High Resolution Melting (HRM) for High-Throughput Genotyping—Limitations and Caveats in Practical Case Studies


Abstract
:1. Introduction
2. Results
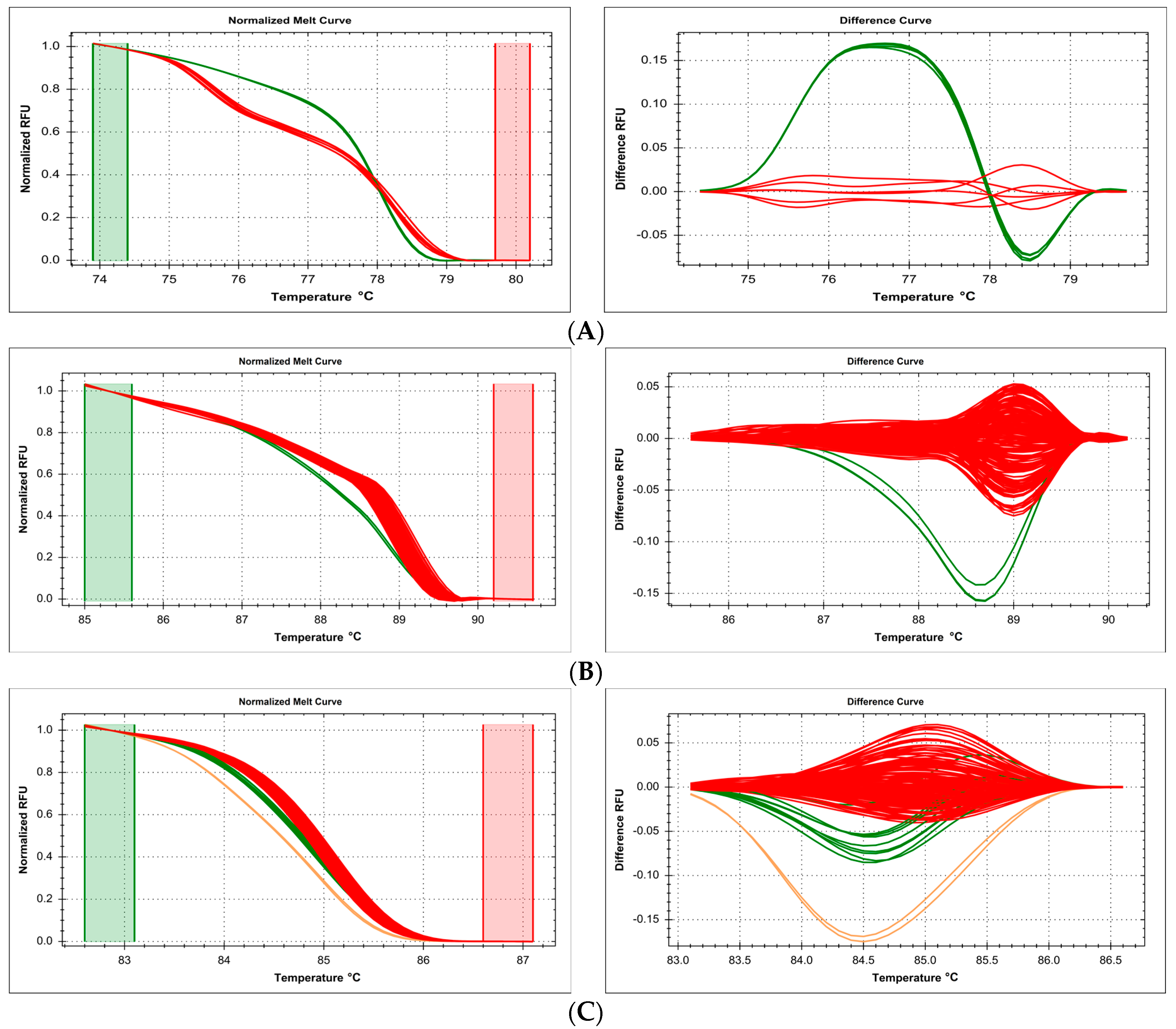
2.1. DNA Source Issues—Problems with Formalin-Fixed, Paraffin-Embedded (FFPE)-Derived Template DNA
2.2. DNA Preparation Issues—Different Isolation Methods and Sample Composition Make Parallel Analysis Difficult
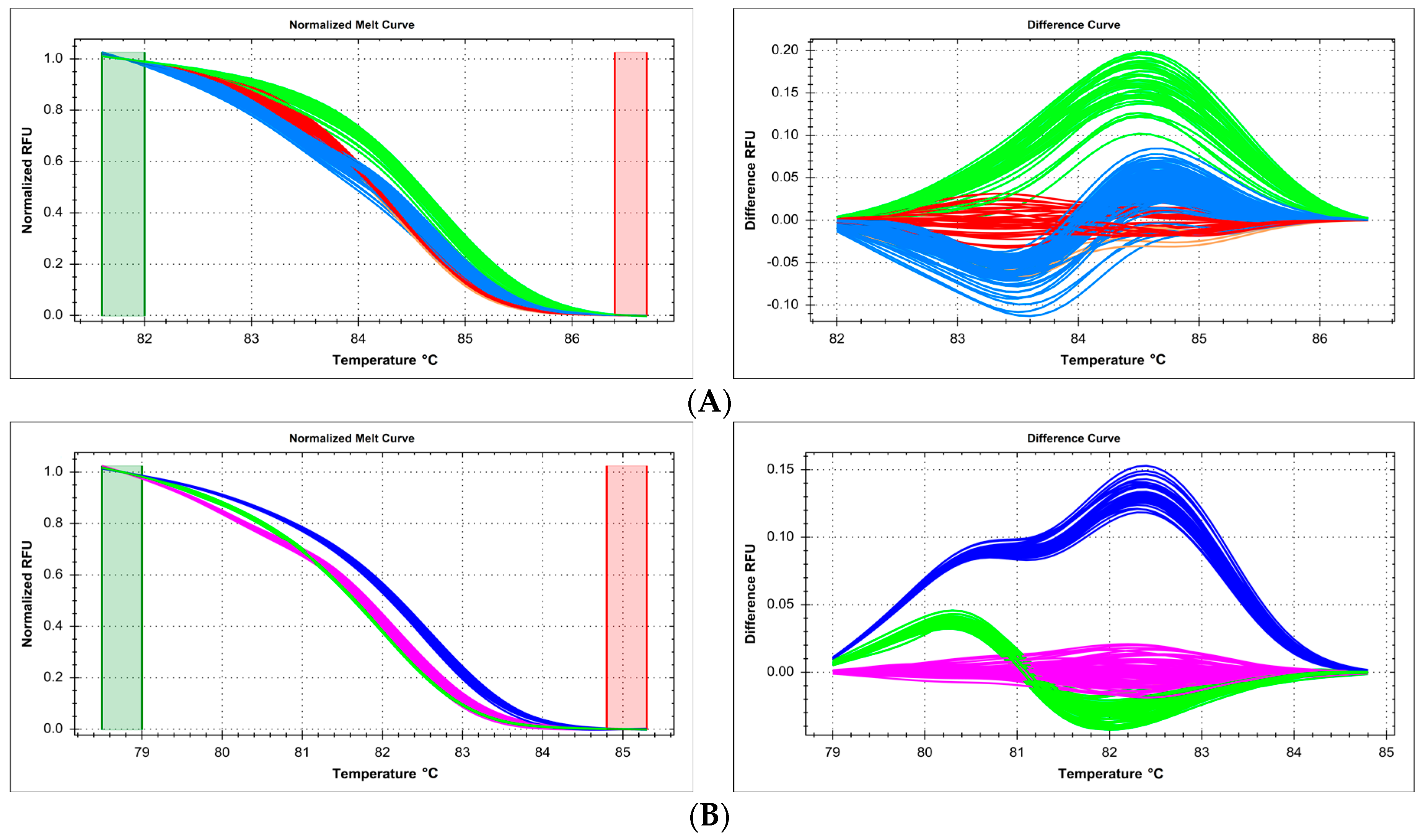
2.3. Primer Design Issues—Shorter Amplicon Length Is Better
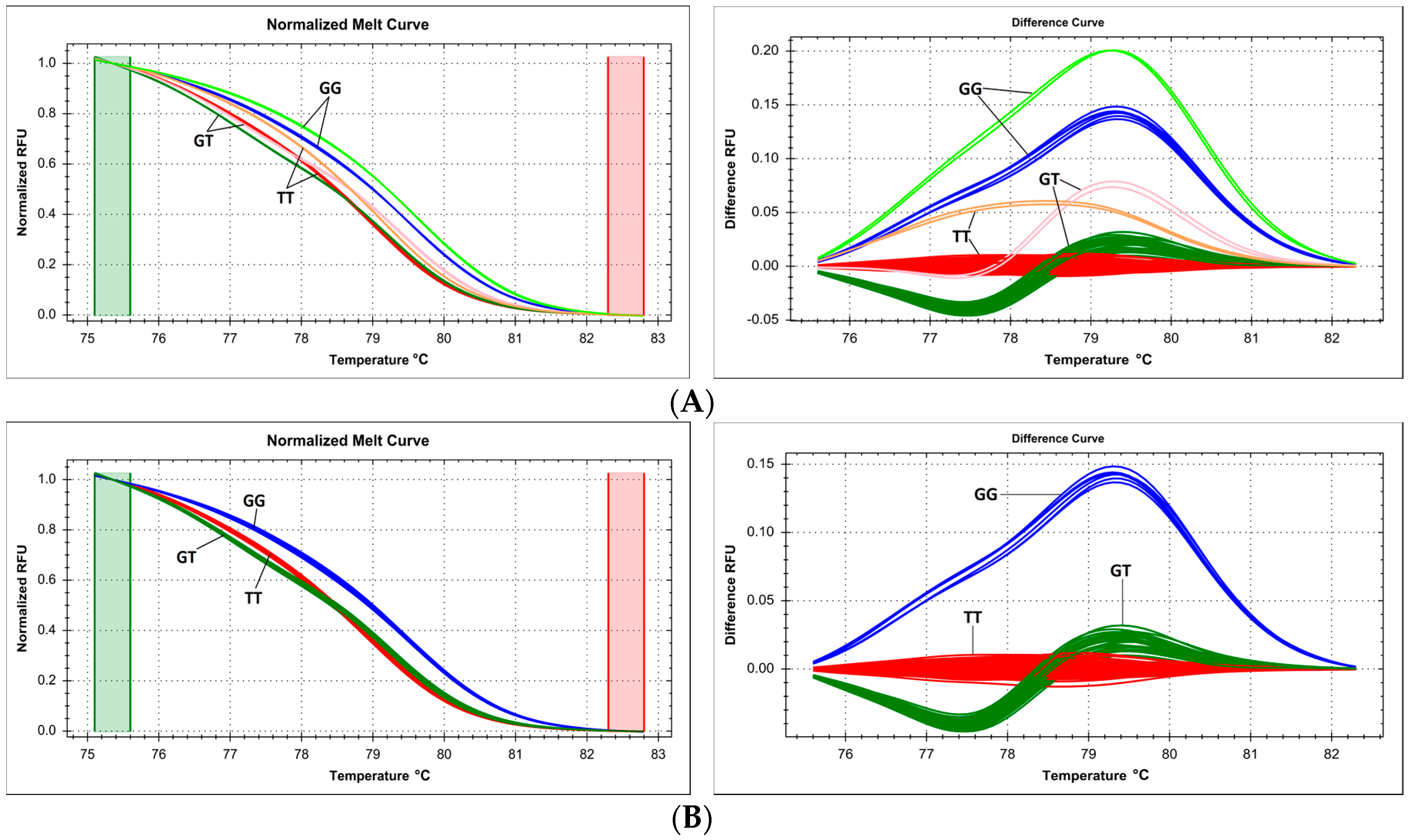
2.4. Primer Design Issues—Unexpected Variation within Primer Sequence
2.5. Technical Issues with Experimental Mixture Preparation—Master Mix Reproducibility and Systematic Errors of Automated Pipetting
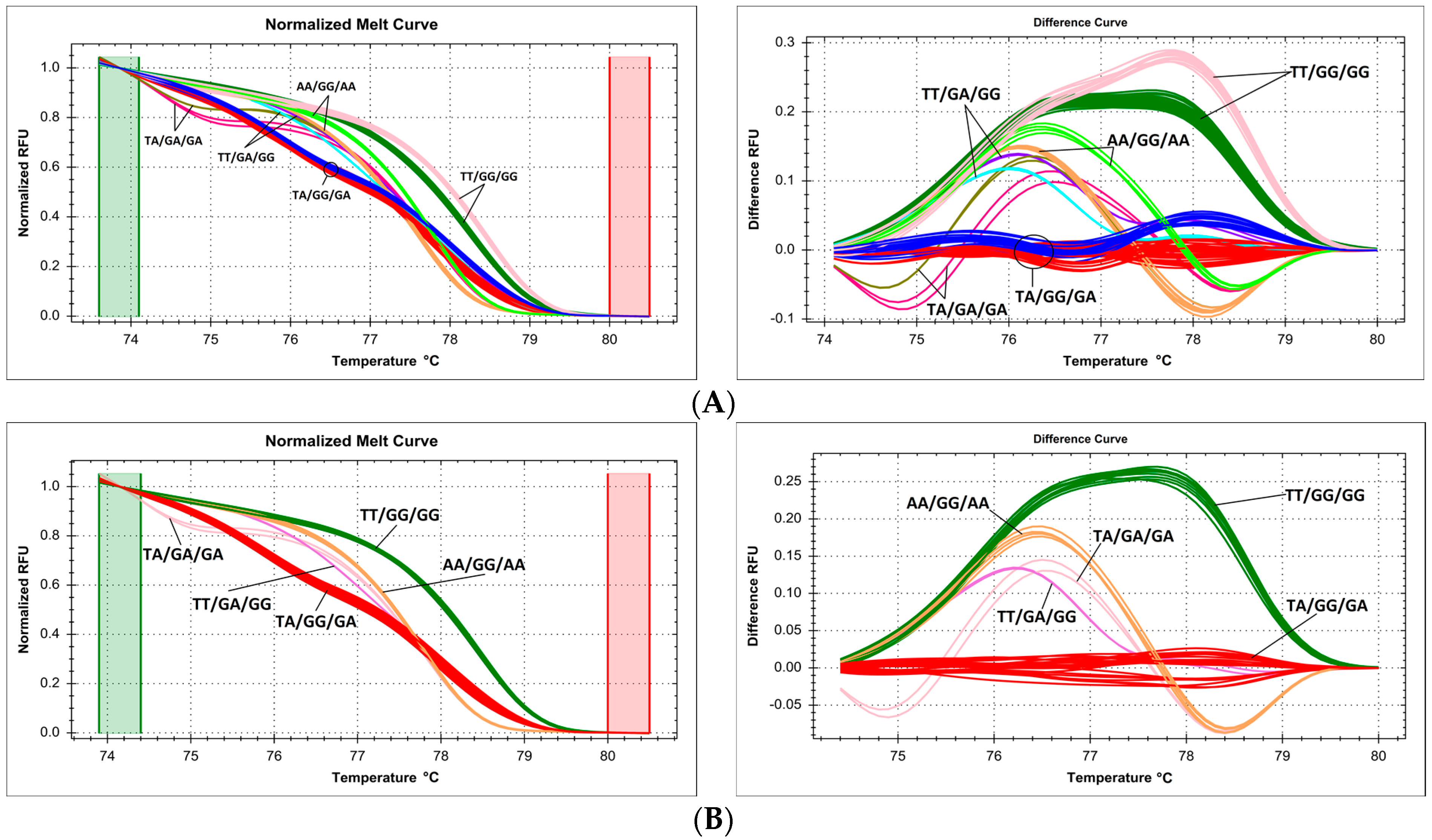
2.6. Issues with Multiple Proximate Polymorphic Sites—Complex Melting Curves Lead to Underestimation of the Prevalence of Variants within a Single Amplicon
3. Discussion
4. Materials and Methods
4.1. Materials and Genomic DNA Samples
4.2. Detection of Polymorphisms Using High Resolution Melting (HRM) Scanning and Genotyping
4.3. HRM Conditions and Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Er, T.K.; Chang, J.G. High-resolution melting: Applications in genetic disorders. Clin. Chim. Acta 2012, 414, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.; Giffard, P.M. Microbiological applications of high-resolution melting analysis. J. Clin. Microbiol. 2012, 50, 3418–3421. [Google Scholar] [CrossRef] [PubMed]
- Caban, M.; Strapagiel, D.; Bartosz, G.; Staczek, P.; Ciesielska, A.; Gadzalski, M.; Grzelak, A. Method for identification of pathogenic fungi species contained in a sample taken from a patient. Patent Number: PL408734-A1, 2014-07-02. Biuletyn Urzędu Patentowego 2016. [Google Scholar] [CrossRef]
- Ninghui, G.; Bing, W.; Wei, R.; Mengmeng, L.; Meiling, C.; Dongya, M.; Liqiong, Y.; Wencheng, X. Application of PCR and high-resolution melting for rapid identification of yeasts routinely isolated in a clinical microbiology laboratory. Ann. Clin. Lab. Sci. 2015, 45, 680–685. [Google Scholar] [PubMed]
