M.; Luca, E.; Desnick, R.J. Fabry disease: 45 novel mutations in the α-galactosidase a gene causing the classical phenotype. Mol. Genet. Metab. 2002, 76, 23–30. [Google Scholar] [CrossRef]
- Ferreira, S.; Ortiz, A.; Germain, D.P.; Viana-Baptista, M.; Caldeira-Gomes, A.; Camprecios, M.; Fenollar-Cortes, M.; Gallegos-Villalobos, A.; Garcia, D.; Garcia-Robles, J.A.; et al. The α-galactosidase a p.Arg118cys variant does not cause a Fabry disease phenotype: Data from individual patients and family studies. Mol. Genet. Metab. 2015, 114, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Pockrandt, A.M.; Seemann, S.; Sharif, M.; Runge, F.; Pohlers, S.; Zheng, C.; Glaser, A.; Beller, M.; Rolfs, A.; et al. Enzyme enhancers for the treatment of Fabry and pompe disease. Mol. Ther. 2015, 23, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Weidemann, F.; Kurschat, C.; Canaan-Kuhl, S.; Duning, T.; Stypmann, J.; Schmitz, B.; Reiermann, S.; Kramer, J.; Blaschke, D.; et al. α-Galactosidase A p.A143T, a non-Fabry disease-causing variant. Orphanet. J. Rare Dis. 2016, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Xu, X.; Zhang, L.; Zhang, X.; Zheng, Y.; Luo, S.; Guo, H.; Xia, K.; Li, J.; Yao, H.; et al. GLA variation p.E66Q identified as the genetic etiology of Fabry disease using exome sequencing. Gene 2016, 575, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, M.; Sakamoto, N.; Kobayashi, A.; Suzuki, S.; Yoshihisa, A.; Yamaki, T.; Nakazato, K.; Suzuki, H.; Saitoh, S.; Kiko, Y.; et al. Familial hypertrophic obstructive cardiomyopathy with the GLA E66Q mutation and zebra body. BMC Cardiovasc. Disord. 2016, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Sekijima, Y.; Hattori, K.; Nagamatsu, K.; Shimizu, Y.; Yazaki, M.; Sakurai, A.; Endo, F.; Fukushima, Y.; Ikeda, S.I. P.E66Q mutation in the GLA gene is associated with a high risk of cerebral small-vessel occlusion in elderly Japanese males. Eur. J. Neurol. 2014, 21, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Satomura, A.; Yanai, M.; Fujita, T.; Nakayama, T. Comment on ‘p.E66q mutation in the GLA gene is associated with a high risk of cerebral small-vessel occlusion in elderly japanese males’. Eur. J. Neurol. 2014, 21, e62. [Google Scholar] [CrossRef] [PubMed]
- Shimotori, M.; Maruyama, H.; Nakamura, G.; Suyama, T.; Sakamoto, F.; Itoh, M.; Miyabayashi, S.; Ohnishi, T.; Sakai, N.; Wataya-Kaneda, M.; et al. Novel mutations of the GLA gene in Japanese patients with Fabry disease and their functional characterization by active site specific chaperone. Hum. Mutat. 2008, 29, 331. [Google Scholar] [CrossRef] [PubMed]
- Niemann, M.; Rolfs, A.; Giese, A.; Mascher, H.; Breunig, F.; Ertl, G.; Wanner, C.; Weidemann, F. Lyso-Gb3 indicates that the α-galactosidase a mutation D313Y is not clinically relevant for Fabry disease. JIMD Rep. 2013, 7, 99–102. [Google Scholar] [PubMed]
- Rigoldi, M.; Concolino, D.; Morrone, A.; Pieruzzi, F.; Ravaglia, R.; Furlan, F.; Santus, F.; Strisciuglio, P.; Torti, G.; Parini, R. Intrafamilial phenotypic variability in four families with anderson-Fabry disease. Clin. Genet. 2014, 86, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, Y.A.; Zeidner, K.M.; Grace, M.E.; Desnick, R.J. Human α-galactosidase A: Glycosylation site 3 is essential for enzyme solubility. Biochem. J. 1998, 332, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Baker, R.; Mehta, A.; Hughes, D. The N215S mutation results in a distinct subtype of Fabry disease. Mol. Genet. Metab. 2015, 114, S113. [Google Scholar] [CrossRef]
