
Radical-Mediated Enzymatic Polymerizations

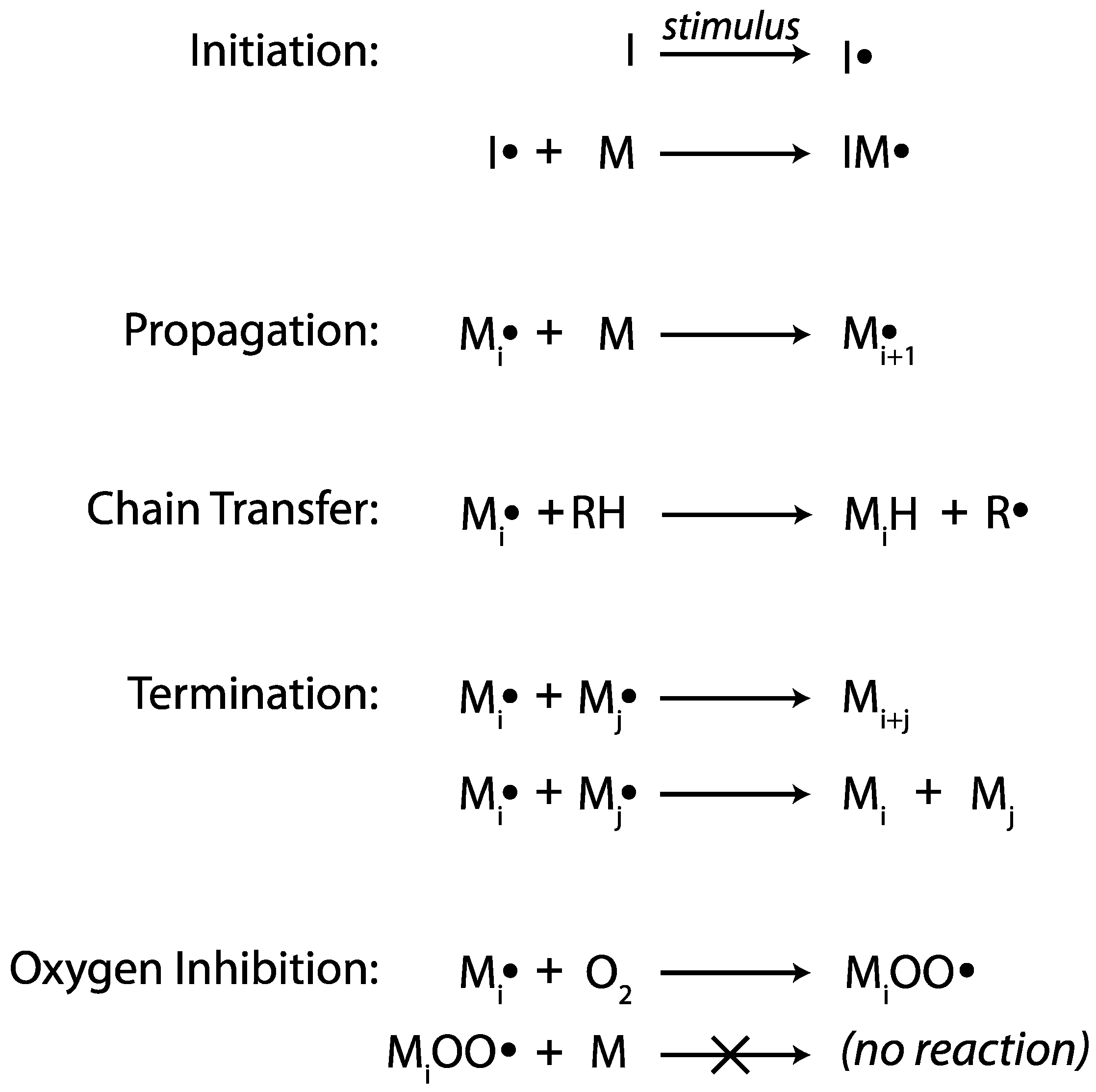{kind=link}
{kind=link}
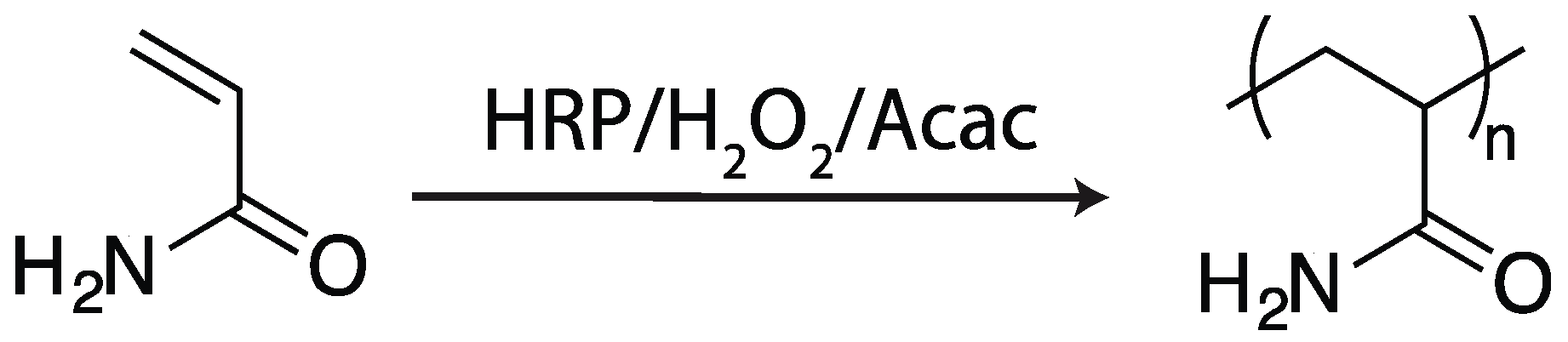{kind=link}
{kind=link}
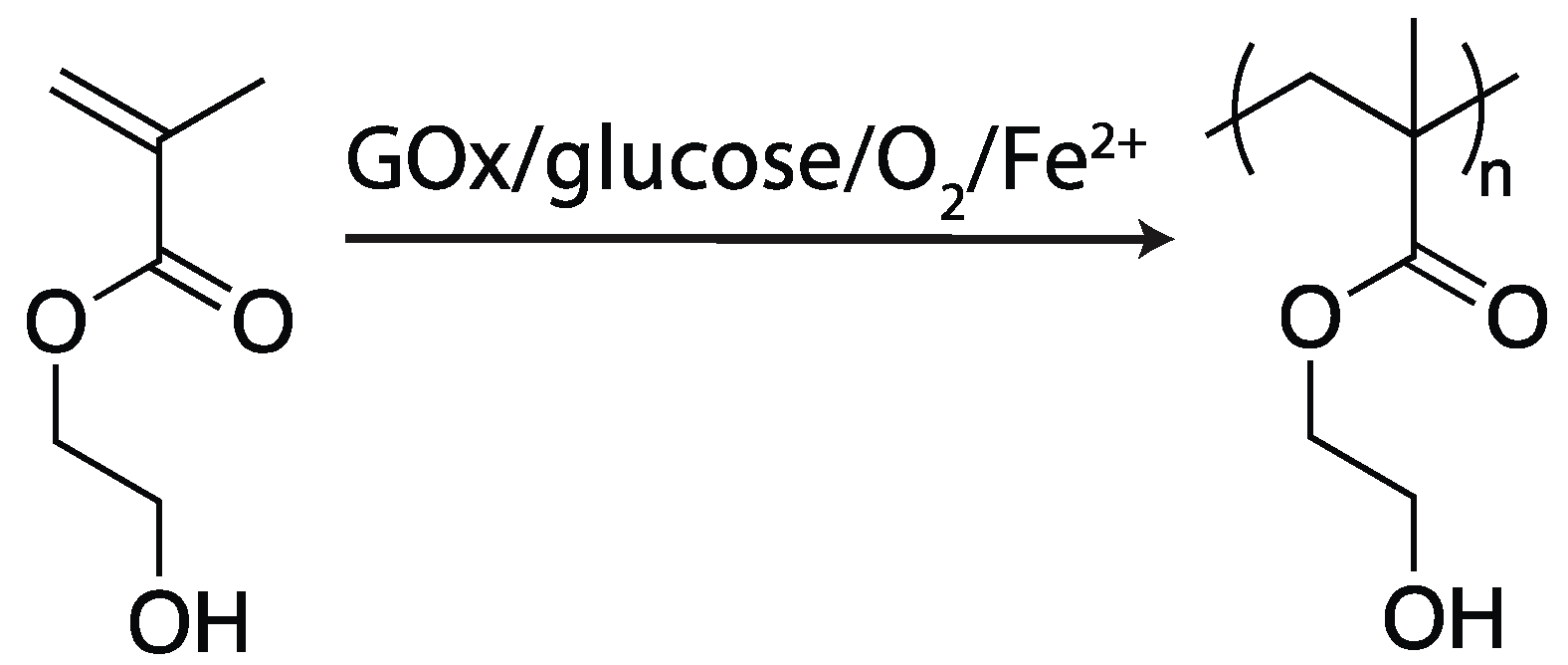{kind=link}
{kind=link}
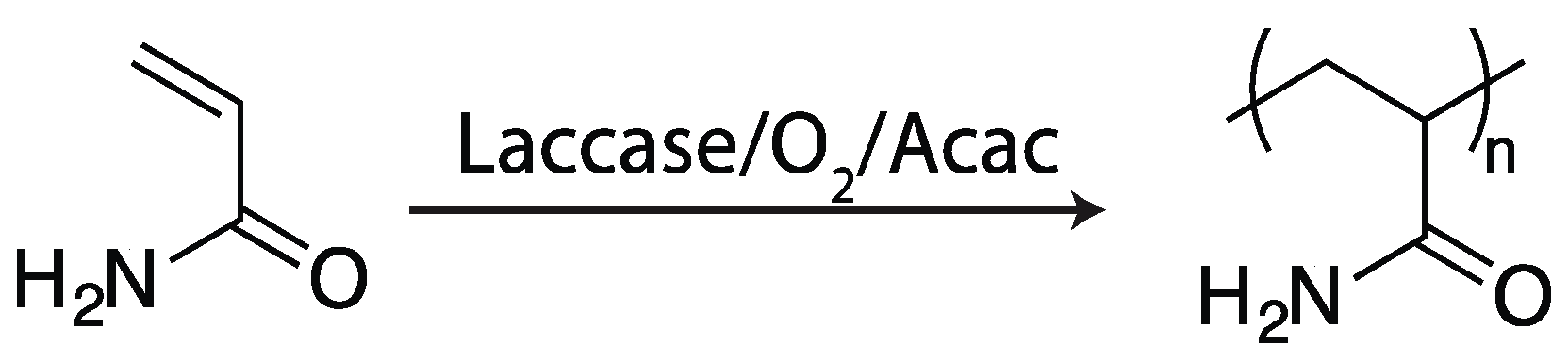{kind=link}
{kind=link}
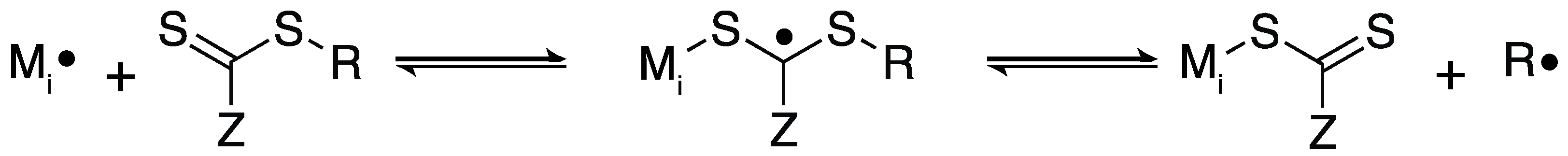{kind=link}
{kind=link}
{kind=link}
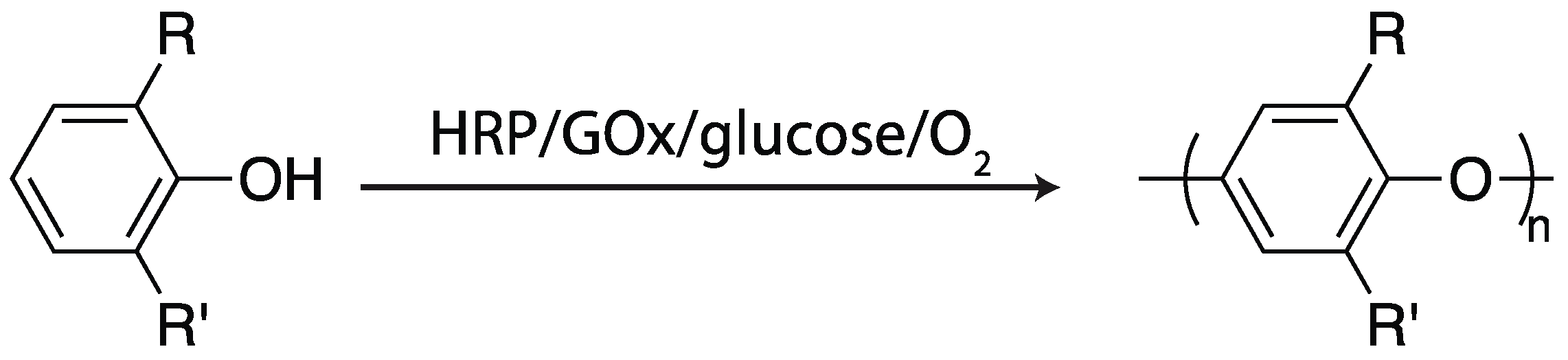{kind=link}
Abstract
:1. Introduction
2. Enzyme-Mediated Polymerization Reactions
2.1. Chain-Growth Free Radical Polymerization

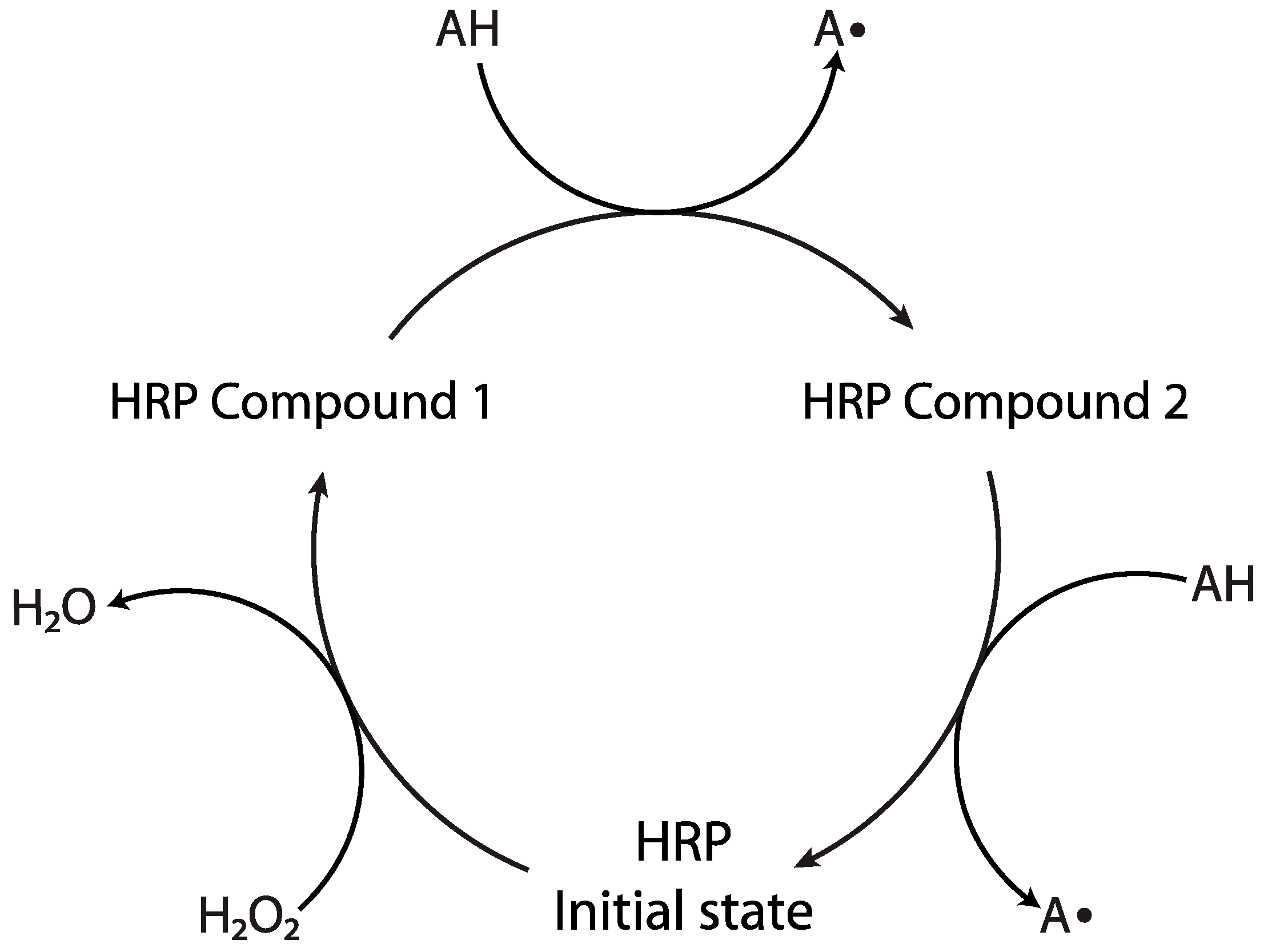
2.1.1. Horseradish Peroxidase


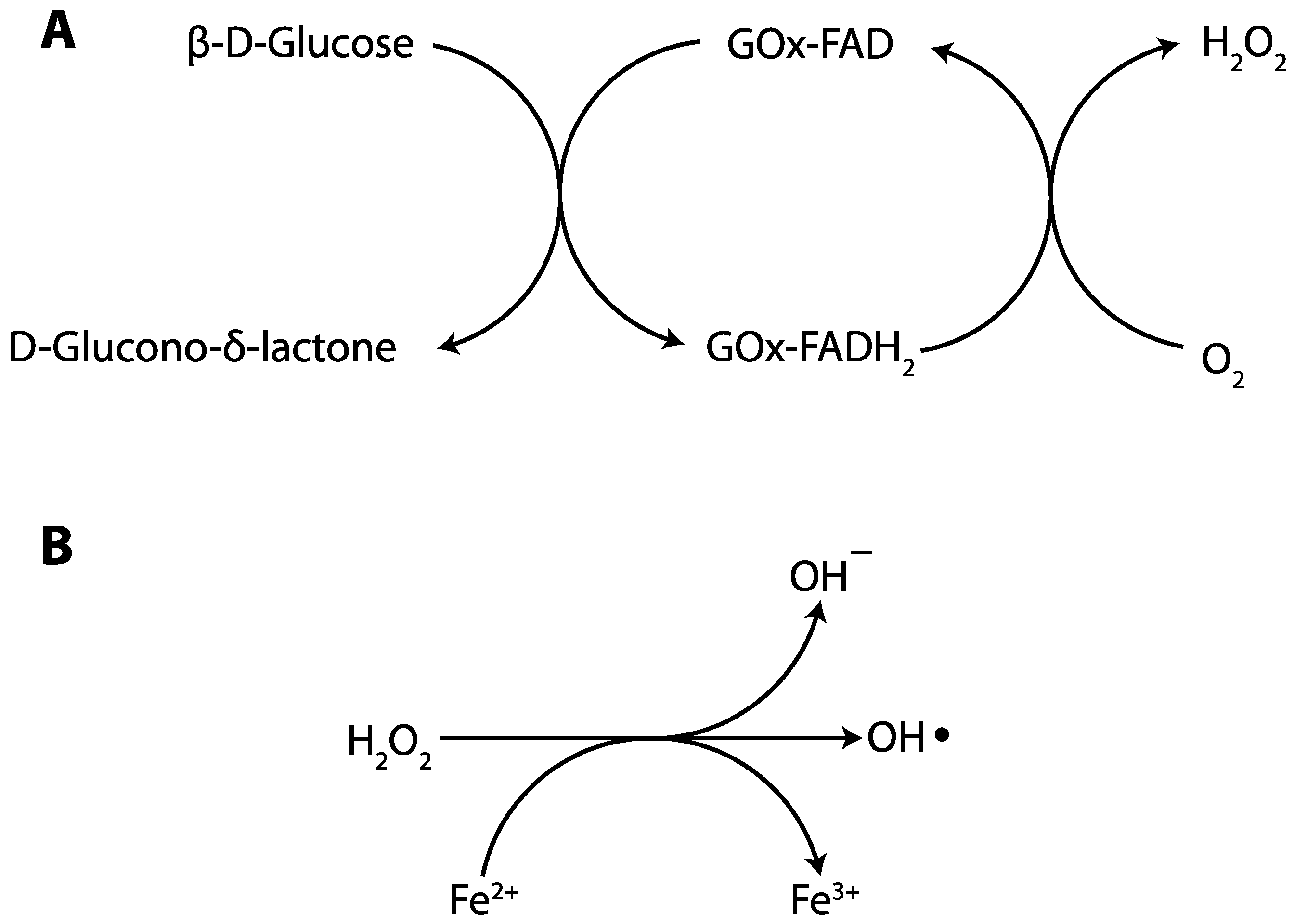
2.1.2. Glucose Oxidase


2.1.3. Laccase


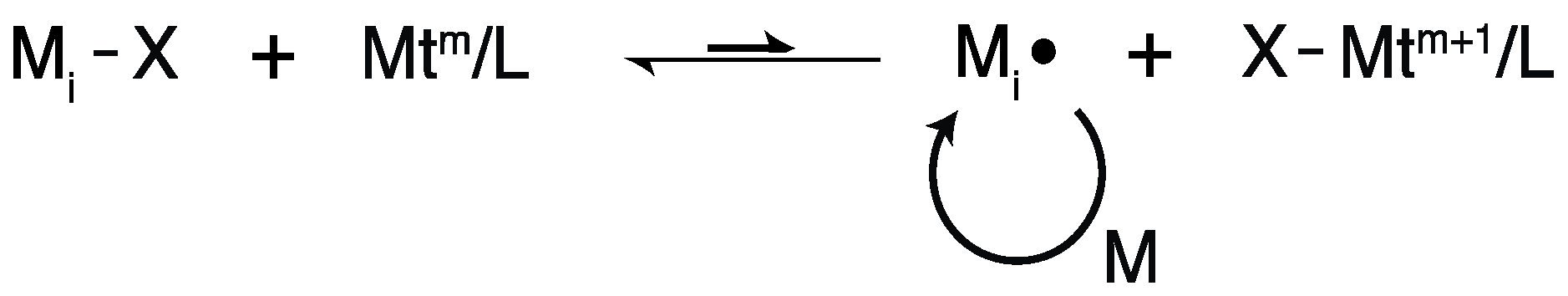
2.2. Living Radical Polymerization


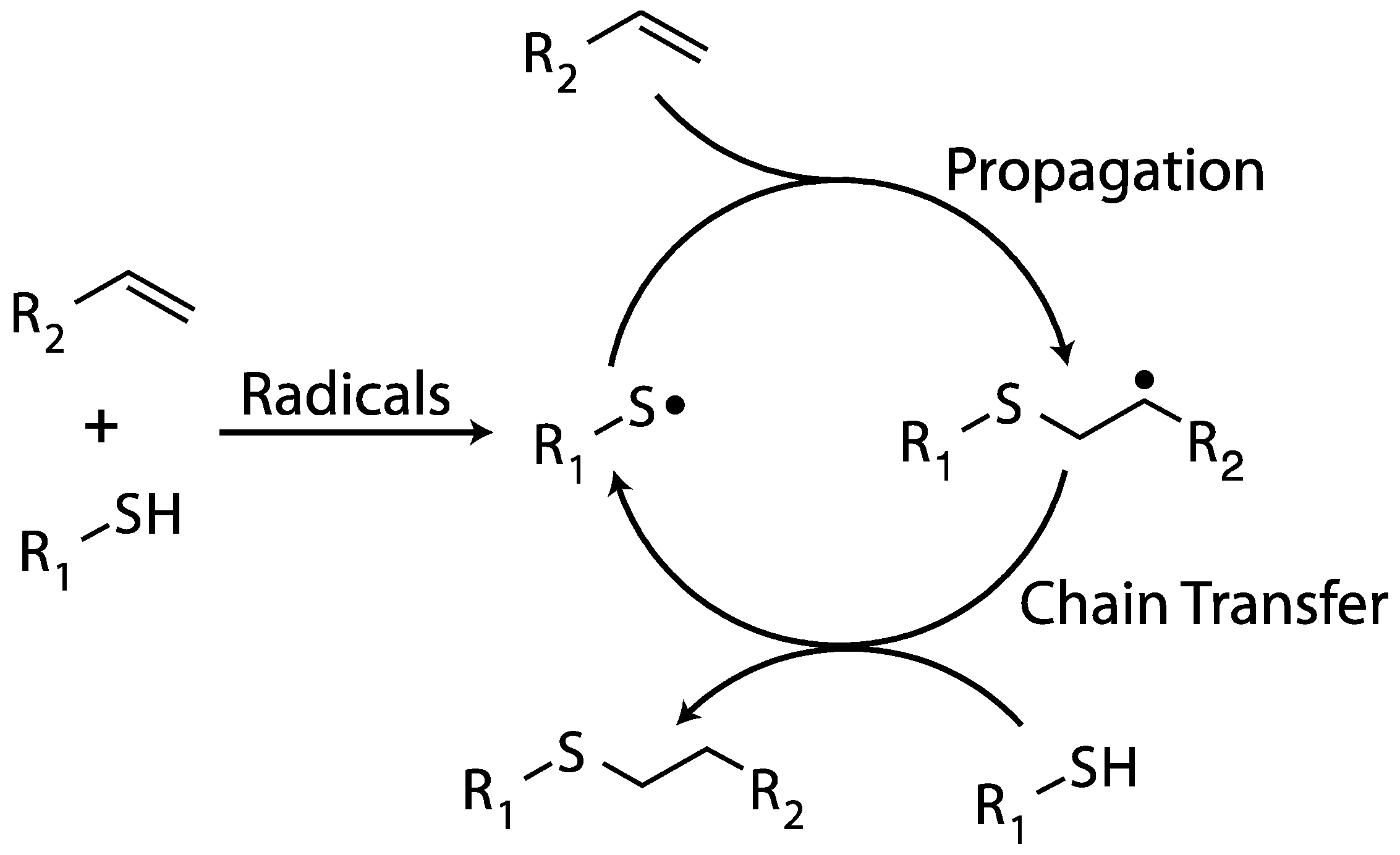
2.3. Thiol–ene Polymerization

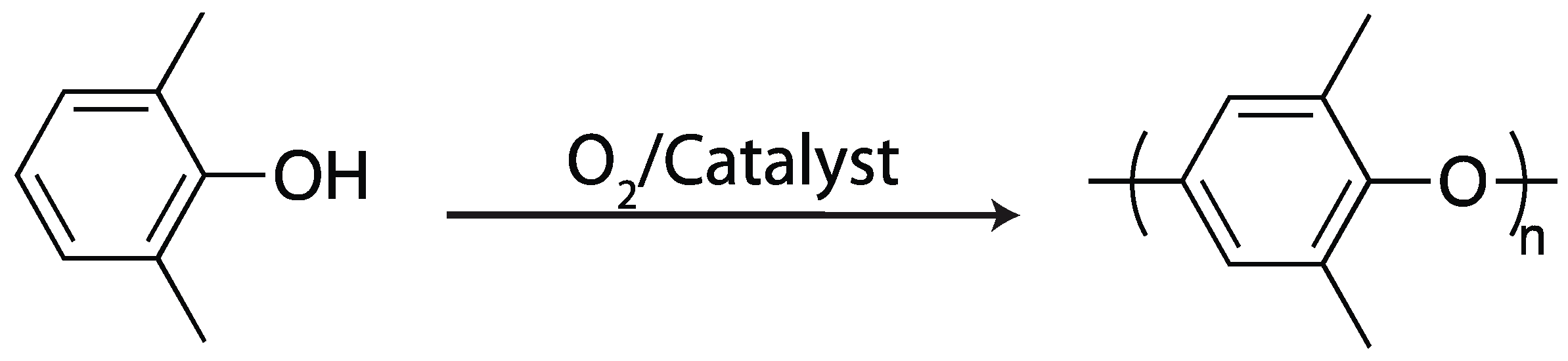
2.4. Oxidative Coupling

2.4.1. Horseradish Peroxidase
2.4.2. Glucose Oxidase

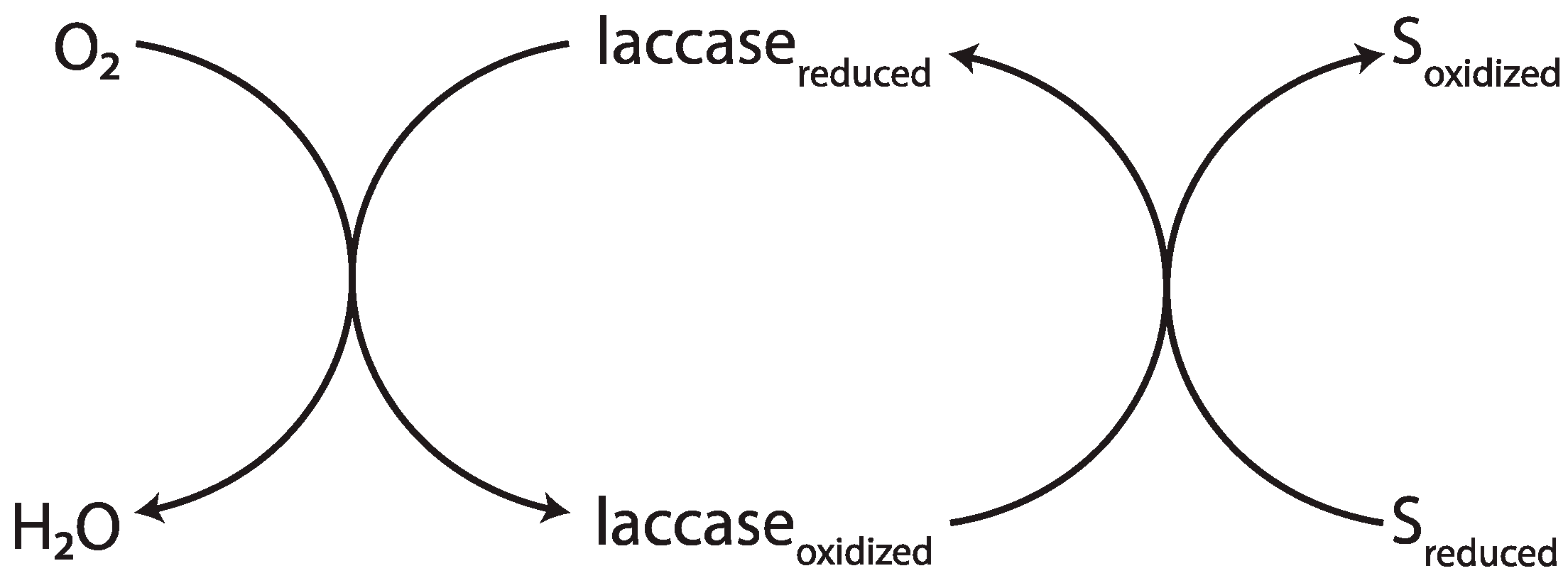
2.4.3. Laccase
3. Formulation Considerations for Enzymatic Polymerizations
3.1. Enzyme Deactivation
3.2. Enzyme Promiscuity
3.3. The Role of Mediators
4. Applications Utilizing Enzyme-Mediated in Situ Polymerization
4.1. Adhesives/Biomaterials
4.2. Sensors
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Radzicka, A.; Wolfenden, R. A proficient enzyme. Science 1995, 267, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Parravano, G. Chain reactions induced by enzymic systems. J. Am. Chem. Soc. 1951, 73, 183–184. [Google Scholar] [CrossRef]
- Mena, M.; Shirai, K.; Tecante, A.; Barzana, E.; Gimeno, M. Enzymatic syntheses of linear and hyperbranched poly-l-lactide using compressed R134a—ionic liquid media. J. Supercrit. Fluids 2015, 103, 77–82. [Google Scholar] [CrossRef]
- Jaros, D.; Schwarzenbolz, U.; Raak, N.; Lobner, J.; Henle, T.; Rohm, H. Cross-linking with microbial transglutaminase: Relationship between polymerisation degree and stiffness of acid casein gels. Int. Dairy J. 2014, 38, 174–178. [Google Scholar] [CrossRef]
- Kavitha, V.; Mandal, A.B.; Gnanamani, A. Microbial mediated dimerization of fattyacids of sunflower oil: An effective role of lipase and biosurfactant. J. Appl. Polym. Sci. 2014, 131, 40555. [Google Scholar] [CrossRef]
- Hunley, M.T.; Sari, N.; Beers, K.L. Microstructure analysis and model discrimination of enzyme-catalyzed copolyesters. ACS Macro Lett. 2013, 2, 375–379. [Google Scholar] [CrossRef]
- Yung, C.W.; Wu, L.Q.; Tullman, J.A.; Payne, G.F.; Bentley, W.E.; Barbari, T.A. Transglutaminase crosslinked gelatin as a tissue engineering scaffold. J. Biomed. Mater. Res. A 2007, 83, 1039–1046. [Google Scholar] [CrossRef]
- Fuchs, S.; Kutscher, M.; Hertel, T.; Winter, G.; Pietzsch, M.; Coester, C. Transglutaminase: New insights into gelatin nanoparticle cross-linking. J. Microencapsul. 2010, 27, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kopelman, D.; Wu, L.Q.; Hijji, K.; Attar, I.; Preiss-Bloom, O.; Payne, G.F. Biomimetic sealant based on gelatin and microbial transglutaminase: An initial in vivo investigation. J. Biomed. Mater. Res. B 2009, 91, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Colombani, D. Chain-growth control in free radical polymerization. Prog. Polym. Sci. 1997, 22, 1649–1720. [Google Scholar] [CrossRef]
