
Integrative Analysis of Metabolomic, Proteomic and Genomic Data to Reveal Functional Pathways and Candidate Genes for Drip Loss in Pigs

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Biological Pathways Involved in the Metabolite and Protein Abundance
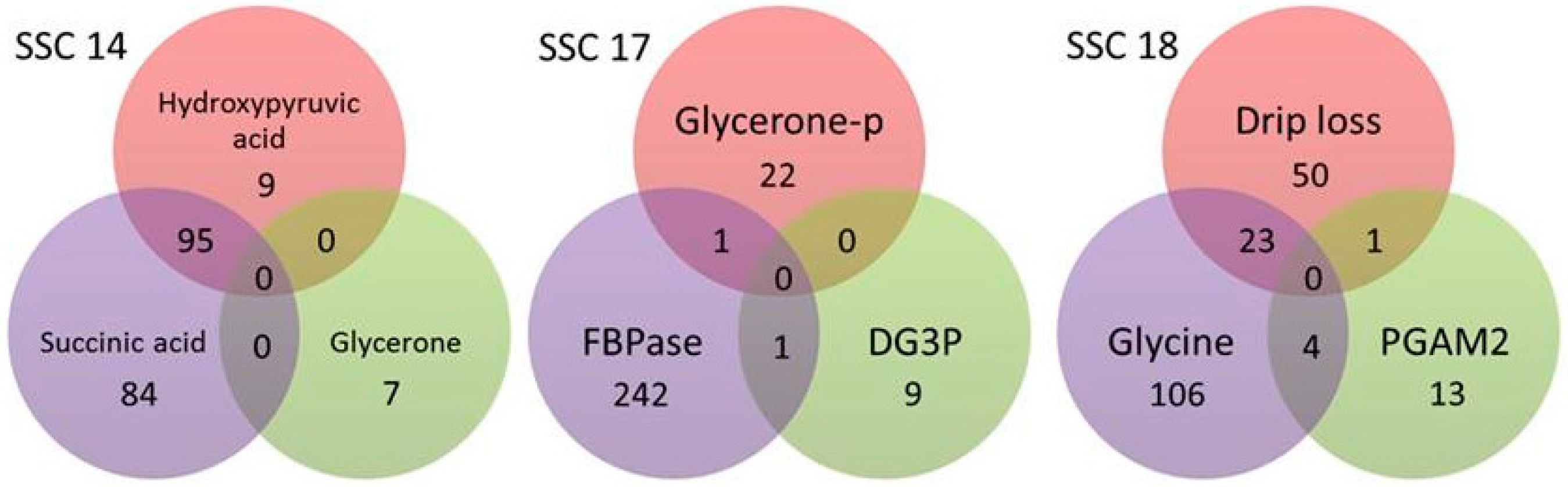
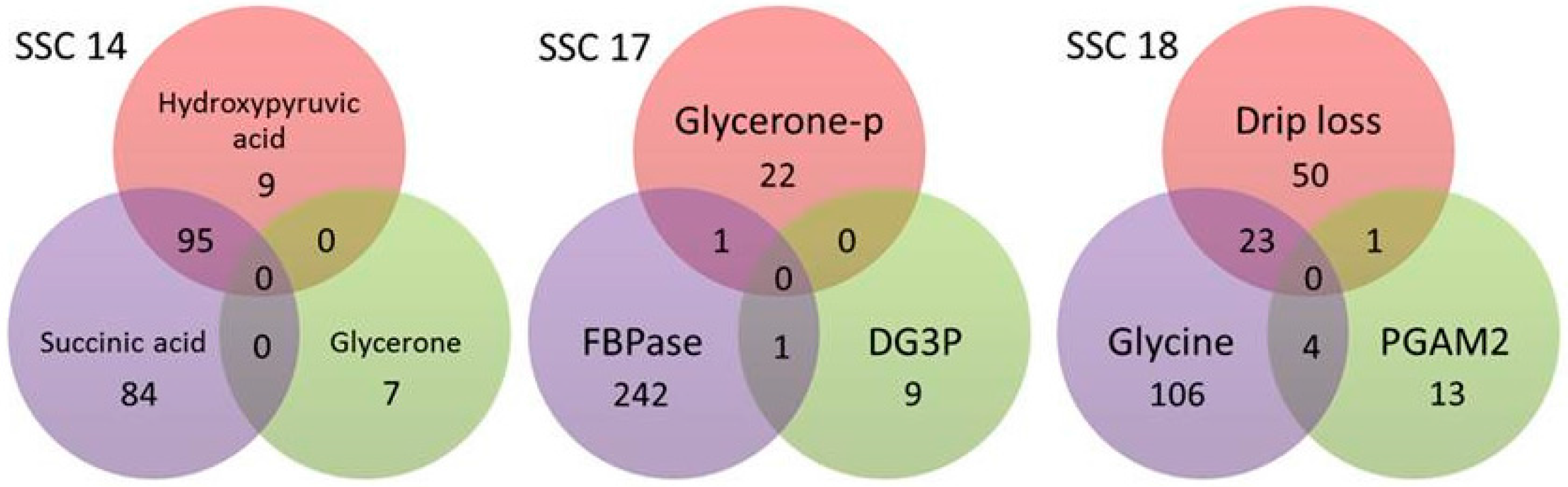
2.2. Whole-Genome Association Analysis for Drip Loss and Metabolites and Proteins of Selected Biological Pathways
- Chromosome-wide significant (at least q ≤ 0.1);
- within the “Top 10” or “Top 25” of significant SNPs for metabolic traits or drip loss;
- exonic or intronic.
3. Discussion
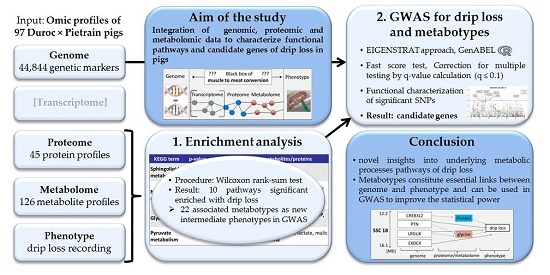
3.1. Systems Biological Approach or Integrated Analysis of Genome, Proteome and Metabolome to Elucidate the “Muscle to Meat” Black Box
3.2. Impact of Metabolic Pathways and Involved Metabolites and Proteins for Drip Loss
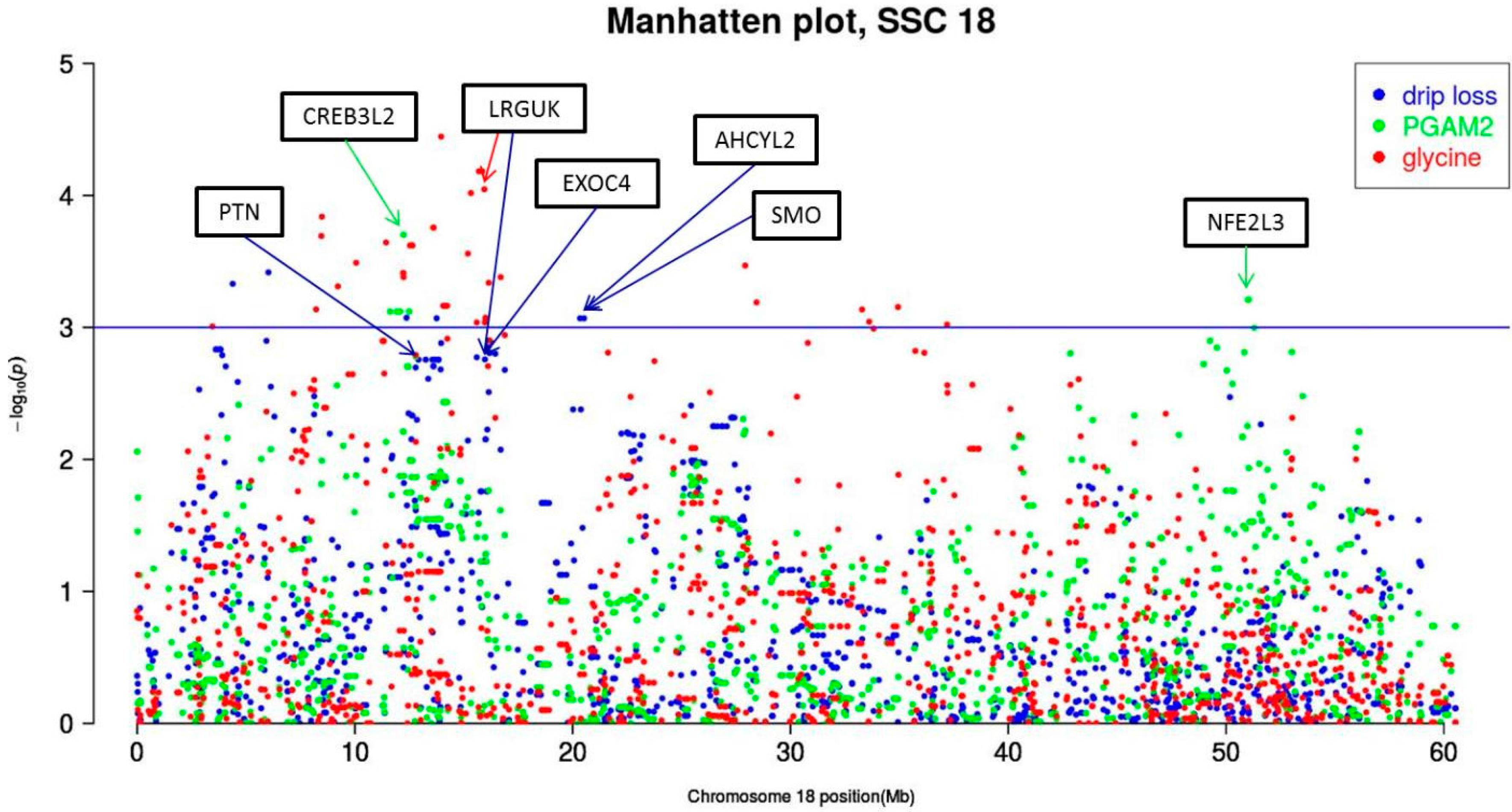
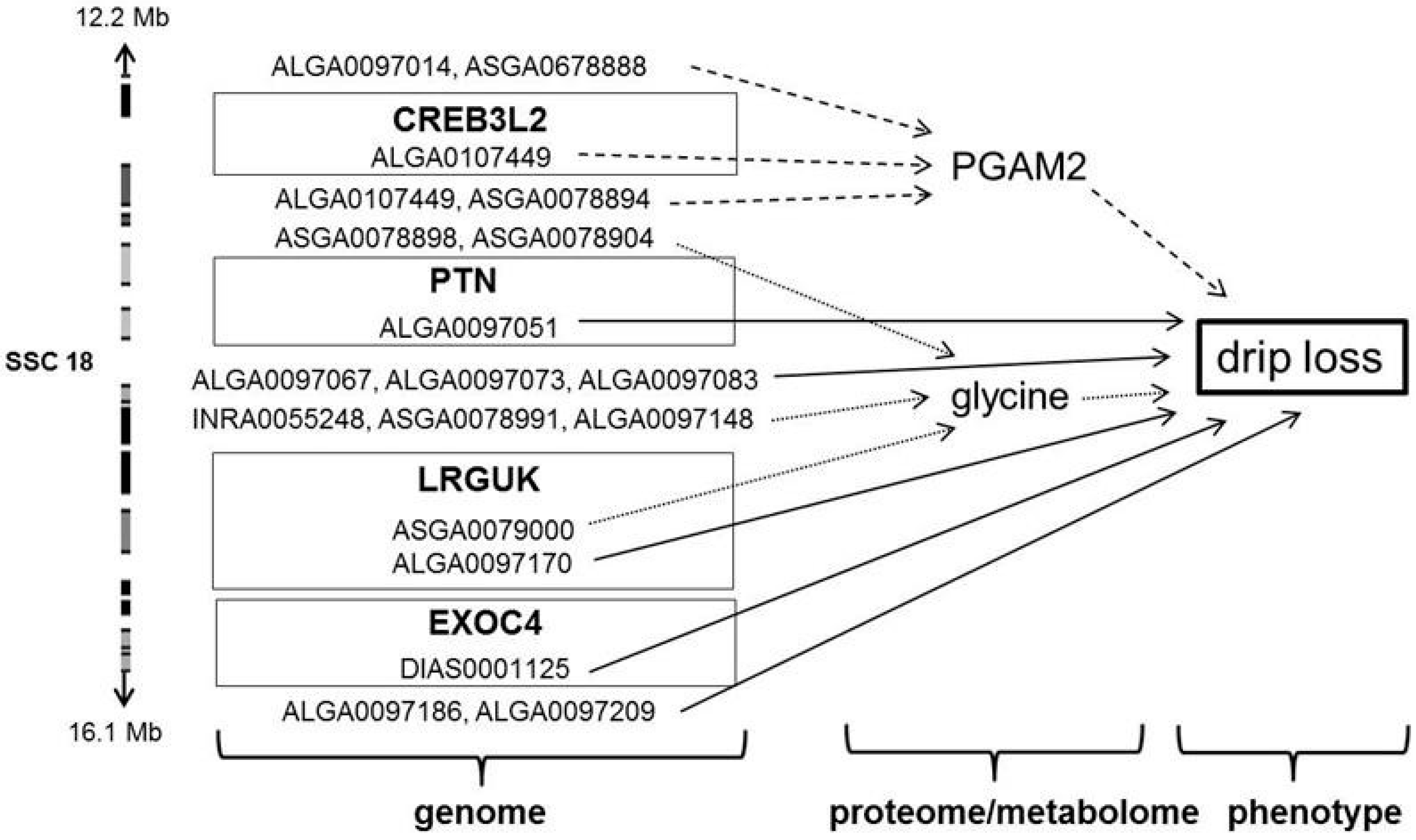
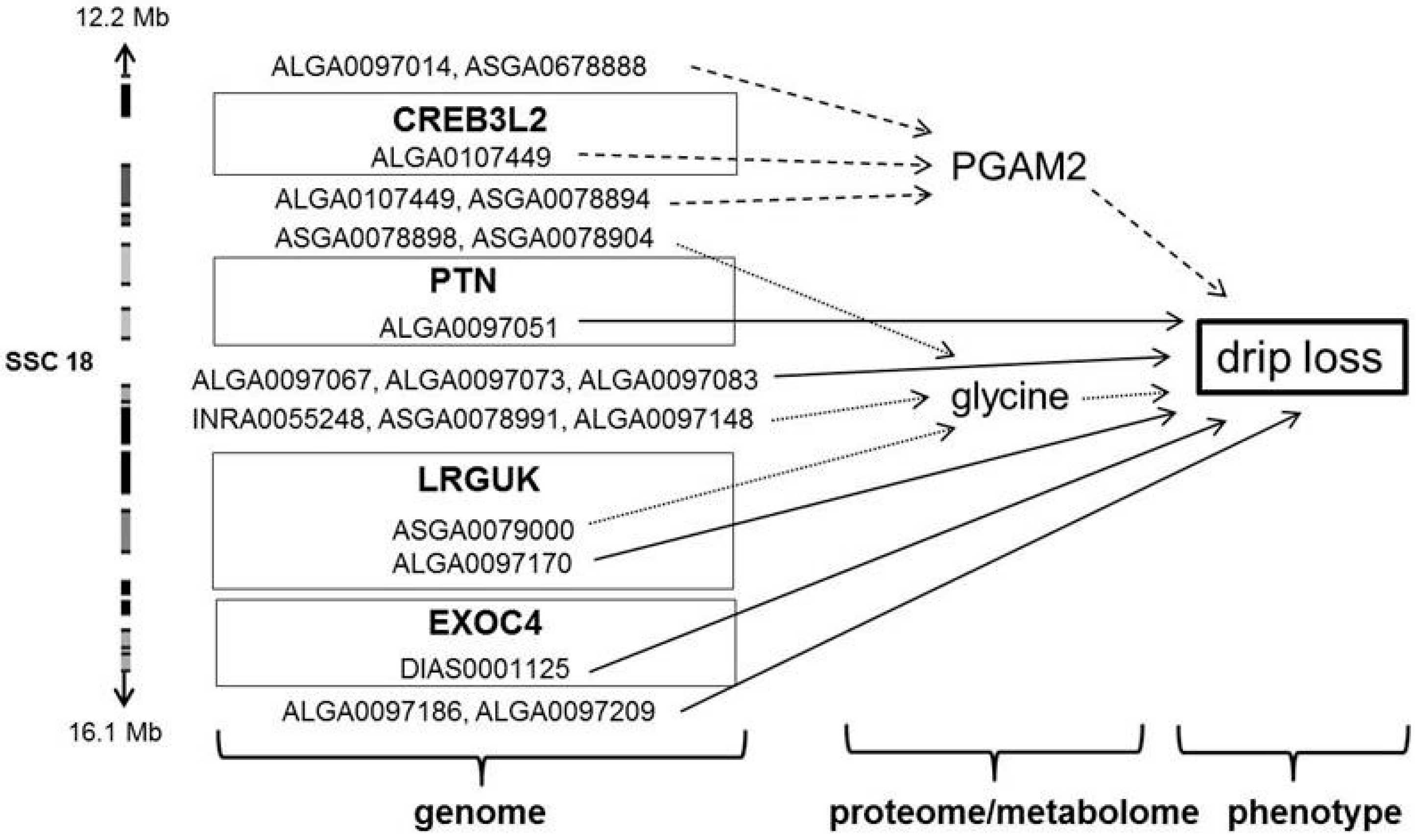
3.3. Significant Markers and Candidate Genes for Drip Loss and Associated Metabolic Traits
3.4. Challenges and Perspectives
4. Materials and Methods
4.1. Animals, Tissue Collection, Phenotyping
4.2. Untargeted Metabolite Profiling
4.3. Targeted Protein Profiling
4.4. Genome Profiling
4.5. Statistical Analysis
4.5.1. Quality Control and Annotation of Genetic Data
4.5.2. Metabolite and Protein Enrichment and Pathway Analysis
4.5.3. Genome-Wide Association (GWA) Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bertram, H.C.; Oksbjerg, N.; Young, J.F. NMR-based metabonomics reveals relationship between pre-slaughter exercise stress, the plasma metabolite profile at time of slaughter, and water-holding capacity in pigs. Meat Sci. 2010, 84, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Offer, G. Modelling of the formation of pale, soft and exudative meat: Effects of chilling regime and rate and extent of glycolysis. Meat Sci. 1991, 30, 157–184. [Google Scholar] [CrossRef]
- Schafer, A.; Rosenvold, K.; Purslow, P.P.; Andersen, H.J.; Henckel, P. Physiological and structural events postmortem of importance for drip loss in pork. Meat Sci. 2002, 61, 355–366. [Google Scholar] [CrossRef]