- Odell, I.D.; Cloud, J.L.; Seipp, M.; Wittwer, C.T. Rapid species identification within the Mycobacterium chelonae-abscessus group by high-resolution melting analysis of hsp65 PCR products. Am. J. Clin. Pathol. 2005, 123, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Issa, R.; Abdul, H.; Hashim, S.H.; Seradja, V.H.; Shaili, N.; Hassan, N.A. High resolution melting analysis for the differentiation of Mycobacterium species. J. Med. Microbiol. 2014, 63, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Huang, C.L.; Lin, C.C.; Chen, C.C.; Chang, Y.C.; Chang, S.S.; Tseng, C.P. Rapid detection and identification of clinically important bacteria by high-resolution melting analysis after broad-range ribosomal RNA real-time PCR. Clin. Chem. 2006, 52, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.P.; Chua, K.H.; Thong, K.L. One-step species-specific high resolution melting analysis for nosocomial bacteria detection. J. Microbiol. Methods 2014, 107, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Iacumin, L.; Ginaldi, F.; Manzano, M.; Anastasi, V.; Reale, A.; Zotta, T.; Rossi, F.; Coppola, R.; Comi, G. High resolution melting analysis (HRM) as a new tool for the identification of species belonging to the Lactobacillus casei group and comparison with species-specific PCRs and multiplex PCR. Food Microbiol. 2015, 46, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Simko, I. High-resolution dna melting analysis in plant research. Trends Plant Sci. 2016, 21, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Druml, B.; Cichna-Markl, M. High resolution melting (HRM) analysis of DNA—Its role and potential in food analysis. Food Chem. 2014, 158, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Erali, M.; Voelkerding, K.V.; Wittwer, C.T. High resolution melting applications for clinical laboratory medicine. Exp. Mol. Pathol. 2008, 85, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.L.; Sanford, L.N.; Wittwer, C.T. High-resolution DNA melting analysis in clinical research and diagnostics. Expert Rev. Mol. Diagn. 2010, 10, 219–240. [Google Scholar] [CrossRef] [PubMed]
- Pompanon, F.; Bonin, A.; Bellemain, E.; Taberlet, P. Genotyping errors: Causes, consequences and solutions. Nat. Rev. Genet. 2005, 6, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Rosset, I.; Strapagiel, D.; Sitek, A.; Majewska, M.; Ostrowska-Nawarycz, L.; Żądzińska, E. Association of FTO and TMEM18 polymorphisms with overweight and obesity in the population of Polish children. Anthropol. Rev. 2016, 79, 17–33. [Google Scholar] [CrossRef]