- Achilias, D.S. A review of modeling of diffusion controlled polymerization reactions. Macromol. Theory Simul. 2007, 16, 319–347. [Google Scholar] [CrossRef]
- O’Brien, A.K.; Bowman, C.N. Impact of oxygen on photopolymerization kinetics and polymer structure. Macromolecules 2006, 39, 2501–2506. [Google Scholar] [CrossRef]
- Louie, B.M.; Carratt, G.M.; Soong, D.S. Modeling the free radical solution and bulk polymerization of methyl methacrylate. J. Appl. Polym. Sci. 1985, 30, 3985–4012. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; Hesse, P.; Lacik, I. Free-radical propagation rate coefficient of nonionized methacrylic acid in aqueous solution from low monomer concentrations to bulk polymerization. Macromolecules 2006, 39, 184–193. [Google Scholar] [CrossRef]
- Li, W.H.; Stover, H.D.H. Porous monodisperse poly(divinylbenzene) microspheres by precipitation polymerization. J. Polym. Sci. Pol. Chem. 1998, 36, 1543–1551. [Google Scholar] [CrossRef]
- VivaldoLima, E.; Wood, P.E.; Hamielec, A.E.; Penlidis, A. An updated review on suspension polymerization. Ind. Eng. Chem. Res. 1997, 36, 939–965. [Google Scholar] [CrossRef]
- Chern, C.S. Emulsion polymerization mechanisms and kinetics. Prog. Polym. Sci. 2006, 31, 443–486. [Google Scholar] [CrossRef]
- Thickett, S.C.; Gilbert, R.G. Emulsion polymerization: State of the art in kinetics and mechanisms. Polymer 2007, 48, 6965–6991. [Google Scholar] [CrossRef]
- Derango, R.A.; Chiang, L.C.; Dowbenko, R.; Lasch, J.G. Enzyme-mediated polymerization of acrylic monomers. Biotechnol. Tech. 1992, 6, 523–526. [Google Scholar] [CrossRef]
- Lavery, C.B.; MacInnis, M.C.; MacDonald, M.J.; Williams, J.B.; Spencer, C.A.; Burke, A.A.; Irwin, D.J.G.; D’Cunha, G.B. Purification of peroxidase from horseradish (Armoracia rusticana) roots. J. Agric. Food. Chem. 2010, 58, 8471–8476. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Ma, D.C.; Kaplan, D.L. Enzyme-mediated free radical polymerization of styrene. Biomacromolecules 2000, 1, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Durand, A.; Lalot, T.; Brigodiot, M.; Marechal, E. Enzyme-mediated initiation of acrylamide polymerization: Reaction mechanism. Polymer 2000, 41, 8183–8192. [Google Scholar] [CrossRef]
- Berglund, G.I.; Carlsson, G.H.; Smith, A.T.; Szoke, H.; Henriksen, A.; Hajdu, J. The catalytic pathway of horseradish peroxidase at high resolution. Nature 2002, 417, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Veitch, N.C. Horseradish peroxidase: A modern view of a classic enzyme. Phytochemistry 2004, 65, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Taylor, K.E.; Zou, H.X.; Biswas, N.; Bewtra, J.K. Phenol conversion and dimeric intermediates in horseradish peroxidase-catalyzed phenol removal from water. Environ. Sci. Technol. 1994, 28, 2154–2160. [Google Scholar] [CrossRef] [PubMed]
- Cooper, V.A.; Nicell, J.A. Removal of phenols from a foundry wastewater using horseradish peroxidase. Water Res. 1996, 30, 954–964. [Google Scholar] [CrossRef]
- Huang, J.; Chang, Q.; Ding, Y.B.; Han, X.Y.; Tang, H.Q. Catalytic oxidative removal of 2,4-dichlorophenol by simultaneous use of horseradish peroxidase and graphene oxide/Fe3O4 as catalyst. Chem. Eng. J. 2014, 254, 434–442. [Google Scholar] [CrossRef]