- Borchers, N.; Otto, G.; Kalm, E. Genetic relationship of drip loss to further meat quality traits in purebred Piétrains. Arch. Tierz. 2007, 50, 84–91. [Google Scholar]
- Ciobanu, D.C.; Lonergan, S.M.; Huff-Lonergan, E.J. Genetics of Meat Quality and Carcass Traits. In The Genetics of the Pig; Rothschild, M.F., Ruvinsky, A., Eds.; CAB International: Oxfordshire, UK, 2011; p. 358. [Google Scholar]
- Huff-Lonergan, E.; Lonergan, S.M. Mechanisms of water-holding capacity of meat: The role of postmortem biochemical and structural changes. Meat Sci. 2005, 71, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Huff-Lonergan, E.; Lonergan, S.M. New frontiers in understanding drip loss in pork: Recent insights on the role of postmortem muscle biochemistry. J. Anim. Breed. Genet. 2007, 124, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Rosenvold, K.; Andersen, H.J. Factors of significance for pork quality—A review. Meat Sci. 2003, 64, 219–237. [Google Scholar] [CrossRef]
- Terenina, E.; Babigumira, B.M.; Le Mignon, G.; Bazovkina, D.; Rousseau, S.; Salin, F.; Bendixen, C.; Mormede, P. Association study of molecular polymorphisms in candidate genes related to stress responses with production and meat quality traits in pigs. Domest. Anim. Endocrinol. 2013, 44, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Ponsuksili, S.; Murani, E.; Phatsara, C.; Jonas, E.; Walz, C.; Schwerin, M.; Schellander, K.; Wimmers, K. Expression profiling of muscle reveals transcripts differentially expressed in muscle that affect water-holding capacity of pork. J. Agric. Food Chem. 2008, 56, 10311–10317. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Jennen, D.G.J.; Tholen, E.; Juengst, H.; Kleinwachter, T.; Holker, M.; Tesfaye, D.; Un, G.; Schreinemachers, H.J.; Murani, E.; et al. A genome scan reveals QTL for growth, fatness, leanness and meat quality in a Duroc-Pietrain resource population. Anim. Genet. 