- Sitek, A.; Rosset, I.; Strapagiel, D.; Majewska, M.; Ostrowska-Nawarycz, L.; Żądzińska, E. Association of FTO gene with obesity in Polish schoolchildren. Anthropol. Rev. 2014, 77, 33–44. [Google Scholar] [CrossRef]
- Ratajewski, M.; Slomka, M.; Karas, K.; Sobalska-Kwapis, M.; Korycka-Machala, M.; Salkowska, A.; Dziadek, J.; Strapagiel, D.; Dastych, J. Functional Analysis of the rs774872314, rs116171003, rs200231898 and rs201107751 Polymorphisms in the Human RORγT Gene Promoter Region. Genes 2017, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, C.T.; Reed, G.H.; Gundry, C.N.; Vandersteen, J.G.; Pryor, R.J. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin. Chem. 2003, 49, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.G.; Durtschi, J.D.; Bromley, L.K.; Wittwer, C.T.; Voelkerding, K.V. Amplicon DNA melting analysis for mutation scanning and genotyping: Cross-platform comparison of instruments and dyes. Clin. Chem. 2006, 52, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.G.; Durtschi, J.D.; Wittwer, C.T.; Voelkerding, K.V. Expanded instrument comparison of amplicon DNA melting analysis for mutation scanning and genotyping. Clin. Chem. 2007, 53, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- De Leeneer, K.; Coene, I.; Poppe, B.; de Paepe, A.; Claes, K. Rapid and sensitive detection of BRCA1/2 mutations in a diagnostic setting: Comparison of two high-resolution melting platforms. Clin. Chem. 2008, 54, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Tindall, E.A.; Petersen, D.C.; Woodbridge, P.; Schipany, K.; Hayes, V.M. Assessing high-resolution melt curve analysis for accurate detection of gene variants in complex DNA fragments. Hum. Mutat. 2009, 30, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Ebili, H.O.; Ilyas, M. Cancer mutation screening: Comparison of high-resolution melt analysis between two platforms. Ecancermedicalscience 2015, 9, 522. [Google Scholar] [CrossRef] [PubMed]