- Tang, D.P.; Yuan, R.; Chal, Y.Q. Ultrasensitive electrochemical immunosensor for clinical immunoassay using thionine-doped magnetic gold nanospheres as labels and horseradish peroxidase as enhancer. Anal. Chem. 2008, 80, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Munge, B.; Patel, V.; Jensen, G.; Bhirde, A.; Gong, J.D.; Kim, S.N.; Gillespie, J.; Gutkind, J.S.; Papadimitrakopoulos, F.; et al. Carbon nanotube amplification strategies for highly sensitive immunodetection of cancer biomarkers. J. Am. Chem. Soc. 2006, 128, 11199–11205. [Google Scholar] [CrossRef] [PubMed]
- Bobrow, M.N.; Harris, T.D.; Shaughnessy, K.J.; Litt, G.J. Catalyzed reporter deposition, a novel method of signal amplification—application to immunoassays. J. Immunol. Methods 1989, 125, 279–285. [Google Scholar] [CrossRef]
- Emery, O.; Lalot, T.; Brigodiot, M.; Marechal, E. Free-radical polymerization of acrylamide by horseradish peroxidase-mediated initiation. J. Polym. Sci. Pol. Chem. 1997, 35, 3331–3333. [Google Scholar] [CrossRef]
- Durand, A.; Lalot, T.; Brigodiot, M.; Marechal, E. Enzyme-mediated radical initiation of acrylamide polymerization: Main characteristics of molecular weight control. Polymer 2001, 42, 5515–5521. [Google Scholar] [CrossRef]
- Teixeira, D.; Lalot, T.; Brigodiot, M.; Marechal, E. β-diketones as key compounds in free-radical polymerization by enzyme-mediated initiation. Macromolecules 1999, 32, 70–72. [Google Scholar] [CrossRef]
- Cai, Z.Q.; Wang, W.C.; Ruan, G.; Wen, X.F. Kinetic study of acrylamide radical polymerization initiated by the horseradish peroxidase-mediated system. Int. J. Chem. Kinet. 2012, 44, 475–481. [Google Scholar] [CrossRef]
- Su, T.; Zhang, D.; Tang, Z.; Wu, Q.; Wang, Q.G. HRP-mediated polymerization forms tough nanocomposite hydrogels with high biocatalytic performance. Chem. Commun. 2013, 49, 8033–8035. [Google Scholar] [CrossRef]
- Fukushima, H.; Kohri, M.; Kojima, T.; Taniguchi, T.; Saito, K.; Nakahira, T. Surface-initiated enzymatic vinyl polymerization: Synthesis of polymer-grafted silica particles using horseradish peroxidase as catalyst. Polym. Chem. 2012, 3, 1123–1125. [Google Scholar] [CrossRef]
- Kalra, B.; Gross, R.A. Horseradish peroxidase mediated free radical polymerization of methyl methacrylate. Biomacromolecules 2000, 1, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Sutton, H.C.; Winterbourn, C.C. On the participation of higher oxidation-states of iron and copper in fenton reactions. Free Radic. Biol. Med. 1989, 6, 53–60. [Google Scholar] [CrossRef]
- Nappi, A.J.; Vass, E. Comparative studies of enhanced iron-mediated production of hydroxyl radical by glutathione, cysteine, ascorbic acid, and selected catechols. Biochim. Biophys. Acta 1997, 1336, 295–302. [Google Scholar] [CrossRef]
- Siedlecka, E.M.; Wieckowska, A.; Stepnowski, P. Influence of inorganic ions on MTBE degradation by Fenton’s reagent. J. Hazard. Mater. 2007, 147, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Hata, Y.; Matsuda, T.; Ikada, Y. Initiation of radical polymerization by glucose oxidase utilizing dissolved oxygen. J. Polym. Sci. Pol. Chem. 1991, 29, 1217–1218. [Google Scholar] [CrossRef]
- Johnson, L.M.; Fairbanks, B.D.; Anseth, K.S.; Bowman, C.N. Enzyme-mediated redox initiation for hydrogel generation and cellular encapsulation. Biomacromolecules 2009, 10, 3114–3121. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Kitamura, Y.; Yoshizawa, H. Emulsion polymerization of styrene by horseradish peroxidase-mediated initiation. Colloid Polym. Sci. 2005, 284, 108–111. [Google Scholar] [CrossRef]