2007, 38, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Kim, J.J.; Jonas, E.; Wimmers, K.; Ponsuksili, S.; Murani, E.; Phatsara, C.; Tholen, E.; Juengst, H.; Tesfaye, D.; et al. Combined line-cross and half-sib QTL analysis in Duroc-Pietrain population. Mamm. Genome 2008, 19, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Otto, G.; Roehe, R.; Looft, H.; Thoelking, L.; Knap, P.W.; Rothschild, M.F.; Plastow, G.S.; Kalm, E. Associations of DNA markers with meat quality traits in pigs with emphasis on drip loss. Meat Sci. 2007, 75, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Wu, Y.; Zhou, X.; Xia, J.; Zhang, W.; Song, Y.; Liu, F.; Chen, Y.; Zhang, L.; Gao, X.; et al. Pathway-based genome-wide association studies for two meat production traits in simmental cattle. Sci. Rep. 2015, 5, 18389. [Google Scholar] [CrossRef] [PubMed]
- Bernal Rubio, Y.L.; Gualdrón Duarte, J.L.; Bates, R.O.; Ernst, C.W.; Nonneman, D.; Rohrer, G.A.; King, A.; Shackelford, S.D.; Wheeler, T.L.; Cantet, R.J.C.; et al. Meta-analysis of genome-wide association from genomic prediction models. Anim. Genet. 2016, 47, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Picard, B.; Lebret, B.; Cassar-Malek, I.; Liaubet, L.; Berri, C.; Le Bihan-Duval, E.; Hocquette, J.F.; Renand, G. Recent advances in omic technologies for meat quality management. Meat Sci. 2015, 109, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, S.; Gauba, R.; Gusev, Y.; Clarkeand, R. Integrative analysis workflow for untargeted metabolomics in translational research. Metabolomics 2014, 4, 130. [Google Scholar] [CrossRef]
- Hollung, K.; Veiseth, E.; Jia, X.H.; Faergestad, E.M.; Hildrum, K.I. Application of proteomics to understand the molecular mechanisms behind meat quality. Meat Sci. 2007, 77, 97–104. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Zolla, L. Meat science: From proteomics to integrated omics towards system biology. J. Proteom. 2013, 78, 558–577. [Google Scholar] [CrossRef] [PubMed]
- Te Pas, M.; Kruijt, L.; Smits, M. Use of Biomarkers As tools for Tracking and Tracing Meat and Meat Products and to Predict and Monitor Meat Quality. First Annual Conference on Body ABND Carcass Evaluation, Meat Quality, Software and Tracebility. Available online: http://library.wur.nl/WebQuery/wurpubs/428956 (accessed on 26 September 2012).