- Radvanszky, J.; Surovy, M.; Nagyova, E.; Minarik, G.; Kadasi, L. Comparison of different DNA binding fluorescent dyes for applications of high-resolution melting analysis. Clin. Biochem. 2015, 48, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Vossen, R.H.; Aten, E.; Roos, A.; den Dunnen, J.T. High-resolution melting analysis (HRMA): More than just sequence variant screening. Hum. Mutat. 2009, 30, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, C.T. High-resolution DNA melting analysis: Advancements and limitations. Hum. Mutat. 2009, 30, 857–859. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Dumont, T.; Calvez-Kelm, F.L.; Forey, N.; McKay-Chopin, S.; Garritano, S.; Gioia-Patricola, L.; de Silva, D.; Weigel, R.; Sangrajrang, S.; Lesueur, F.; et al. Description and validation of high-throughput simultaneous genotyping and mutation scanning by high-resolution melting curve analysis. Hum. Mutat. 2009, 30, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Erali, M.; Wittwer, C.T. High resolution melting analysis for gene scanning. Methods 2010, 50, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.A.; Zadorozhnyaya, O.; Beach, S.R.; Brody, G.H. Comparison of the genotyping results using DNA obtained from blood and saliva. Psychiatr. Genet. 2008, 18, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Martin-Nunez, G.M.; Gomez-Zumaquero, J.M.; Soriguer, F.; Morcillo, S. High resolution melting curve analysis of DNA samples isolated by different DNA extraction methods. Clin. Chim. Acta 2012, 413, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Heideman, D.A.; Lurkin, I.; Doeleman, M.; Smit, E.F.; Verheul, H.M.; Meijer, G.A.; Snijders, P.J.; Thunnissen, E.; Zwarthoff, E.C. KRAS and BRAF mutation analysis in routine molecular diagnostics: Comparison of three testing methods on formalin-fixed, paraffin-embedded tumor-derived DNA. J. Mol. Diagn. 2012, 14, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Do, H.; Krypuy, M.; Mitchell, P.L.; Fox, S.B.; Dobrovic, A. High resolution melting analysis for rapid and sensitive EGFR and KRAS mutation detection in formalin fixed paraffin embedded biopsies. BMC Cancer 2008, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, I.; Kjeldsen, T.E.; Hager, H.; Hansen, L.L.; Wojdacz, T.K. The influence of DNA degradation in formalin-fixed, paraffin-embedded (FFPE) tissue on locus-specific methylation assessment by MS-HRM. Exp. Mol. Pathol. 