- Meehan, S.M.; Junsanto, T.; Rydel, J.J.; Desnick, R.J. Fabry disease: Renal involvement limited to podocyte pathology and proteinuria in a septuagenarian cardiac variant. Pathologic and therapeutic implications. Am. J. Kidney Dis. 2004, 43, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated transport and maturation of lysosomal α-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Okumiya, T.; Ishii, S.; Takenaka, T.; Kase, R.; Kamei, S.; Sakuraba, H.; Suzuki, Y. Galactose stabilizes various missense mutants of α-galactosidase in Fabry disease. Biochem. Biophys. Res. Commun. 1995, 214, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Ishii, S.; Kizu, H.; Ikeda, K.; Yasuda, K.; Kato, A.; Martin, O.R.; Fan, J.Q. In vitro inhibition and intracellular enhancement of lysosomal α-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur. J. Biochem. 2000, 267, 4179–4186. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Mena-Barragan, T.; Higaki, K.; Johnson, J.L.; Drury, J.E.; Lieberman, R.L.; Nakasone, N.; Ninomiya, H.; Tsukimura, T.; Sakuraba, H.; et al. Molecular basis of 1-deoxygalactonojirimycin arylthiourea binding to human α-galactosidase A: Pharmacological chaperoning efficacy on Fabry disease mutants. ACS Chem. Biol. 2014, 9, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Yamashita, Y.; Nakagawa, S.; Koike, Y.; Adachi, I.; Hollinshead, J.; Nash, R.J.; Ikeda, K.; Asano, N. 2,5-dideoxy-2,5-imino-d-altritol as a new class of pharmacological chaperone for Fabry disease. Bioorg. Med. Chem. 2010, 18, 3790–3794. [Google Scholar] [CrossRef] [PubMed]
- Ayers, B.J.; Ngo, N.; Jenkinson, S.F.; Martinez, R.F.; Shimada, Y.; Adachi, I.; Weymouth-Wilson, A.C.; Kato, A.; Fleet, G.W. Glycosidase inhibition by all 10 stereoisomeric 2,5-dideoxy-2,5-iminohexitols prepared from the enantiomers of glucuronolactone. J. Org. Chem. 2012, 77, 7777–7792. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.C.; Wang, J.H.; Li, H.Y.; Lu, S.J.; Hu, J.M.; Yun, W.Y.; Chiu, C.H.; Yang, W.B.; Chien, Y.H.; Hwu, W.L. Bioevaluation of sixteen admdp stereoisomers toward α-galactosidase A: Development of a new pharmacological chaperone for the treatment of Fabry disease and potential enhancement of enzyme replacement therapy efficiency. Eur. J. Med. Chem. 2016, 123, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Mena-Barragan, T.; Narita, A.; Matias, D.; Tiscornia, G.; Nanba, E.; Ohno, K.; Suzuki, Y.; Higaki, K.; Garcia Fernandez, J.M.; Ortiz Mellet, C. pH-responsive pharmacological chaperones for rescuing mutant glycosidases. Angew. Chem. Int. Ed. Engl. 2015, 54, 11696–11700. [Google Scholar] [CrossRef] [PubMed]
- Motabar, O.; Liu, K.; Southall, N.; Marugan, J.J.; Goldin, E.; Sidransky, E.; Zheng, W. High throughput screening for inhibitors of α-galactosidase. Curr. Chem. Genom. 2010, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Peña-García, J.; den-Haan, H.; Pérez-Sánchez, H.; del Prete, R.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Identification of an allosteric binding site on human lysosomal α-galactosidase opens the way to new pharmacological chaperones for Fabry disease. PLoS ONE 2016, 11, e0165463. [Google Scholar] [CrossRef] [PubMed]
- Linear Correlation and Regression. Available online: http://vassarstats.net/corr_big.html (accessed on 30 November 2016).