- Rohart, F.; Paris, A.; Laurent, B.; Canlet, C.; Molina, J.; Mercat, M.J.; Tribout, T.; Muller, N.; Iannuccelli, N.; Villa-Vialaneix, N.; et al. Phenotypic prediction based on metabolomic data for growing pigs from three main European breeds. J. Anim. Sci. 2012, 90, 4729–4740. [Google Scholar] [CrossRef] [PubMed]
- Muroya, S.; Oe, M.; Nakajima, I.; Ojima, K.; Chikuni, K. CE-TOF MS-based metabolomic profiling revealed characteristic metabolic pathways in postmortem porcine fast and slow type muscles. Meat Sci. 2014, 98, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Welzenbach, J.; Neuhoff, C.; Looft, C.; Schellander, K.; Tholen, E.; Große-Brinkhaus, C. Different statistical approaches to investigate porcine muscle metabolome profiles to highlight new biomarkers for pork quality assessment. PLoS ONE 2016, 11, e0149758. [Google Scholar] [CrossRef] [PubMed]
- Heidt, H.; Cinar, M.U.; Uddin, M.J.; Looft, C.; Juengst, H.; Tesfaye, D.; Becker, A.; Zimmer, A.; Ponsuksili, S.; Wimmers, K.; et al. A genetical genomics approach reveals new candidates and confirms known candidate genes for drip loss in a porcine resource population. Mamm. Genome 2013, 24, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.M.; Stapleton, P.C.; Corcoran, D.; Hamill, R.M.; White, A. Understanding meat quality through the application of genomic and proteomic approaches. Meat Sci. 2006, 74, 3–16. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Marrocco, C.; Zolla, V.; D’Andrea, M.; Zolla, L. Meat quality of the longissimus lumborum muscle of Casertana and Large White pigs: Metabolomics and proteomics intertwined. J. Proteom. 2011, 75, 610–627. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.S.; Damon, M.; Guitton, N.; Guisle, I.; Ecolan, P.; Vincent, A.; Cherel, P.; Gondret, F. Differentially-expressed genes in pig longissimus muscles with contrasting levels of fat, as identified by combined transcriptomic, reverse transcription PCR, and proteomic analyses. J. Agric. Food Chem. 2009, 57, 3808–3817. [Google Scholar] [CrossRef] [PubMed]
- Neuhoff, C.; Grosse-Brinkhaus, C.; Heidt, H.; Tesfaye, D.; Tholen, E.; Looft, C.; Schellander, K.; Cinar, M. Proteome analysis of skeletal muscle in high and low drip loss Duroc × Pietrain F2 pigs. In Proceedings of the 33rd Conference of the International Society of Animal Genetics (ISAG), Cairns, Australia, 15–20 July 2012; p. 2044.
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Yu, M.; Zhao, S. Understanding quantitative genetics in the systems biology era. Int. J. Biol. Sci. 2009, 5, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Widmann, P.; Reverter, A.; Fortes Marina, R.S.; Weikard, R.; Suhre, K.; Hammon, H.; Albrecht, E.; Kuehn, C. A systems biology approach using metabolomic data reveals genes and pathways interacting to modulate divergent growth in cattle. BMC Genom. 2013, 14, 798. [Google Scholar] [CrossRef]
- Ala-Korpela, M.; Kangas, A.J.; Inouye, M. Genome-wide association studies and systems biology: Together at last. Trends Genet. 2011, 27, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Kadarmideen, H.N. Genomics to systems biology in animal and veterinary sciences: Progress, lessons and opportunities. Livest. Sci. 2014, 166, 232–248. [Google Scholar] [CrossRef]
- Chagoyen, M.; Pazos, F. Tools for the functional interpretation of metabolomic experiments. Brief. Bioinform. 2013, 14, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.; Simpson, K.M.; Escher, R.; Buchet-Poyau, K.; Beissbarth, T.; Carmichael, C.; Ritchie, M.E.; Schütz, F.; Cannon, P.; Liu, M.; et al. Integrative analysis of RUNX1 downstream pathways and target genes. BMC Genom. 2008, 9, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Greenberg, S.A.; Kong, S.W.; Altschuler, J.; Kohane, I.S.; Park, P.J. Discovering statistically significant pathways in expression profiling studies. Proc. Natl. Acad. Sci. USA 2005, 102, 13544–13549. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Strimmer, K. A general modular framework for gene set enrichment analysis. BMC Bioinform. 2009, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Binke, R.; Schwagele, F. The quantification of animal and plant species in meat products—Development of an efficient method by means of PCR. FleischWirtschaft 2003, 83, 135–139. [Google Scholar]
- Scheffler, T.L.; Gerrard, D.E. Mechanisms controlling pork quality development: The biochemistry controlling postmortem energy metabolism. Meat Sci. 2007, 77, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Honikel, K.O.; Kim, C.J. Causes of the development of PSE pork. Fleischwirtschaft 1986, 66, 349–353. [Google Scholar]
- Binke, R. From muscle to meat. Fleischwirtschaft 2004, 84, 224–227. [Google Scholar]
- Lambert, I.H.; Nielsen, J.H.; Andersen, H.J.; Ortenblad, N. Cellular model for induction of drip loss in meat. J. Agric. Food Chem. 2001, 49, 4876–4883. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, K.A.; Young, J.F.; Theil, P.; Kolko, M.; Oksbjerg, N.; Lambert, I.H. Role of phospholipase A2 in the induction of drip loss in porcine muscle. J. Agric. Food Chem. 2007, 55, 1970–1976. [Google Scholar] [CrossRef]
- Lim, D.G.; Jo, C.; Seo, K.S.; Nam, K.C. Comparison of meat quality of loins and butts in different two-way crossbred pigs. Livest. Sci. 2014, 161, 210–217. [Google Scholar] [CrossRef]