2015, 99, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, L.L.; Er, T.K.; Chen, C.C.; Hsieh, J.S.; Chang, J.G.; Liu, T.C. Characteristics and prevalence of KRAS, BRAF, and PIK3CA mutations in colorectal cancer by high-resolution melting analysis in Taiwanese population. Clin. Chim. Acta 2012, 413, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Destouni, A.; Poulou, M.; Kakourou, G.; Vrettou, C.; Tzetis, M.; Traeger-Synodinos, J.; Kitsiou-Tzeli, S. Single-cell high resolution melting analysis: A novel, generic, pre-implantation genetic diagnosis (PGD) method applied to cystic fibrosis (HRMA CF-PGD). J. Cyst. Fibros. 2016, 15, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Poulou, M.; Destouni, A.; Kakourou, G.; Kanavakis, E.; Tzetis, M. Prenatal diagnosis for CF using High Resolution Melting Analysis and simultaneous haplotype analysis through QF-PCR. J. Cyst. Fibros. 2014, 13, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.; Wittwer, C.T.; Palais, R.; Zhou, L. Simultaneous mutation scanning and genotyping by high-resolution DNA melting analysis. Nat. Protoc. 2007, 2, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Seipp, M.T.; Durtschi, J.D.; Liew, M.A.; Williams, J.; Damjanovich, K.; Pont-Kingdon, G.; Lyon, E.; Voelkerding, K.V.; Wittwer, C.T. Unlabeled oligonucleotides as internal temperature controls for genotyping by amplicon melting. J. Mol. Diagn. 2007, 9, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Tucker, E.J.; Huynh, B.L. Genotyping by high-resolution melting analysis. Methods Mol. Biol. 2014, 1145, 59–66. [Google Scholar] [PubMed]
- Reed, G.H.; Wittwer, C.T. Sensitivity and specificity of single-nucleotide polymorphism scanning by high-resolution melting analysis. Clin. Chem. 2004, 50, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Wojdacz, T.K.; Dobrovic, A.; Hansen, L.L. Methylation-sensitive high-resolution melting. Nat. Protoc. 2008, 3, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Slomka, M.; Sobalska-Kwapis, M.; Korycka-Machala, M.; Bartosz, G.; Dziadek, J.; Strapagiel, D. Genetic variation of the ABC transporter gene ABCC1 (Multidrug resistance protein 1-MRP1) in the Polish population. BMC Genet. 2015, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Van der Stoep, N.; van Paridon, C.D.; Janssens, T.; Krenkova, P.; Stambergova, A.; Macek, M.; Matthijs, G.; Bakker, E. Diagnostic guidelines for high-resolution melting curve (HRM) analysis: An interlaboratory validation of BRCA1 mutation scanning using the 96-well LightScanner. Hum. Mutat. 