- Andreotti, G.; Guarracino, M.R.; Cammisa, M.; Correra, A.; Cubellis, M.V. Prediction of the responsiveness to pharmacological chaperones: Lysosomal human α-galactosidase, a case of study. Orphanet. J. Rare Dis. 2010, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Cammisa, M.; Correra, A.; Andreotti, G.; Cubellis, M.V. Fabry_cep: A tool to identify Fabry mutations responsive to pharmacological chaperones. Orphanet. J. Rare Dis. 2013, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Cammisa, M.; Correra, A.; Andreotti, G.; Cubellis, M.V. Identification and analysis of conserved pockets on protein surfaces. BMC Bioinform. 2013, 14, S9. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
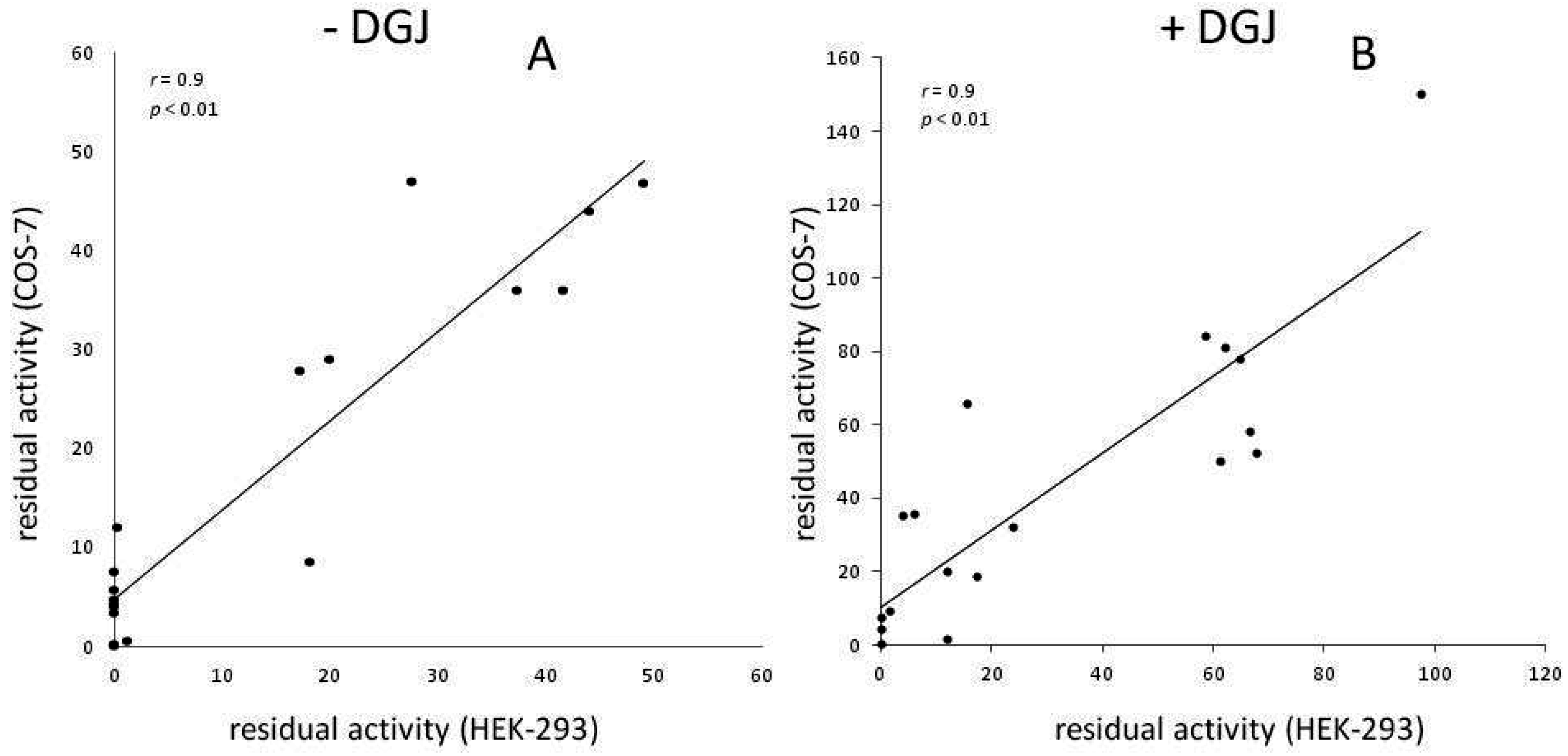
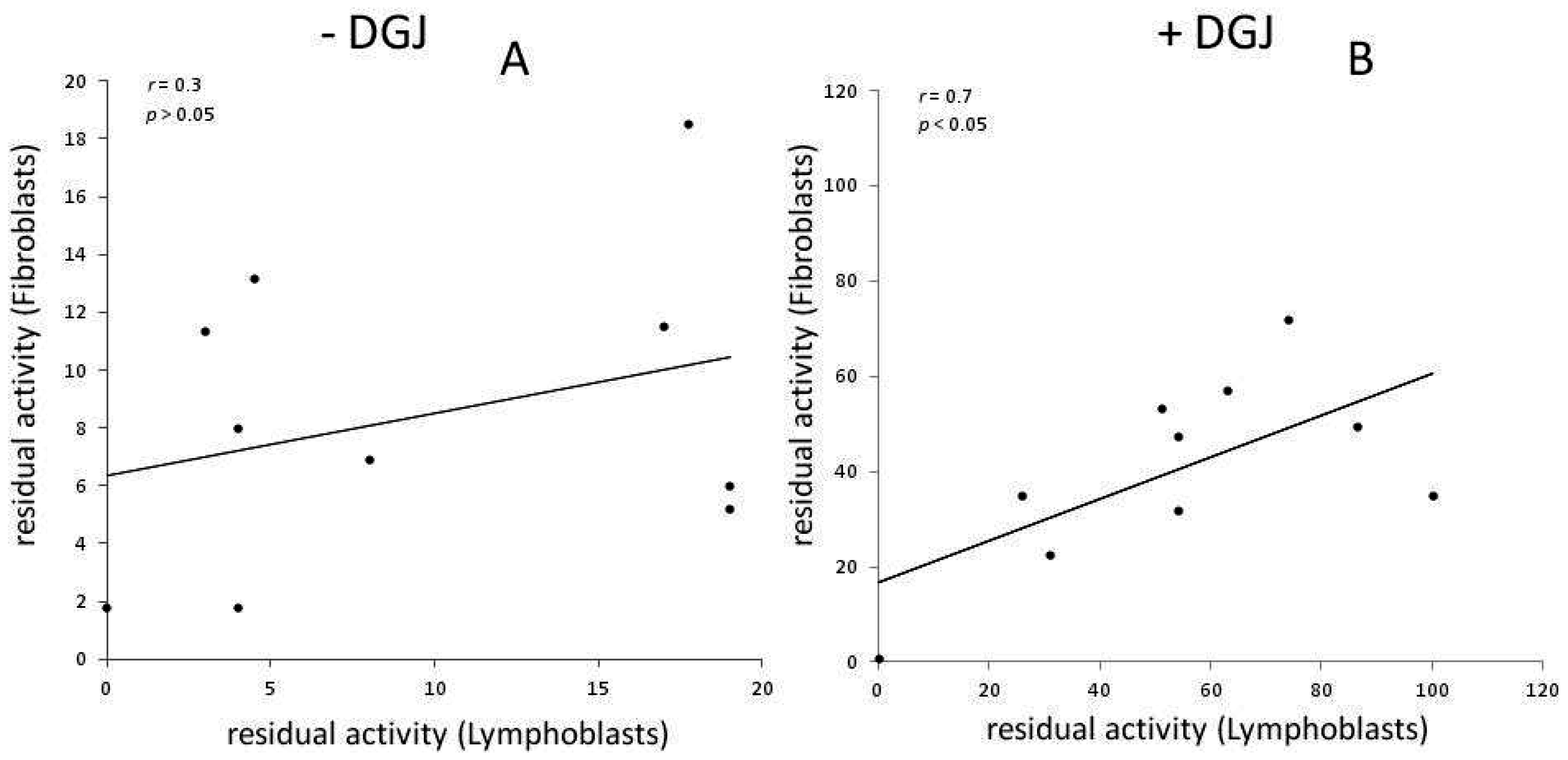
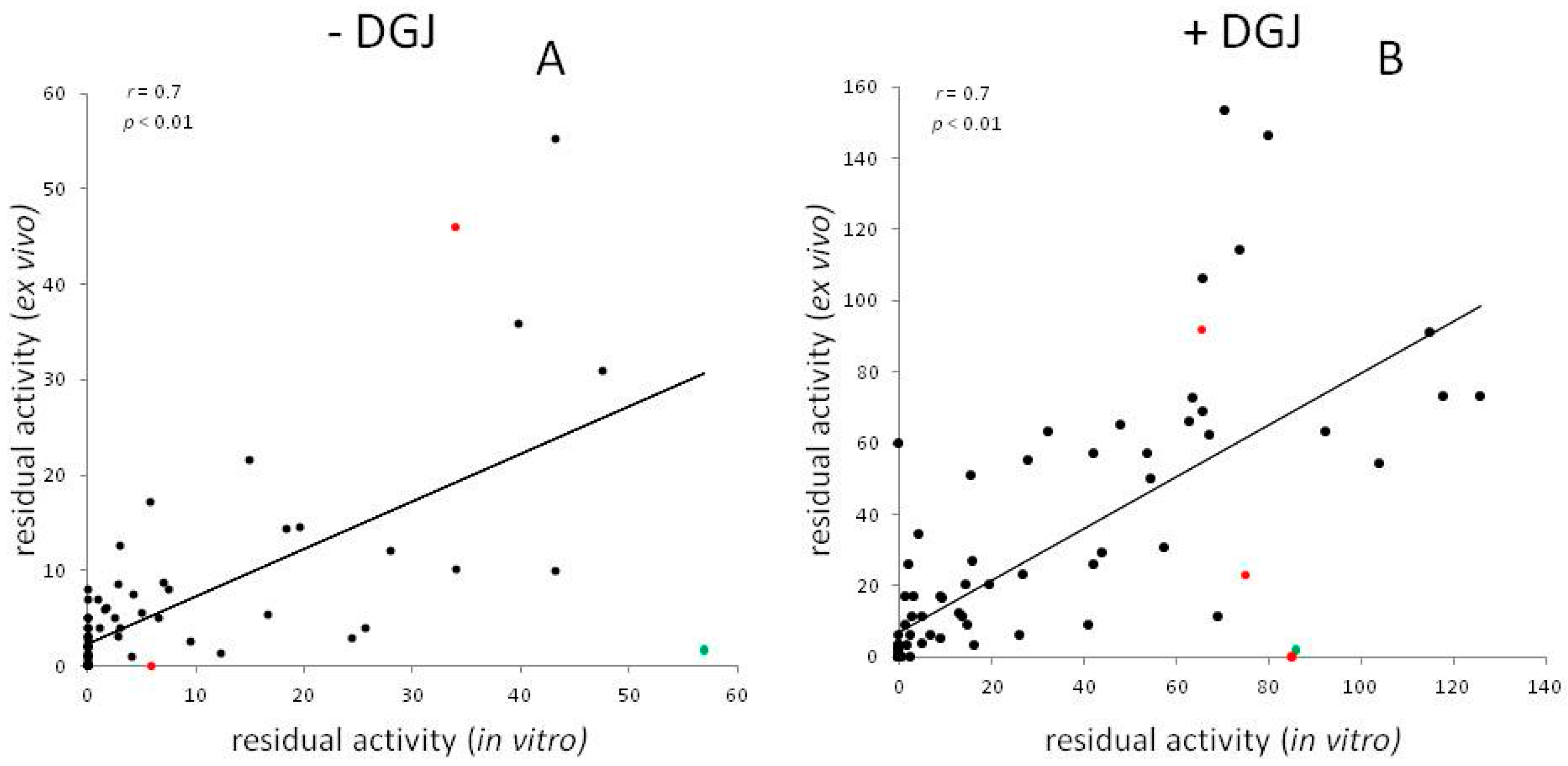

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Cell Type | Concentration and Incubation Time |
|---|---|---|