- Paredi, G.; Sentandreu, M.A.; Mozzarelli, A.; Fadda, S.; Hollung, K.; de Almeida, A.M. Muscle and meat: New horizons and applications for proteomics on a farm to fork perspective. J. Proteom. 2013, 88, 58–82. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Larsen, M.R.; Karlsson, A.H.; Pomponio, L.; Costa, L.N.; Lametsch, R. Gel-based phosphoproteomics analysis of sarcoplasmic proteins in postmortem porcine muscle with pH decline rate and time differences. Proteomics 2011, 11, 4063–4076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davoli, R.; Bigi, D.; Fontanesi, L.; Zambonelli, P.; Yerle, M.; Zijlstra, C.; Bosma, A.A.; Robic, A.; Russo, V. Mapping of 14 expressed sequence tags (ESTs) from porcine skeletal muscle by somatic cell hybrid analysis. Anim. Genet. 2000, 31, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Laville, E.; Sayd, T.; Terlouw, C.; Chambon, C.; Damon, M.; Larzul, C.; Leroy, P.; Glénisson, J.; Chérel, P. Comparison of sarcoplasmic proteomes between two groups of pig muscles selected for shear force of cooked meat. J. Agric. Food Chem. 2007, 55, 5834–5841. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.; Lee, D.-G.; Kwon, J.; Choi, C.-W.; Park, S.H.; Kwon, S.-O.; Jung, J.H.; Park, H.C.; Park, B.Y.; Jang, I.S.; et al. Comparative proteome analysis of porcine Longissimus dorsi on the basis of pH24 of post-mortem muscle. J. Agric. Sci. 2012. [Google Scholar] [CrossRef]
- Corcoran, M.P.; Lamon-Fava, S.; Fielding, R.A. Skeletal muscle lipid deposition and insulin resistance: Effect of dietary fatty acids and exercise. Am. J. Clin. Nutr. 2007, 85, 662–677. [Google Scholar] [PubMed]
- Dobrowsky, R.T.; Kolesnick, R.N. Analysis of sphingomyelin and ceramide levels and the enzymes regulating their metabolism in response to cell stress. Methods Cell Biol. 2001, 66, 135–165. [Google Scholar] [PubMed]
- Aulchenko, Y.S.; Ripke, S.; Isaacs, A.; Van Duijn, C.M. GenABEL: An R library for genome-wide association analysis. Bioinformatics 2007, 23, 1294–1296. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.; Wimmers, K.; Luther, H.; Hofer, A.; Leeb, T. A genome-wide association study to detect QTL for commercially important traits in Swiss Large White boars. PLoS ONE 2013, 8, e55951. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, Y.T.; do Carmo, A.S.; Carvalheiro, R.; Neves, H.H.; Matos, M.C.; Zavarez, L.B.; Pérez O’Brien, A.M.; Sölkner, J.; McEwan, J.C.; Cole, J.B.; et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013, 14, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Kang, H.M.; Eskin, E. Rapid and accurate multiple testing correction and power estimation for millions of correlated markers. PLoS Genet. 2009, 5, e1000456. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful appraoch to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar]
- Jennen, D.G.; Brings, A.D.; Liu, G.; Jüngst, H.; Tholen, E.; Jonas, E.; Tesfaye, D.; Schellander, K.; Phatsara, C. Genetic aspects concerning drip loss and water-holding capacity of porcine meat. J. Anim. Breed. Genet. 2007, 124, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.M.; Lei, T.; Zhou, L.; Chen, X.D.; Long, Q.Q.; Long, H.; Jin, D.; Gan, L.; Yang, Z.Q. Genomic organization, alternative splicing and tissues expression of porcine CREB3L4 gene. Mol. Biol. Rep. 2009, 36, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Laramie, J.M.; Wilk, J.B.; Williamson, S.L.; Nagle, M.W.; Latourelle, J.C.; Tobin, J.E.; Province, M.A.; Borecki, I.B.; Myers, R.H. Polymorphisms near EXOC4 and LRGUK on chromosome 7q32 are associated with type 2 diabetes and fasting glucose; the NHLBI family heart study. BMC Med. Genet. 2008, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Leheska, J.M.; Wulf, D.M.; Maddock, R.J. Effects of fasting and transportation on pork quality development and extent of postmortem metabolism. J. Anim. Sci. 2002, 80, 3194–3202. [Google Scholar] [CrossRef] [PubMed]
- Hamet, P.; Tremblay, J.; Seda, O.; Macmahon, S.; Chalmers, J. Genetic Component of Complications in Type 2 Diabetes. Patent U.S. 20110158979 A1, 15 June 2009. [Google Scholar]
- Casas-Agustench, P.; Arnett, D.K.; Smith, C.E.; Lai, C.-Q.; Parnell, L.D.; Borecki, I.B.; Frazier-Wood, A.C.; Allison, M.; Chen, Y.-D.I.; Taylor, K.D.; et al. Saturated fat intake modulates the association between an obesity genetic risk score and body mass index in two US populations. J. Acad. Nutr. Diet. 2014, 114, 1954–1966. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, J.; Guo, W.; Zhao, Y.; Hu, X.; Li, N. Identifying lipid metabolism genes in pig liver after clenbuterol administration. Front. Biosci. 2012, 4, 2605–2616. [Google Scholar]
- Griffiths, B.; Lewis, C.A.; Bensaad, K.; Ros, S.; Zhang, Q.; Ferber, E.C.; Konisti, S.; Peck, B.; Miess, H.; East, P.; et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Volsky, D.J. PAGE: Parametric analysis of gene set enrichment. BMC Bioinform. 2005, 6, 144. [Google Scholar]
- Hirose, K.; Ito, T.; Fukawa, K.; Arakawa, A.; Mikawa, S.; Hayashi, Y.; Tanaka, K. Evaluation of effects of multiple candidate genes (LEP, LEPR, MC4R, PIK3C3, and VRTN) on production traits in Duroc pigs. J. Anim. Sci. 2014, 85, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, Z.-Y.; Liu, Q.-X. Gene coexpression networks reveal key drivers of phenotypic divergence in porcine muscle. BMC Genom. 2015, 16, 50. [Google Scholar] [CrossRef] [PubMed]
- Fontanesi, L.; Schiavo, G.; Galimberti, G.; Calò, D.G.; Russo, V. A genomewide association study for average daily gain in Italian Large White pigs. J. Anim. Sci. 2014, 92, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Scott, A.; Wang, Y.; Wang, L.; Zhao, Y.; Doerner, S.; Satake, M.; Croniger, C.M.; Wang, Z. PTPRT regulates high-fat diet-induced obesity and insulin resistance. PLoS ONE 2014, 9, e100783. [Google Scholar] [CrossRef] [PubMed]