2009, 30, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Wang, C.; Yin, H.; Tian, Y.; Li, J. Evaluation of different reaction systems for HRM analysis in apple. Biosci. Methods 2012, 3, 1–6. [Google Scholar] [CrossRef]
- Ebili, H.; Ilyas, M. High resolution melt analysis, DNA template quantity disparities and result reliability. Clin. Lab. 2015, 61, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Cousins, M.M.; Donnell, D.; Eshleman, S.H. Impact of mutation type and amplicon characteristics on genetic diversity measures generated using a high-resolution melting diversity assay. J. Mol. Diagn. 2013, 15, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Martino, A.; Mancuso, T.; Rossi, A.M. Application of high-resolution melting to large-scale, high-throughput SNP genotyping: A comparison with the TaqMan method. J. Biomol. Screen. 2010, 15, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Valisheva, I.; Harris, R.J.; Zhu-Shimoni, J. A sensitive mutation screening method supporting cell line development for biotherapeutics. Anal. Biochem. 2016, 505, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Garritano, S.; Gemignani, F.; Voegele, C.; Nguyen-Dumont, T.; Le Calvez-Kelm, F.; De Silva, D.; Lesueur, F.; Landi, S.; Tavtigian, S.V. Determining the effectiveness of High Resolution Melting analysis for SNP genotyping and mutation scanning at the TP53 locus. BMC Genet. 2009, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Thomas, V.; Mazard, B.; Garcia, C.; Lacan, P.; Gagnieu, M.C.; Joly, P. UGT1A1 (TA)n genotyping in sickle-cell disease: High resolution melting (HRM) curve analysis or direct sequencing, what is the best way? Clin. Chim. Acta 2013, 424, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Gorniak, P.; Ejduk, A.; Borg, K.; Makuch-Lasica, H.; Nowak, G.; Lech-Maranda, E.; Prochorec-Sobieszek, M.; Warzocha, K.; Juszczynski, P. Comparison of high-resolution melting analysis with direct sequencing for the detection of recurrent mutations in DNA methyltransferase 3A and isocitrate dehydrogenase 1 and 2 genes in acute myeloid leukemia patients. Eur. J. Haematol. 2016, 96, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Strapagiel, D.; Sobalska-Kwapis, M.; Slomka, M.; Marciniak, B. Biobank Lodz—DNA based biobank at the University of Lodz, Poland. Open J. Bioresour. 2016, 3. [Google Scholar] [CrossRef]