| Ishii_2000 [50] | Transfection COS1 | 20 μM DGJ 1 day |
| Spada_2006 [10] | Transfection COS7 | 20 μM DGJ 72 h |
| Shin_2007 [51] | T-cells and fibroblasts | 20 μM DGJ 3 or 4 days |
| Ishii_2007 [52] | Lymphoblasts and fibroblasts | 20 μM DGJ 5 days |
| Shin_2008 [53] | T-cells | 20 μM DGJ 3 days |
| Park_2009 [54] | Transfection COS7 | 20 μM DGJ 2 days |
| Benjamin_2009 [55] | Lymphoblasts and fibroblasts | Depending on EC50 5 days |
| Filoni_2010 [56] | Transfection COS1 and lymphocytes | 20 μM DGJ 72 h |
| Wu_2011 [57] | Transfection HEK293 | Depending on EC50 4 to 5 days |
| Andreotti_2011 [58] | Transfection COS7 | 20 μM DGJ 48 h |
| Lukas_2013 [2] | Transfection HEK293H | 20 μM DGJ 60 h |
| Giugliani_2013 [59] | Transfection HEK293 | 10 μM DGJ |
| Lukas_2016 [60] | Transfection HEK293 | 20 μM DGJ 60 h |
| Mutation | No. Hemiz | −DGJ | +DGJ | PSSM | Humdiv | Humvar | Reference |
|---|---|---|---|---|---|---|---|
| L3P | 4 | 117.7 | 129.4 | −3 | Probably damaging | Probably damaging | [60] |