- Scott, A. Role of PTPRT in Obesity and Its Substrate Paxillin Tyrosine-88 in Colorectal Cancer. Ph.D. Thesis, Case Western Reserve University, Cleveland, OH, USA, 2014. [Google Scholar]
- Tizioto, P.C.; Decker, J.E.; Taylor, J.F.; Schnabel, R.D.; Mudadu, M.A.; Silva, F.L.; Mourão, G.B.; Coutinho, L.L.; Tholon, P.; Sonstegard, T.S.; et al. Genome scan for meat quality traits in Nelore beef cattle. Physiol. Genom. 2013, 45, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Krastanov, A. Metabolomics—The state of art. Biotechnol. Biotechnol. Equip. 2010, 24, 1537–1543. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Genomic selection: A paradigm shift in animal breeding. Anim. Front. 2016, 6, 6–14. [Google Scholar] [CrossRef]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Berlin & Heidelberg, Germany, 2005; pp. 397–420. [Google Scholar]
- Zentralverband der Deutschen Schweineproduktion (ZDS). Richtlinien für die Stationsprüfung auf Mastleistung, Schlachtkörperwert und Fleischbeschaffenheit beim Schwein. Available online: http://www.zds-bonn.de/ (accessed on 15 July 2016).
- Lisec, J.; Schauer, N.; Kopka, J.; Willmitzer, L.; Fernie, A.R. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 2006, 1, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Kellermann, J. ICPL—Isotope-coded protein label. In 2D Page: Sample Preparation and Fragmentation; Humana Press: New York, NY, USA, 2008; pp. 113–123. [Google Scholar]
- Paradela, A.; Marcilla, M.; Navajas, R.; Ferreira, L.; Ramos-Fernandez, A.; Fernández, M.; Mariscotti, J.F.; García-del Portillo, F.; Albar, J.P. Evaluation of isotope-coded protein labeling (ICPL) in the quantitative analysis of complex proteomes. Talanta 2010, 80, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Gallien, S.; Duriez, E.; Domon, B. Selected reaction monitoring applied to proteomics. J. Mass Spectrom. 2011, 46, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Archibald, A.L.; Bolund, L.; Churcher, C.; Fredholm, M.; Groenen Martien, A.M.; Harlizius, B.; Lee, K.-T.; Milan, D.; Rogers, J.; Rothschild, M.F.; et al. Pig genome sequence—Analysis and publication strategy. BMC Genom. 2010, 11, 438. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Stuart, A.; Ord, J.K.; Kendall, M.G. Kendall’s Advanced Theory of Statistics, 5th ed.; Edward Arnold: London, UK, 1991. [Google Scholar]
- Van der Sijde, M.R.; Ng, A.; Fu, J. Systems genetics: From GWAS to disease pathways. Biochim. Biophys. 2014, 1842, 1903–1909. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Mean ± SD 1 | Min 2 | Max 3 | Correlation to Drip Loss 4 |
|---|---|---|---|---|
| drip loss, % | 1.97 ± 1.40 | 0.40 | 5.30 | 1 |
| pH1 | 6.53 ± 0.22 | 5.89 | 6.94 | −0.31 ** |
| pH24 | 5.52 ± 1.12 | 5.32 | 6.06 | −0.35 *** |
| PKM | 26,454.10 ± 17,829.55 | 13.47 | 88,251.64 | −0.20 * |
| PGAM2 | 5600.37 ± 4985.98 | −10.77 | 32,935.16 | −0.19 |
| FBPase | 27,407.08 ± 20,231.70 | 809.35 | 114,192.30 | −0.11 |
| TPI1 | 1754.68 ± 1526.65 | 32.13 | 7802.84 | −0.21 * |
| pyruvic acid | 4.32 × 10−2 ± 3.62 × 10−2 | 6.16 × 10−3 | 2.11 × 10−1 | 0.22 * |
| lactic acid | 6.49 × 10−1 ± 3.28 × 10−1 | 1.88 × 10−1 | 1.64 | 0.08 |
| glucose | 9.02 × 10−3 ± 1.32 × 10−2 | 1.21 × 10−4 | 8.41 × 10−2 | 0.19 |
| phosphoenol pyruvate | 5.59 × 10−2 ± 8.95 × 10−2 | 1.80 × 10−3 | 0.53 | 0.13 |
| glycerone-p | 1.86 ± 1.10 | 2.48 × 10−1 | 5.85 | 0.07 |
| DG3P | 2.56 × 10−1 ± 4.09 × 10−1 | 2.61 × 10−3 | 2.61 | 0.14 |
| fumaric acid | 2.67 × 10−3 ± 1.25 × 10−3 | 5.50 × 10−4 | 7.23 × 10−3 | 0.12 |
| succinic acid | 1.38 × 10−2 ± 5.02 × 10−3 | 3.23 × 10−3 | 3.23 × 10−2 | −0.02 |
| malic acid | 6.03 × 10−3 ± 2.92 × 10−3 | 8.85 × 10−4 | 1.64 × 10−2 | 0.11 |
| methylglyoxal | 9.62 × 10−3 ± 5.44 × 10−3 | 2.61 × 10−4 | 2.89 × 10−2 | 0.22 * |
| glycine | 8.59 × 10−2 ± 2.39 × 10−2 | 4.84 × 10−2 | 1.62 × 10−1 | 0.11 |
| hydroxypyruvic acid | 1.06 × 10−2 ± 6.81 × 10−3 | 1.76 × 10−3 | 4.98 × 10−2 | 0.02 |
| F6P | 2.17 × 10−2 ± 3.43 × 10−2 | 2.91 × 10−4 | 2.25 × 10−1 | 0.12 |
| serine | 6.04 × 10−3 ± 2.99 × 10−3 | 1.76 × 10−3 | 2.15 × 10−2 | −0.01 |
| glycerone | 1.41 × 10−1 ± 8.36 × 10−2 | 2.17 × 10−2 | 4.37 × 10−1 | 0.20 |
| ceramide | 1.68 × 10−4 ± 1.24 × 10−3 | 2.33 × 10−6 | 6.59 × 10−4 | 0.05 |
| glucosylceramide | 2.46 × 10−3 ± 4.72 × 10−3 | 1.69 × 10−4 | 2.72 × 10−2 | 0.21 * |
| phosphoethanolamine | 8.57 × 10−4 ± 5.01 × 10−4 | 2.28 × 10−4 | 3.52 × 10−3 | 0.12 |
| Pathway | KEGG-ID | p-Value * | Involved Metabolites and Proteins |
|---|---|---|---|
| Sphingolipid metabolism | 00600 | 0.014 | ceramide, glucosylceramide, phosphoethanolamine, serine |
| Type II diabetes mellitus | 04930 | 0.018 | pyruvic acid, glucose, PKM |
| Methane metabolism | 00680 | 0.020 | glycine, pyruvic acid, hydroxypyruvic acid, F6P, malic acid, serine, phosphoenol pyruvate, glycerone-p, glycerone, DG3P |
| Renal cell carcinoma | 05211 | 0.027 | fumaric acid, malic acid |
| Insulin secretion | 04911 | 0.043 | pyruvic acid, glucose |
| Meiosis yeast | 04113 | 0.045 | glucose |
| NAFLD | 04932 | 0.045 | glucose |
| Glycolysis/Gluconeogenesis | 00010 | 0.045 | pyruvic acid, lactic acid, glucose, phosphoenol pyruvate, glycerone-p, DG3P, FBPase, TPI1, PKM, PGAM2 |
| Pyruvate metabolism | 00620 | 0.053 | fumaric acid, pyruvic acid, succinic acid, lactic acid, malic acid, phosphoenol pyruvate, methylglyoxal, PKM |
