1. Introduction
Myrothecium roridum is a plant pathogenic fungus widespread in the soils of tropical and subtropical regions.
M. roridum infects different economical crops, including soybean, eggplant, pepper, tomato, and cotton, decreasing their yields [
1]. This fungus shows strong infectivity, and the spread of
M. roridum infection is very fast. Currently, no effective treatment and hazard prevention measure for
M. roridum exists. Trichothecenes are sesquiterpene compounds isolated from different kinds of fungi, including
Stachybotrys atra [
2],
Trichoderma brevicompactum [
3], and
M. roridum [
4]. Trichothecenes are the main plant pathogenic factors of
Fusarium graminearum; the treatment of trichothecene mycotoxin with wheat could cause gibberellic disease, and a substantial amount of trichothecene mycotoxin was found in infected wheat ears [
5,
6,
7,
8]. However, the main pathogenic mechanism of
M. roridum remains unclear. Elucidating the molecular mechanism for the pathogenicity of
M. roridum by transcriptome sequencing is necessary to facilitate the prevention of diseases caused by
M. roridum.
The genes related to the trichothecene biosynthesis in
Fusarium species were identified [
9], and the trichodiene synthase encoded by the
Tri5 gene was considered a key enzyme in trichothecene biosynthesis. The structural and mechanism analysis of engineered trichodiene synthase from
Trichoderma harzianum has been investigated, demonstrating that this enzyme could catalyze the conversion of farnesyl pyrophosphate (FPP) to trichodiene [
10]. The
Tri5-deleted
F. graminearum almost lost its infectivity toward wheat [
7]. The
Tri6 gene, encoding transcriptional proteins, could regulate trichothecene mycotoxin biosynthesis [
6]. The
Tri4 gene encoded trichodiene oxygenase, the
Tri11 gene encoded isotrichodermin C-15 hydroxylase. The
Tri12 gene encoded a kind of membrane transport protein responsible for the exportation of trichothecene mycotoxins [
11]. The
Tri5 gene from
M. roridum was inserted into pMD18T vector and sequenced [
12], but this gene has not been heterologously expressed and enzymatic characterized. However, genes related to trichothecene mycotoxin biosynthesis in