- Naves, A.F.; Carmona-Ribeiro, A.M.; Petri, D.F.S. Immobilized horseradish peroxidase as a reusable catalyst for emulsion polymerization. Langmuir 2007, 23, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Hatzinikolaou, D.G.; Hansen, O.C.; Macris, B.J.; Tingey, A.; Kekos, D.; Goodenough, P.; Stougaard, P. A new glucose oxidase from Aspergillus niger: Characterization and regulation studies of enzyme and gene. Appl. Microbiol. Biotechnol. 1996, 46, 371–381. [Google Scholar] [PubMed]
- Kalisz, H.M.; Hendle, J.; Schmid, R.D. Structural and biochemical properties of glycosylated and deglycosylated glucose oxidase from Penicillium amagasakiense. Appl. Microbiol. Biotechnol. 1997, 47, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Rando, D.; Kohring, G.W.; Giffhorn, F. Production, purification and characterization of glucose oxidase from a newly isolated strain of Penicillium pinophilum. Appl. Microbiol. Biotechnol. 1997, 48, 34–40. [Google Scholar] [CrossRef]
- Pluschkell, S.; Hellmuth, K.; Rinas, U. Kinetics of glucose oxidase excretion by recombinant Aspergillus niger. Biotechnol. Bioeng. 1996, 51, 215–220. [Google Scholar] [CrossRef]
- Witt, S.; Wohlfahrt, G.; Schomburg, D.; Hecht, H.J.; Kalisz, H.M. Conserved arginine-516 of Penicillium amagasakiense glucose oxidase is essential for the efficient binding of β-d-glucose. Biochem. J. 2000, 347, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.M.; Deforest, C.A.; Pendurti, A.; Anseth, K.S.; Bowman, C.N. Formation of three-dimensional hydrogel multilayers using enzyme-mediated redox chain initiation. ACS Appl. Mater. Interfaces 2010, 2, 1963–1972. [Google Scholar] [CrossRef]
- Berron, B.J.; Johnson, L.M.; Ba, X.; McCall, J.D.; Alvey, N.J.; Anseth, K.S.; Bowman, C.N. Glucose oxidase-mediated polymerization as a platform for dual-mode signal amplification and biodetection. Biotechnol. Bioeng. 2011, 108, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, R.; Bowman, C.N. Kinetics of interfacial radical polymerization initiated by a glucose-oxidase mediated redox system. Biomaterials 2012, 33, 6909–6914. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, R.; Tibbitt, M.W.; Anseth, K.S.; Bowman, C.N. Formation of core-shell particles by interfacial radical polymerization initiated by a glucose oxidase-mediated redox system. Chem. Mater. 2013, 25, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.A.; Wu, Q.; Wei, Q.C.; Wang, Q.G. Bioinorganic nanocomposite hydrogels formed by HRP-GOx-cascade-catalyzed polymerization and exfoliation of the layered composites. Chem. Eur. J. 2015, 21, 12620–12626. [Google Scholar] [CrossRef] [PubMed]
- Gormley, A.J.; Chapman, R.; Stevens, M.M. Polymerization amplified detection for nanoparticle-based biosensing. Nano Lett. 2014, 14, 6368–6373. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Tang, Z.; He, H.J.; Li, W.J.; Wang, X.; Liao, C.N.; Sun, Y.; Wang, Q.G. Glucose oxidase triggers gelation of N-hydroxyimide-heparin conjugates to form enzyme-responsive hydrogels for cell-specific drug delivery. Chem. Sci. 2014, 5, 4204–4209. [Google Scholar] [CrossRef]