- DNA Genotek. Laboratory Protocol for Manual Purification od DNA from 0.5 mL Using a Deep 96-Well Plate. Available online: http://www.dnagenotek.com/US/pdf/PD-PR-052.pdf (accessed on 3 November 2017).
- Caban, M.; Strapagiel, D.; Dziadek, J.; Korycka-Machala, M.; Grzelak, A. Principles of a new protocol for prediction of azole resistance in Candida albicans infections on the basis of ERG11 polymorphisms. Curr. Microbiol. 2016, 73, 172–182. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. GenBank Database. Available online: http://www.ncbi.nlm.nih.gov (accessed on 3 November 2017).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Issue | Troubleshooting | Solutions |
|---|---|---|
| DNA source | poor quality results from DNA isolated from FFPE tissue samples |
|
| DNA preparation | different isolation methods and sample composition make parallel analysis difficult |
|
| Primer design | poor melting curve resolution and PCR optimization |
|
| unexpected variation within primer sequence and incorrect clustering |
| |
| Technical issues with reproducibility | reagents, equipment, handling |
|
| reproducibility of manual pipetting |
| |
| reproducibility of automated liquid handling instrument |
| |
| Multiple proximate polymorphic sites | underestimation of the prevalence of variants within a single amplicon |
|
| HRM | Direct Sequencing of the Area of Interest | ||||||
|---|---|---|---|---|---|---|---|
| Gene | dbSNP ID | Forward Primer (5′→3′) | Reverse Primer (5′→3′) | Size (bp) | Forward Primer (5′→3′) | Reverse Primer (5′→3′) | Size (bp) |
| ABCC1 | rs4148337 | AGCCTGGGTGACAAGAGTGA | TGGATCTCAGGATGGCTAGG | 189 | GTGGTGAAACCCCGTCTTTA | CCTTGGAGCAACACAGACAA | 604 |
| rs4148337 (new) | AAGCTGAGGCAGGAGAATCA | AAGGTAGCAAGCAGCTGAGG | 163 | ||||
| rs2074087 | CTCACACATGTGCACTCACG | TCTGTGCTGGCATAGGTACG | 204 | CATGTCCCACCTTCAGACCT | CCAGCTTAACTCCGTGCTTC | 748 | |
| rs2074087 (new) rs45492500 rs45607032 | GCCAAGCTAGGCAGTCTCAC | GGCAAGTAGCTCATGCTGTG | 99 | ||||