| Steptomycin biosynthesis | 00521 | 0.056 | glucose, myo-inositol |
| Trait | ID | PC 1 | λ 2 | Number of Significant SNP/QTL per Porcine Chromosome 3 | ∑SNP 4 | Min p-Value 5 | Min q-Value 6 | Max 7 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 6 | 7 | 8 | 10 | 13 | 14 | 16 | 17 | 18 | ||||||||
| drip loss | - | 10 | 1.007 | 4/4 | 74/20 | 78 # | 6.58 | 6.26 | 8.8 | |||||||||||
| PKM | 100158154 | 10 | 1 | 33/13 | 33 # | 10.7 | 7.84 | 14.3 | ||||||||||||
| PGAM2 | 100188980 | 10 | 1.06 | 18/7 | 18 # | 19.9 | 8.67 | 13.9 | ||||||||||||
| FBPase | 100134828 | 10 | 1 | 5/1 | 244/92 | 118 *, 131 # | 1.98 | 2.27 | 16.6 | |||||||||||
| glucose | C00031 | 10 | 1 | 2/2 | 2 # | 5.86 | 8.80 | 15.3 | ||||||||||||
| glycerone-p | C00111 | 10 | 1.046 | 4/1 | 7/4 | 23/10 | 34 # | 2.35 | 5.07 | 17.3 | ||||||||||
| DG3P | C00197 | 10 | 1 | 10/5 | 2 *, 8 # | 1.50 | 2.19 | 17.3 | ||||||||||||
| succinic acid | C00042 | 2 | 1.03 | 179/64 | 122 *, 57 # | 29.3 | 5.07 | 13.3 | ||||||||||||
| glycine | C00037 | 10 | 1.05 | 97/41 | 2/2 | 133/48 | 102 *, 130 # | 3.39 | 4.67 | 17.1 | ||||||||||
| hydroxyl-pyruvic acid | C00168 | 10 | 1 | 104/28 | 76 *, 28 # | 3.44 | 1.88 | 16.1 | ||||||||||||
| F6P | C00085 | 10 | 1 | 12/9 | 12 # | 8.00 | 7.69 | 14.8 | ||||||||||||
| glycerone | C00184 | 10 | 1 | 7/4 | 7 # | 7.95 | 8.56 | 14.8 | ||||||||||||
| ceramide | C00195 | 4 | 1.006 | 20/8 | 20 # | 11.8 | 8.02 | 14.4 | ||||||||||||
| glucosyl-ceramide | C01190 | 10 | 1.012 | 3/3 | 1/1 | 4 # | 1.59 | 6.64 | 17.4 | |||||||||||
| phosphor-ethanolamine | C00346 | 10 | 1.08 | 15/8 | 11 *, 4 # | 15.4 | 3.81 | 14.5 | ||||||||||||
| ∑SNP/QTL excluding double counting | 7/4 | 97/41 | 1/1 | 33/13 | 15/8 | 13/10 | 27/12 | 2/2 | 5/1 | 195/80 | 4/4 | 275/100 | 197/54 | |||||||
| ∑overlapping SNP/QTL 8 | 1/1 | 95/16 | 2/7 | 28/21 | ||||||||||||||||
| SSC 1 | 1 | 2 | 3 | 4 | 6 | 7 | 8 | 10 | 13 | 14 | 16 | 17 | 18 | ∑ |
| Genes 2 | 30 | 148 | 4 | 65 | 31 | 48 | 70 | 15 | 12 | 375 | 13 | 367 | 252 | 1430 |
| SNP 3 | 2/7 | 30/97 | -/1 | 15/33 | 2/15 | -/13 | 2/27 | 1/2 | 5/5 | 83/195 | -/4 | 54/275 | 63/197 | 257/871 |
| SSC 1 | Trait | Gene 2 | SNP 3 | Position 4 | Mut 5 | MAF 6 | eEff (se) 7 | Chi2 | Emp. p-Value 8 | q-Value 9 | Var 10 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | glycerone-p | ENPP3 | INRA0001633 | 35387799 | G/A | 0.47 | −4.00 × 10−2 | (1.00 × 10−2) | 18.68 | 0.22 | 5.07 | 17.35 |
| glucosylceramide | SAMD4A | ALGA0007238 | 204522804 | C/A | 0.47 | −9.32 × 10−5 | (2.15 × 10−5) | 18.80 | 0.16 | 6.64 | 17.44 | |
| 4 | PKM | NTNG1 | INRA0016801 | 123080603 | G/A | 0.27 | −9.21 × 102 | (2.57 × 102) | 12.88 | 3.32 | 7.84 | 13.21 |
| GBP4 | ASGA0023322 | 139599066 | G/A | 0.38 | −6.43 × 102 | (1.77 × 102) | 13.26 | 2.71 | 7.84 | 12.72 | ||
| PKN2 | M1GA0006779 | 139861416 | C/A | 0.43 | 6.76 × 102 | (1.89 × 102) | 12.88 | 3.32 | 7.84 | 12.40 | ||
| ZNHIT6 | ALGA0029718 | 142789911 | A/G | 0.46 | 8.52 × 102 | (2.20 × 102) | 15.01 | 1.07 | 7.84 | 14.29 | ||
| ALGA0029732 | 142739989 | G/A | 0.39 | 9.23 × 102 | (2.49 × 102) | 13.70 | 2.14 | 7.84 | 13.09 | |||
| ALGA0029741 | 142730172 | G/A | 0.46 | 8.13 × 102 | (2.15 × 102) | 14.20 | 1.64 | 7.84 | 13.50 | |||
| DDAH1 | ASGA0023626 | 143204232 | A/G | 0.40 | 9.05 × 102 | (2.43 × 102) | 13.86 | 1.97 | 7.84 | 13.21 | ||
| WDR63 | INRA0018033 | 143449789 | A/G | 0.40 | 9.05 × 102 | (2.43 × 102) | 13.86 | 1.97 | 7.84 | 10.77 | ||
| 6 | phosphor-ethanolamine | PIK3C3 | DRGA0006746 | 118055075 | G/A | 0.26 | 2.91 × 10−5 | (7.54 × 10−6) | 14.93 | 1.76 | 3.81 | 14.36 |
| TTLL5 | INRA0022204 | 120225026 | C/A | 0.26 | 2.91 × 10−5 | (7.54 × 10−6) | 14.93 | 1.76 | 3.81 | 14.36 | ||
| 10 | glycine | AKT3 | MARC0098464 | 18065301 | C/A | 0.34 | −1.55 × 10−3 | (3.80 × 10−4) | 16.56 | 0.69 | 5.11 | 15.69 |
| 13 | FBPase | HLCS | MARC0019610 | 210504370 | G/A | 0.49 | 6.54 × 102 | (1.70 × 102) | 14.71 | 1.25 | 8.64 | 13.92 |
| MARC0005075 | 210516458 | A/C | 0.49 | 6.54 × 102 | (1.70 × 102) | 14.71 | 1.25 | 8.64 | 13.92 | |||
| ASGA0089689 | 210516937 | G/A | 0.49 | 6.54 × 102 | (1.70 × 102) | 14.71 | 1.25 | 8.64 | 13.92 | |||
| ASGA0089950 | 210531047 | A/G | 0.49 | 6.54 × 102 | (1.70 × 102) | 14.71 | 1.25 | 8.64 | 13.92 | |||
| ASGA0097399 | 210534054 | G/C | 0.49 | 6.54 × 102 | (1.70 × 102) | 14.71 | 1.25 | 8.64 | 13.92 | |||
| 14 | succinic acid | ANK3 | MARC0033238 | 68550413 | G/A | 0.52 | 1.69 × 10−4 | (4.59 × 10−5) | 13.60 | 2.93 | 2.82 | 13.26 |
| ASGA0064107 | 68604989 | A/G | 0.52 | 1.69 × 10−4 | (4.59 × 10−5) | 13.60 | 2.93 | 2.82 | 13.26 | |||
| RASGEF1A | ALGA0078235 | 66284845 | G/A | 0.52 | 1.69 × 10−4 | (4.59 × 10−4) | 13.60 | 2.93 | 2.82 | 13.26 | ||
| ALGA0078240 | 66320818 | A/C | 0.52 | 1.69 × 10−4 | (4.59 × 10−5) | 13.60 | 2.93 | 2.82 | 13.26 | |||
| ALGA0078243 | 66332408 | G/A | 0.52 | 1.69 × 10−4 | (4.59 × 10−5) | 13.60 | 2.93 | 2.82 | 13.26 | |||
| 17 | DG3P | PTPRT | MARC0016232 | 50694545 | A/G | 0.41 | −1.96 × 10−2 | (5.27 × 10−3) | 13.88 | 1.94 | 6.53 | 13.49 |
| VAPB | H3GA0049968 | 65818274 | A/G | 0.48 | 1.71 × 10−2 | (4.79 × 10−3) | 12.78 | 3.51 | 6.53 | 12.55 | ||
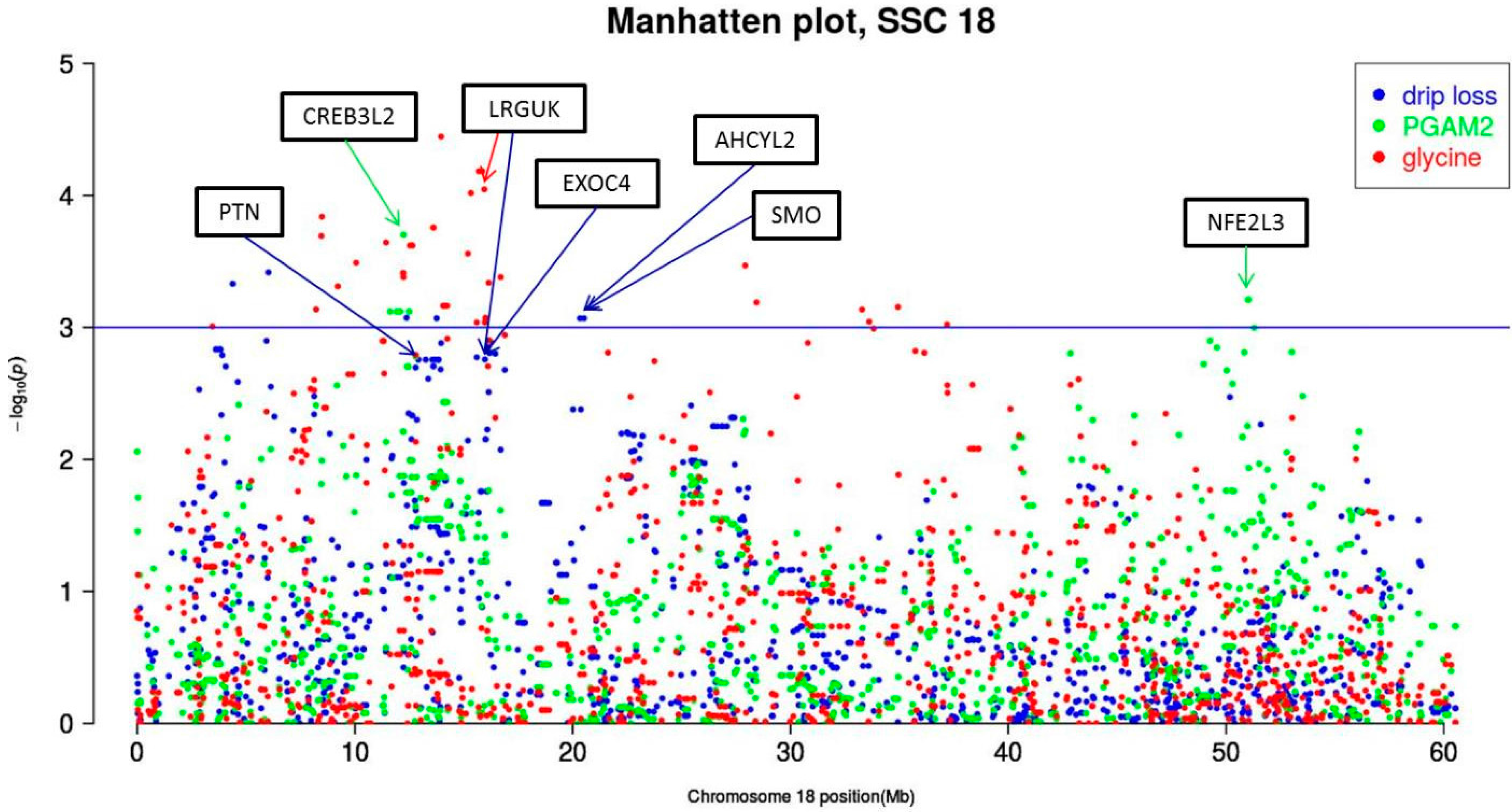
| 18 | PGAM2 | CREB3L2 | ALGA0107449 | 12234417 | G/A | 0.41 | 1.88 × 102 | (4.90 × 101) | 14.79 | 1.99 | 8.67 | 13.98 |
| drip loss | PTN | ALGA0097051 | 12921061 | A/G | 0.25 | −7.81 × 10−2 | (2.49 × 10−2) | 9.87 | 17.6 | 6.26 | 5.61 | |