| E66Q | 3 | 47.6 | 53.66 | −2 | Probably damaging | Probably damaging | [68] |
| R118C | 8 | 24.5 | 27.8 | −2 | Probably damaging | Possibly damaging | [10] |
| N139S | 7 | 147.8 | 176.4 | −1 | Benign | Benign | [69] |
| S126G | 18 | 51.3 | 67.4 | −2 | Benign | Benign | [70] |
| A143T | 19 | 39.7 | 63.7 | −1 | Probably damaging | Possibly damaging | [71] |
| I289V | 3 | 79.9 | 95 | 0 | Probably damaging | Possibly damaging | |
| D313Y | 129 | 75.5 | 100.3 | −1 | Probably damaging | Possibly damaging | [71] |
| R363H | 3 | 28 | 65.7 | −1 | Benign | Benign | [72] |
| A368T | 3 | 103.7 | 93.3 | 0 | Benign | Benign | [2] |
| T385A | 36 | 45 | 48.9 | −2 | Possibly damaging | Benign | [2] |
| W399S | 5 | 53 | 51.5 | −4 | Possibly damaging | Benign | [60] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Citro, V.; Cammisa, M.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. The Large Phenotypic Spectrum of Fabry Disease Requires Graduated Diagnosis and Personalized Therapy: A Meta-Analysis Can Help to Differentiate Missense Mutations. Int. J. Mol. Sci. 2016, 17, 2010. https://doi.org/10.3390/ijms17122010
Citro V, Cammisa M, Liguori L, Cimmaruta C, Lukas J, Cubellis MV, Andreotti G. The Large Phenotypic Spectrum of Fabry Disease Requires Graduated Diagnosis and Personalized Therapy: A Meta-Analysis Can Help to Differentiate Missense Mutations. International Journal of Molecular Sciences. 2016; 17(12):2010. https://doi.org/10.3390/ijms17122010
Chicago/Turabian StyleCitro, Valentina, Marco Cammisa, Ludovica Liguori, Chiara Cimmaruta, Jan Lukas, Maria Vittoria Cubellis, and Giuseppina Andreotti. 2016. "The Large Phenotypic Spectrum of Fabry Disease Requires Graduated Diagnosis and Personalized Therapy: A Meta-Analysis Can Help to Differentiate Missense Mutations" International Journal of Molecular Sciences 17, no. 12: 2010. https://doi.org/10.3390/ijms17122010