M. roridum have not been fully identified.
Transcriptome sequencing in different kinds of infected plants or pathogenic fungi was performed to investigate the pathogenic mechanisms of fungi [
13]. In this study, the transcriptome of
M. roridum was sequenced using the Illumina sequencing platform 2000. Genes related to trichothecene mycotoxin biosynthesis in
M. roridum including
Tri4,
Tir5 and
Tri11 were identified. This is the first attempt to characterize the complete transcriptome of
M. roridum and reveal genes related to trichothecene mycotoxin biosynthesis, which would provide a useful theoretical guide for the hazard prevention of
M. roridum toward economical crops. Moreover, the elucidation of genes related to trichothecene mycotoxin biosynthesis establishes a foundation for the prevention of crop diseases caused by
M. roridum. 2. Results
2.1. Sequencing and De Novo Assembly
In summary, 44,992,952 sequence reads with Q20 of 92.49% and GC percent of 57.57% were generated, and a total of 8,997,597,886 (8.79 GB) nucleotides were obtained (
Table 1). Moreover, raw data were deposited in NCBI with accession number SRP080968. The Trinity software (
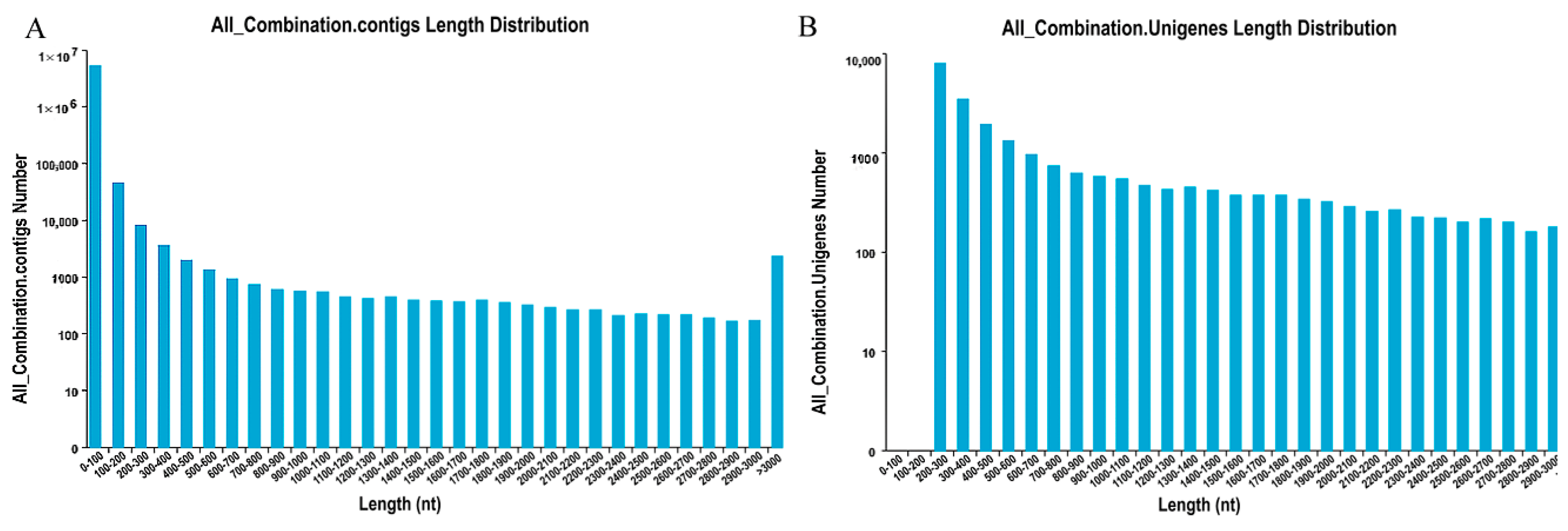
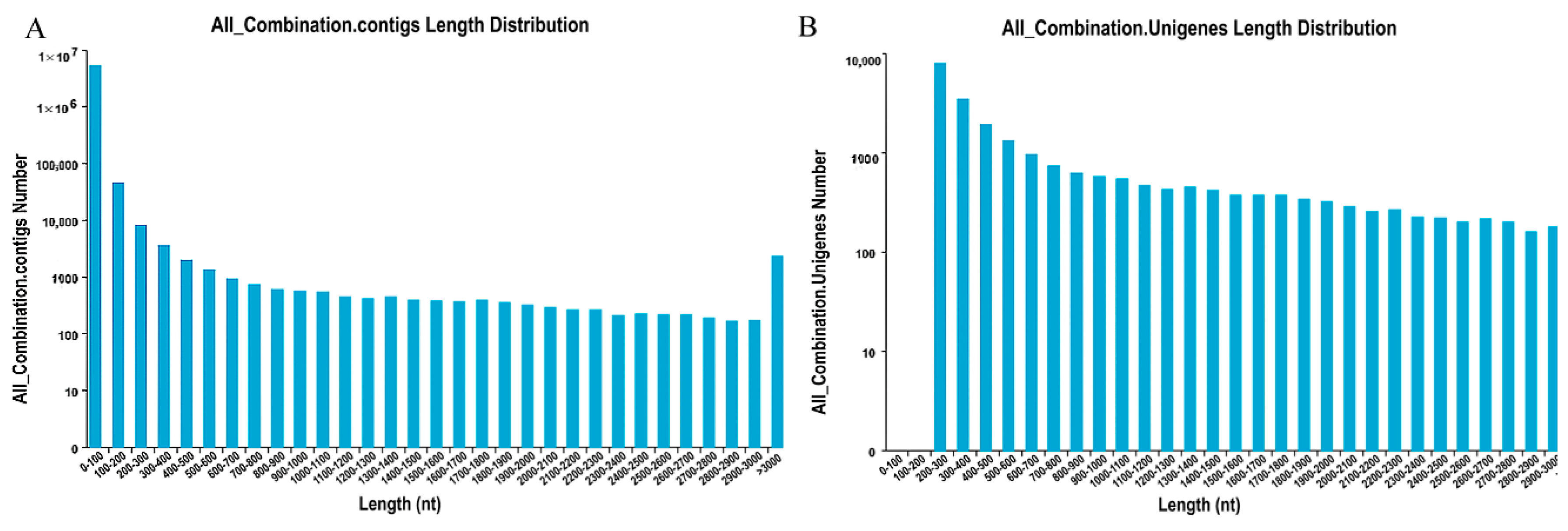
http://trinityrnaseq.sourceforge.net/) was used to assemble short reads through a step-wise strategy, which yielded 5,247,474 contigs with a mean length of 39.83 bp (
Figure 1A) and 25,996 unigenes with a mean length of 1094 bp (
Figure 1B). A total of 2299 unigenes with lengths greater than 3000 bp were found, indicating the abundance of secondary metabolites in
M. roridum. 2.2. Functional Annotation and Classification
All the unigenes were annotated by public databases, including Nr, Nt, Swissprot, KEGG (Kyoto Encyclopedia of Genes and Genomes), COG (Cluster of Orthologous Groups of proteins), GO (Gene Ontology Consortium); 18,199 unigenes were annotated, while 7797 unigenes remained unannotated. A total of 18,160 unigenes matched sequences in the Nr databases. Furthermore, the unigene number of
M. roridum annotated in different public databases is shown in
Table 2.