| glycine | LRGUK | ASGA0079000 | 15942579 | A/G | 0.31 | −1.63 × 10−3 | (4.07 × 10−4) | 16.06 | 0.90 | 1.66 | 15.28 | |
| drip loss | ALGA0097170 | 15969549 | G/A | 0.45 | −4.34 × 10−2 | (1.38 × 10−2) | 9.87 | 17.5 | 6.26 | 5.61 | ||
| EXOC4 | DIAS0001125 | 16179365 | G/A | 0.48 | 4.15 × 10−2 | (1.31 × 10−2) | 10.08 | 15.6 | 6.26 | 5.72 | ||
| AHCYL2 | H3GA0050495 | 20338092 | A/G | 0.28 | −7.16 × 10−2 | (2.14 × 10−2) | 11.21 | 8.54 | 6.26 | 6.32 | ||
| SMO | ASGA0079098 | 20520014 | G/A | 0.30 | −7.16 × 10−2 | (2.14 × 10−2) | 11.21 | 8.54 | 6.26 | 6.32 | ||
| PGAM2 | NFE2L3 | ASGA0100894 | 51012467 | C/A | 0.42 | 1.89 × 102 | (5.35 × 101) | 12.53 | 6.18 | 8.67 | 12.10 | |
| SNP | SSC 1 | Position 2 | Mut 3 | MAF 4 | eEff (se) 5 | Chi2 | Emp. p-Value 6 | q-Value 7 | Var 8 | Located within a Gene 9 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ALGA0089069 | 16 | 11629284 | C/A | 0.08 | 2.26 × 10−1 | (5.65 × 10−2) | 16.05 | 6.58 × 10−5 | 7.02 × 10−2 | 8.82 | × |
| 2 | CASI0008411 | 16 | 23115634 | G/A | 0.10 | 1.89 × 10−1 | (4.86 × 10−2) | 15.03 | 1.12 × 10−4 | 7.02 × 10−2 | 8.30 | × |
| 3 | MARC0097282 | 16 | 10946289 | G/A | 0.33 | 7.45 × 10−2 | (1.95 × 10−2) | 14.65 | 1.38 × 10−4 | 7.02 × 10−2 | 8.15 | × |
| 4 | ASGA0072217 | 16 | 9183890 | A/G | 0.34 | 7.25 × 10−2 | (1.93 × 10−2) | 14.16 | 1.78 × 10−4 | 7.02 × 10−2 | 7.90 | × |
| 5 | ALGA0111681 | 18 | 6026724 | G/A | 0.15 | 1.35 × 10−1 | (3.78 × 10−2) | 12.71 | 3.83 × 10−4 | 6.26 × 10−2 | 7.11 | × |
| 6 | ASGA0104044 | 18 | 4388048 | A/C | 0.15 | 1.27 × 10−1 | (3.61 × 10−2) | 12.34 | 4.68 × 10−4 | 6.26 × 10−2 | 6.92 | × |
| 7 | MARC0003904 | 18 | 12368984 | G/A | 0.35 | −6.37 × 10−2 | (1.90 × 10−2) | 11.23 | 8.45 × 10−4 | 6.26 × 10−2 | 6.33 | × |
| 8 | ASGA0078921 | 18 | 13751595 | G/A | 0.29 | −7.46 × 10−2 | (2.23 × 10−2) | 11.21 | 8.53 × 10−4 | 6.26 × 10−2 | 6.33 | × |
| 9 | H3GA0050495 | 18 | 20338092 | G/A | 0.30 | −7.16 × 10−2 | (2.14 × 10−2) | 11.21 | 8.54 × 10−4 | 6.26 × 10−2 | 6.33 | AHCYL2 |
| 10 | ASGA0079098 | 18 | 20520014 | A/G | 0.30 | −7.16 × 10−2 | (2.14 × 10−2) | 11.21 | 8.54 × 10−4 | 6.26 × 10−2 | 6.33 | SMO |
| 11 | ALGA0105391 | 18 | 5935981 | G/A | 0.31 | 6.69 × 10−2 | (2.07 × 10−2) | 10.48 | 1.26 × 10−3 | 6.26 × 10−2 | 5.94 | × |
| 12 | INRA0055248 | 18 | 13959002 | G/A | 0.47 | −4.24 × 10−2 | (1.31 × 10−2) | 10.40 | 1.32 × 10−3 | 6.26 × 10−2 | 5.90 | × |
| 13 | MARC0036783 | 18 | 16113241 | A/G | 0.47 | −4.23 × 10−2 | (1.32 × 10−2) | 10.27 | 1.41 × 10−3 | 6.26 × 10−2 | 5.82 | × |
| 14 | ASGA0098607 | 18 | 3614625 | A/G | 0.38 | −5.35 × 10−2 | (1.68 × 10−2) | 10.20 | 1.47 × 10−3 | 6.26 × 10−2 | 5.79 | × |
| 15 | ALGA0104874 | 18 | 3620895 | A/G | 0.38 | −5.35 × 10−2 | (1.68 × 10−2) | 10.20 | 1.47 × 10−3 | 6.26 × 10−2 | 5.79 | × |
| 16 | ASGA0088995 | 18 | 3741888 | G/G | 0.38 | −5.35 × 10−2 | (1.68 × 10−2) | 10.20 | 1.47 × 10−3 | 6.26 × 10−2 | 5.79 | × |
| 17 | H3GA0050278 | 18 | 3808173 | A/G | 0.38 | −5.35 × 10−2 | (1.68 × 10−2) | 10.20 | 1.47 × 10−3 | 6.26 × 10−2 | 5.79 | × |
| 18 | ASGA0078689 | 18 | 3833808 | G/A | 0.38 | −5.35 × 10−2 | (1.68 × 10−2) | 10.20 | 1.47 × 10−3 | 6.26 × 10−2 | 5.79 | × |
| 19 | DIAS0001125 | 18 | 16179365 | G/A | 0.48 | 4.15 × 10−2 | (1.31 × 10−2) | 10.08 | 1.56 × 10−3 | 6.26 × 10−2 | 5.72 | EXOC4 |
| 20 | ALGA0097186 | 18 | 16444813 | G/A | 0.47 | 4.20 × 10−2 | (1.32 × 10−2) | 10.06 | 1.58 × 10−3 | 6.26 × 10−2 | 5.71 | × |
| 21 | ALGA0096804 | 18 | 3907848 | G/A | 032 | 5.58 × 10−2 | (1.77 × 10−2) | 10.01 | 1.63 × 10−3 | 6.26 × 10−2 | 5.69 | × |
| 22 | ALGA0116114 | 18 | 15594213 | A/G | 0.46 | −4.22 × 10−2 | (1.34 × 10−2) | 9.94 | 1.69 × 10−3 | 6.26 × 10−2 | 5.65 | × |
| 23 | ALGA0097170 | 18 | 15969549 | G/A | 0.45 | −4.34 × 10−2 | (1.38 × 10−2) | 9.88 | 1.75 × 10−3 | 6.26 × 10−2 | 5.62 | LRGUK |
| 24 | ALGA0097051 | 18 | 12921061 | A/G | 0.25 | −7.81 × 10−2 | (2.49 × 10−2) | 9.87 | 1.76 × 10−3 | 6.26 × 10−2 | 5.61 | PTN |
| 25 | ALGA0097067 | 18 | 13674866 | A/G | 0.25 | −7.81 × 10−2 | (2.49 × 10−2) | 9.87 | 1.76 × 10−3 | 6.26 × 10−2 | 5.61 | × |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welzenbach, J.; Neuhoff, C.; Heidt, H.; Cinar, M.U.; Looft, C.; Schellander, K.; Tholen, E.; Große-Brinkhaus, C. Integrative Analysis of Metabolomic, Proteomic and Genomic Data to Reveal Functional Pathways and Candidate Genes for Drip Loss in Pigs. Int. J. Mol. Sci. 2016, 17, 1426. https://doi.org/10.3390/ijms17091426
Welzenbach J, Neuhoff C, Heidt H, Cinar MU, Looft C, Schellander K, Tholen E, Große-Brinkhaus C. Integrative Analysis of Metabolomic, Proteomic and Genomic Data to Reveal Functional Pathways and Candidate Genes for Drip Loss in Pigs. International Journal of Molecular Sciences. 2016; 17(9):1426. https://doi.org/10.3390/ijms17091426
Chicago/Turabian StyleWelzenbach, Julia, Christiane Neuhoff, Hanna Heidt, Mehmet Ulas Cinar, Christian Looft, Karl Schellander, Ernst Tholen, and Christine Große-Brinkhaus. 2016. "Integrative Analysis of Metabolomic, Proteomic and Genomic Data to Reveal Functional Pathways and Candidate Genes for Drip Loss in Pigs" International Journal of Molecular Sciences 17, no. 9: 1426. https://doi.org/10.3390/ijms17091426
APA StyleWelzenbach, J., Neuhoff, C., Heidt, H., Cinar, M. U., Looft, C., Schellander, K., Tholen, E., & Große-Brinkhaus, C. (2016). Integrative Analysis of Metabolomic, Proteomic and Genomic Data to Reveal Functional Pathways and Candidate Genes for Drip Loss in Pigs. International Journal of Molecular Sciences, 17(9), 1426. https://doi.org/10.3390/ijms17091426