- Simizu, S.; Ishida, K.; Wierzba, M.K.; Sato, T.A.; Osada, H. Expression of heparanase in human tumor cell lines and human head and neck tumors. Cancer Lett. 2003, 193, 83–89. [Google Scholar] [CrossRef]
- Ikeda, R.; Tanaka, H.; Uyama, H.; Kobayashi, S. Laccase-catalyzed polymerization of acrylamide. Macromol. Rapid Commun. 1998, 19, 423–425. [Google Scholar] [CrossRef]
- Hollmann, F.; Gumulya, Y.; Tolle, C.; Liese, A.; Thum, O. Evaluation of the laccase from Myceliophthora thermophila as industrial biocatalyst for polymerization reactions. Macromolecules 2008, 41, 8520–8524. [Google Scholar] [CrossRef]
- Ai, M.Q.; Wang, F.F.; Huang, F. Purification and characterization of a thermostable laccase from Trametes trogii and its ability in modification of kraft lignin. J. Microbiol. Biotechnol. 2015, 25, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Aktas, N.; Cicek, H.; Unal, A.T.; Kibarer, G.; Kolankaya, N.; Tanyolac, A. Reaction kinetics for laccase-catalyzed polymerization of 1-naphthol. Bioresour. Technol. 2001, 80, 29–36. [Google Scholar] [CrossRef]
- Yang, H.; Sun, H.F.; Zhang, S.J.; Wu, B.D.; Pan, B.C. Potential of acetylacetone as a mediator for Trametes versicolor laccase in enzymatic transformation of organic pollutants. Environ. Sci. Pollut. Res. 2015, 22, 10882–10889. [Google Scholar] [CrossRef] [PubMed]
- Junker, K.; Zandomeneghi, G.; Schuler, L.D.; Kissner, R.; Walde, P. Enzymatic polymerization of pyrrole with Trametes versicolor laccase and dioxygen in the presence of vesicles formed from AOT (sodium bis-(2-ethylhexyl) sulfosuccinate) as templates. Synth. Met. 2015, 200, 123–134. [Google Scholar] [CrossRef]
- Bajpai, P. Application of enzymes in the pulp and paper industry. Biotechnol. Progr. 1999, 15, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Kanagaraj, J.; Senthilvelan, T.; Panda, R.C. Degradation of azo dyes by laccase: Biological method to reduce pollution load in dye wastewater. Clean Technol. Environ. Policy 2015, 17, 1443–1456. [Google Scholar] [CrossRef]
- Escalona, I.; Grooth, J.; Font, J.; Nijmeijer, K. Removal of BPA by enzyme polymerization using NF membranes. J. Membr. Sci. 2014, 468, 192–201. [Google Scholar] [CrossRef]
- Nieto, M.; Nardecchia, S.; Peinado, C.; Catalina, F.; Abrusci, C.; Gutierrez, M.C.; Ferrer, M.L.; del Monte, F. Enzyme-induced graft polymerization for preparation of hydrogels: Synergetic effect of laccase-immobilized-cryogels for pollutants adsorption. Soft Matter 2010, 6, 3533–3540. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Niu, D.C.; Li, P.; Wu, Q.; Bo, X.W.; Liu, B.J.; Bao, S.; Su, T.; Xu, H.X.; Wang, Q.G. Dual-enzyme-loaded multifunctional hybrid nanogel system for pathological responsive ultrasound imaging and T2-weighted magnetic resonance imaging. ACS Nano 2015, 9, 5646–5656. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.H.; di Lena, F.; Chai, C.L.L. Metalloenzymatic radical polymerization using alkyl halides as initiators. Polym. Chem. 2011, 2, 589–594. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom transfer radical polymerization (ATRP): Current status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Ng, Y.H.; di Lena, F.; Chai, C.L.L. PolyPEGA with predetermined molecular weights from enzyme-mediated radical polymerization in water. Chem. Commun. 2011, 47, 6464–6466. [Google Scholar] [CrossRef] [PubMed]