| rs201533167 rs200922662 | GGGGAGTCACAGCTTTACCA | GGGAATGGGTGAGGGAAT | 248 | AGGGGACAGAGGGACACAG | AAATCTGTGGGGCTCATTTG | 618 | |
| ABCG2 | rs771435451 rs2231153 rs781367109 | TGTGGAAAGAGTTTTGTGGGTA | CTAACCAATAGCCCCTGCTG | 229 | AGGGCCCATCTTCAAATACC | TTGCTTGCTCTCTCCAACATT | 732 |
| RORC | rs116171003 | GTGAATGGGGCCACCTG | GACGACAGGGTCCAGGCT | 45 | CTCGGGGGTAGGAGGAGTAG | CCATCTCCCAACAGATCTTGA | 602 |
| ERG11 | Exon | TTAGGGAAAATTATGACGGTTTAT | CTTTCATCAGTAACAAAATAATTCAAA | 263 | TTTTCTTCATCTTACTTCTTTCTTTCA | TTGACCACCCATAAGAATACCA | 1082 |
| TMEM18 | rs4854344 | TGTTTAGATACACACTCTCCACTGT | GATGGCTGTGCTGGAACTG | 58 | TGCGATGAACTGAGTGTTGC | ACCATTTCTGGAACGTGGAG | 638 |
| FTO | rs9939609 rs76804286 rs9926289 | CATCAGTTATGCATTTAGAATGTCTG | AGAGTAACAGAGACTATCCAAGTGC | 95 | TGGTTTCAGAGGCTTGTGTG | GCCCAAGGATGGTGTTTCTA | 695 |
| XRCC3 | rs861539 | TTCCGCTGTGAATTTGACAG | CTCACCTGGTTGATGCACAG | 125 | - | - | - |
| BRCA1 | rs799917 | AAAGCGCCAGTCATTTGC | CTTCTGCATTTCCTGGATTTGA | 47 | - | - | - |
| rs16941 | GCCGTAATAACATTAGAGAAAATG | TTAATATTGCTTGAGCTGGC | 55 | - | - | - | |
| TGFB2 | rs6684205 | AGTGAACCAAGTGTGAAGGGA | TTCAGAGGAATTTTGGGGAA | 61 | TGGCACCTCCACATATACCA | ACGGCATTCTTCTGCTGTCT | 612 |
| ABCB1 | rs1045642 | CCTGTTTGACTGCAGCATTG | AAGGCATGTATGTTGGCCTC | 108 | TGTTTTCAGCTGCTTGATGG | GTGGGGACCCAGACTCTGTA | 604 |
| rs1045642 (new) | GGGTGGTGTCACAGGAAGAG | AGGCAGTGACTCGATGAAGG | 74 | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Słomka, M.; Sobalska-Kwapis, M.; Wachulec, M.; Bartosz, G.; Strapagiel, D. High Resolution Melting (HRM) for High-Throughput Genotyping—Limitations and Caveats in Practical Case Studies. Int. J. Mol. Sci. 2017, 18, 2316. https://doi.org/10.3390/ijms18112316
Słomka M, Sobalska-Kwapis M, Wachulec M, Bartosz G, Strapagiel D. High Resolution Melting (HRM) for High-Throughput Genotyping—Limitations and Caveats in Practical Case Studies. International Journal of Molecular Sciences. 2017; 18(11):2316. https://doi.org/10.3390/ijms18112316
Chicago/Turabian StyleSłomka, Marcin, Marta Sobalska-Kwapis, Monika Wachulec, Grzegorz Bartosz, and Dominik Strapagiel. 2017. "High Resolution Melting (HRM) for High-Throughput Genotyping—Limitations and Caveats in Practical Case Studies" International Journal of Molecular Sciences 18, no. 11: 2316. https://doi.org/10.3390/ijms18112316