- Sigg, S.J.; Seidi, F.; Renggli, K.; Silva, T.B.; Kali, G.; Bruns, N. Horseradish peroxidase as a catalyst for atom transfer radical polymerization. Macromol. Rapid Commun. 2011, 32, 1710–1715. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.B.; Spulber, M.; Kocik, M.K.; Seidi, F.; Charan, H.; Rother, M.; Sigg, S.J.; Renggli, K.; Kali, G.; Bruns, N. Hemoglobin and red blood cells catalyze atom transfer radical polymerization. Biomacromolecules 2013, 14, 2703–2712. [Google Scholar] [CrossRef]
- Yamashita, K.; Yamamoto, K.; Kadokawa, J. Atom transfer radical polymerization of N-isopropylacrylamide by enzyme mimetic catalyst. Polymer 2013, 54, 1775–1778. [Google Scholar] [CrossRef]
- Zhou, H.; Jiang, W.; An, N.; Zhang, Q.P.; Xiang, S.D.; Wang, L.P.; Tang, J. Enzyme mimetic-catalyzed ATRP and its application in block copolymer synthesis combined with enzymatic ring-opening polymerization. RSC Adv. 2015, 5, 42728–42735. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, A.; Arbe, A.; Kohlbrecher, J.; Colmenero, J.; Pomposo, J.A. Efficient synthesis of single-chain globules mimicking the morphology and polymerase activity of metalloenzymes. Macromol. Rapid Commun. 2015, 36, 1592–1597. [Google Scholar] [CrossRef] [PubMed]
- Simakova, A.; Mackenzie, M.; Averick, S.E.; Park, S.; Matyjaszewski, K. Bioinspired iron-based catalyst for atom transfer radical polymerization. Angew. Chem. Int. Ed. 2013, 52, 12148–12151. [Google Scholar] [CrossRef] [PubMed]
- Dinu, M.V.; Spulber, M.; Renggli, K.; Wu, D.L.; Monnier, C.A.; Petri-Fink, A.; Bruns, N. Filling polymersomes with polymers by peroxidase-catalyzed atom transfer radical polymerization. Macromol. Rapid Commun. 2015, 36, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, X.; Zhu, A.; Ma, K.; Lv, Y.; Wang, X.; An, Z. Enzyme-initiated reversible addition–fragmentation chain transfer polymerization. Macromolecules 2015, 48, 7792–7802. [Google Scholar] [CrossRef]
- Chapman, R.; Gormley, A.J.; Herpoldt, K.L.; Stevens, M.M. Highly Controlled Open Vessel RAFT Polymerizations by Enzyme Degassing. Macromolecules 2014, 47, 8541–8547. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Bowman, C.N. Thiol-Ene Click Chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, C.E.; Lowe, A.B.; Bowman, C.N. Thiol-click chemistry: A multifaceted toolbox for small molecule and polymer synthesis. Chem. Soc. Rev. 2010, 39, 1355–1387. [Google Scholar] [CrossRef] [PubMed]
- Zavada, S.R.; McHardy, N.R.; Scott, T.F. Oxygen-mediated enzymatic polymerization of thiol-ene hydrogels. J. Mater. Chem. B 2014, 2, 2598–2605. [Google Scholar] [CrossRef] [PubMed]
- Pichorner, H.; Couperus, A.; Korori, S.A.A.; Ebermann, R. Plant peroxidase has a thiol oxidase function. Phytochemistry 1992, 31, 3371–3376. [Google Scholar] [CrossRef]
- Burner, U.; Obinger, C. Transient-state and steady-state kinetics of the oxidation of aliphatic and aromatic thiols by horseradish peroxidase. FEBS Lett. 1997, 411, 269–274. [Google Scholar] [CrossRef]
- Hay, A.S. Polymerization by oxidative coupling: Discovery and commercialization of PPO ® and Noryl ® resins. J. Polym. Sci. Pol. Chem. 1998, 36, 505–517. [Google Scholar] [CrossRef]
- Rocha-Martin, J.; Velasco-Lozano, S.; Guisan, J.M.; Lopez-Gallego, F. Oxidation of phenolic compounds catalyzed by immobilized multi-enzyme systems with integrated hydrogen peroxide production. Green Chem. 2014, 16, 303–311. [Google Scholar] [CrossRef]
- Dordick, J.S.; Marletta, M.A.; Klibanov, A.M. Polymerization of phenols catalyzed by peroxidase in nonaqueous media. Biotechnol. Bioeng. 1987, 30, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Zhang, L.; Gao, Y.H.; Wu, Y.F.; Zhao, W.S.; Cui, Y.C. Enzymatic oxidative polymerization of pyrogallic acid for preparation of hindered phenol antioxidant. J. Appl. Polym. Sci. 2015, 132, 41591. [Google Scholar] [CrossRef]
- Sakai, S.; Kawakami, K. Synthesis and characterization of both ionically and enzymatically cross-linkable alginate. Acta Biomater. 2007, 3, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Hiemstra, C.; Zhong, Z.Y.; Feijen, J. Enzyme-mediated fast in situ formation of hydrogels from dextran-tyramine conjugates. Biomaterials 2007, 28, 2791–2800. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K.; Minamihata, K.; Wakabayashi, R.; Goto, M.; Kamiya, N. Enzymatic preparation of a redox-responsive hydrogel for encapsulating and releasing living cells. Chem. Commun. 2014, 50, 5895–5898. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K.; Wakabayashi, R.; Goto, M.; Kamiya, N. Enzyme-mediated preparation of hydrogels composed of poly(ethylene glycol) and gelatin as cell culture platforms. RSC Adv. 2015, 5, 3070–3073. [Google Scholar] [CrossRef]
- Wu, C.Z.; Strehmel, C.; Achazi, K.; Chiapisi, L.; Dernedde, J.; Lensen, M.C.; Gradzielski, M.; Ansorge-Schumacher, M.B.; Haag, R. Enzymatically cross-linked hyperbranched polyglycerol hydrogels as scaffolds for living cells. Biomacromolecules 2014, 15, 3881–3890. [Google Scholar] [CrossRef] [PubMed]
- Uyama, H.; Kurioka, H.; Kobayashi, S. Novel bienzymatic catalysis system for oxidative polymerization of phenols. Polym. J. 1997, 29, 190–192. [Google Scholar] [CrossRef]
- Taboada-Puig, R.; Junghanns, C.; Demarche, P.; Moreira, M.T.; Feijoo, G.; Lema, J.M.; Agathos, S.N. Combined cross-linked enzyme aggregates from versatile peroxidase and glucose oxidase: Production, partial characterization and application for the elimination of endocrine disruptors. Bioresour. Technol. 2011, 102, 6593–6599. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Komatani, K.; Taya, M. Glucose-triggered co-enzymatic hydrogelation of aqueous polymer solutions. RSC Adv. 2012, 2, 1502–1507. [Google Scholar] [CrossRef]
- Sakai, S.; Tsumura, M.; Inoue, M.; Koga, Y.; Fukano, K.; Taya, M. Polyvinyl alcohol-based hydrogel dressing gellable on-wound via a co-enzymatic reaction triggered by glucose in the wound exudate. J. Mater. Chem. B 2013, 1, 5067–5075. [Google Scholar] [CrossRef]
- Zhou, H.F.; Chang, Y.; Wu, X.L.; Yang, D.J.; Qiu, X.Q. Horseradish peroxidase modification of sulfomethylated wheat straw alkali lignin to improve its dispersion performance. ACS Sustain. Chem. Eng. 2015, 3, 518–523. [Google Scholar] [CrossRef]
- Liu, W.; Kumar, J.; Tripathy, S.; Senecal, K.J.; Samuelson, L. Enzymatically synthesized conducting polyaniline. J. Am. Chem. Soc. 1999, 121, 71–78. [Google Scholar] [CrossRef]
- Duan, L.P.; Zhao, Y.; Guo, F.H.; Liu, W.C.; Hou, C.P.; Ni, Z.H. Enzymatic-catalyzed polymerization of water-soluble electrically conductive polymer PEDOT:PSS. Polym. Adv. Technol. 2014, 25, 896–899. [Google Scholar] [CrossRef]
- Mita, N.; Tawaki, S.; Uyama, H.; Kobayashi, S. Laccase-catalyzed oxidative polymerization of phenols. Macromol. Biosci. 2003, 3, 253–257. [Google Scholar] [CrossRef]
- Navarra, C.; Goodwin, C.; Burton, S.; Danieli, B.; Riva, S. Laccase-mediated oxidation of phenolic derivatives. J. Mol. Catal. B Enzym. 2010, 65, 52–57. [Google Scholar] [CrossRef]
- Hou, J.W.; Dong, G.X.; Ye, Y.; Chen, V. Enzymatic degradation of bisphenol-A with immobilized laccase on TiO2 sol-gel coated PVDF membrane. J. Membr. Sci. 2014, 469, 19–30. [Google Scholar] [CrossRef]
- Hautphenne, C.; Debaste, F. Harnessing laccases for the synthesis of bisphenol A biopolymers. Chem. Eng. Technol. 2015, 38, 1223–1228. [Google Scholar] [CrossRef]
- Junker, K.; Luginbuhl, S.; Schuttel, M.; Bertschi, L.; Kissner, R.; Schuler, L.D.; Rakvin, B.; Walde, P. Efficient polymerization of the aniline dimer p-aminodiphenylamine (PADPA) with Trametes versicolor laccase/O2 as catalyst and oxidant and AOT vesicles as templates. ACS Catal. 2014, 4, 3421–3434. [Google Scholar] [CrossRef]
- Khersonsky, O.; Tawfik, D.S. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [PubMed]
- Thennarasu, S.; Krishnamurti, N.; Shantha, K.L. Developments and applications of cyanoacrylate adhesives. J. Adhes. Sci. Technol. 1989, 3, 237–260. [Google Scholar]
- Vote, B.J.; Elder, M.J. Cyanoacrylate glue for corneal perforations: A description of a surgical technique and a review of the literature. Clin. Exp. Ophthalmol. 2000, 28, 437–442. [Google Scholar] [CrossRef]
- Radosevich, M.; Goubran, H.A.; Burnouf, T. Fibrin sealant: Scientific rationale, production methods, properties, and current clinical use. Vox Sang. 1997, 72, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Leggat, P.A.; Kedjarune, U.; Smith, D.R. Toxicity of cyanoacrylate adhesives and their occupational impacts for dental staff. Ind. Health 2004, 42, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Spotnitz, W.D.; Burks, S. Hemostats, sealants, and adhesives: Components of the surgical toolbox. Transfusion 2008, 48, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Silver, F.H.; Wang, M.C.; Pins, G.D. Preparation and use of fibrin glue in surgery. Biomaterials 1995, 16, 891–903. [Google Scholar] [CrossRef]
- Eriksen, J.R.; Bech, J.I.; Linnemann, D.; Rosenberg, J. Laparoscopic intraperitoneal mesh fixation with fibrin sealant (Tisseel®) vs. titanium tacks: A randomised controlled experimental study in pigs. Hernia 2008, 12, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Tang, Z.; Su, T.; Li, W.J.; Wang, Q.G. Hydrogel-coated enzyme electrodes formed by GOx-mediated polymerization for glucose detecting. RSC Adv. 2015, 5, 47244–47247. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zavada, S.R.; Battsengel, T.; Scott, T.F. Radical-Mediated Enzymatic Polymerizations. Int. J. Mol. Sci. 2016, 17, 195. https://doi.org/10.3390/ijms17020195
Zavada SR, Battsengel T, Scott TF. Radical-Mediated Enzymatic Polymerizations. International Journal of Molecular Sciences. 2016; 17(2):195. https://doi.org/10.3390/ijms17020195
Chicago/Turabian StyleZavada, Scott R., Tsatsral Battsengel, and Timothy F. Scott. 2016. "Radical-Mediated Enzymatic Polymerizations" International Journal of Molecular Sciences 17, no. 2: 195. https://doi.org/10.3390/ijms17020195