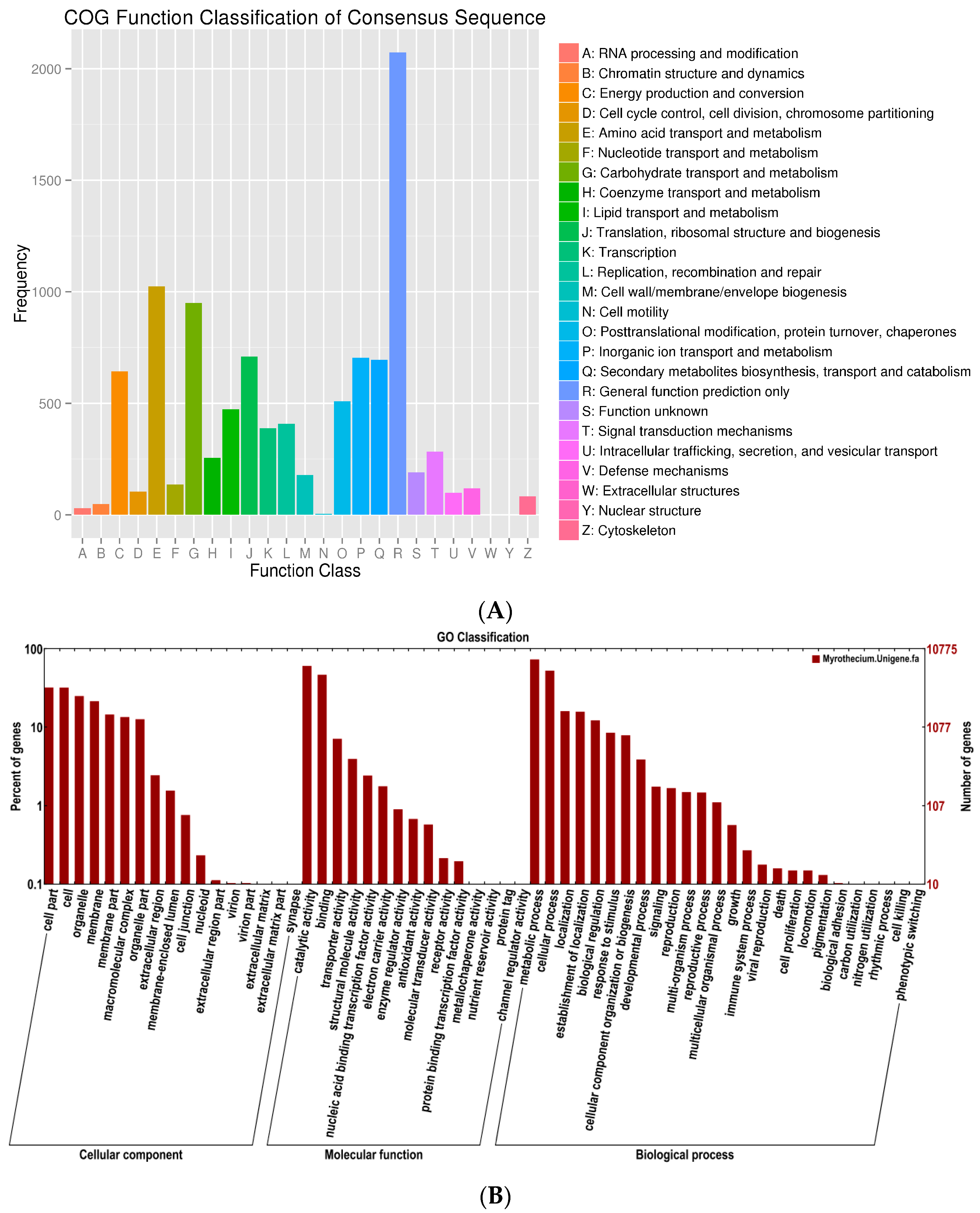
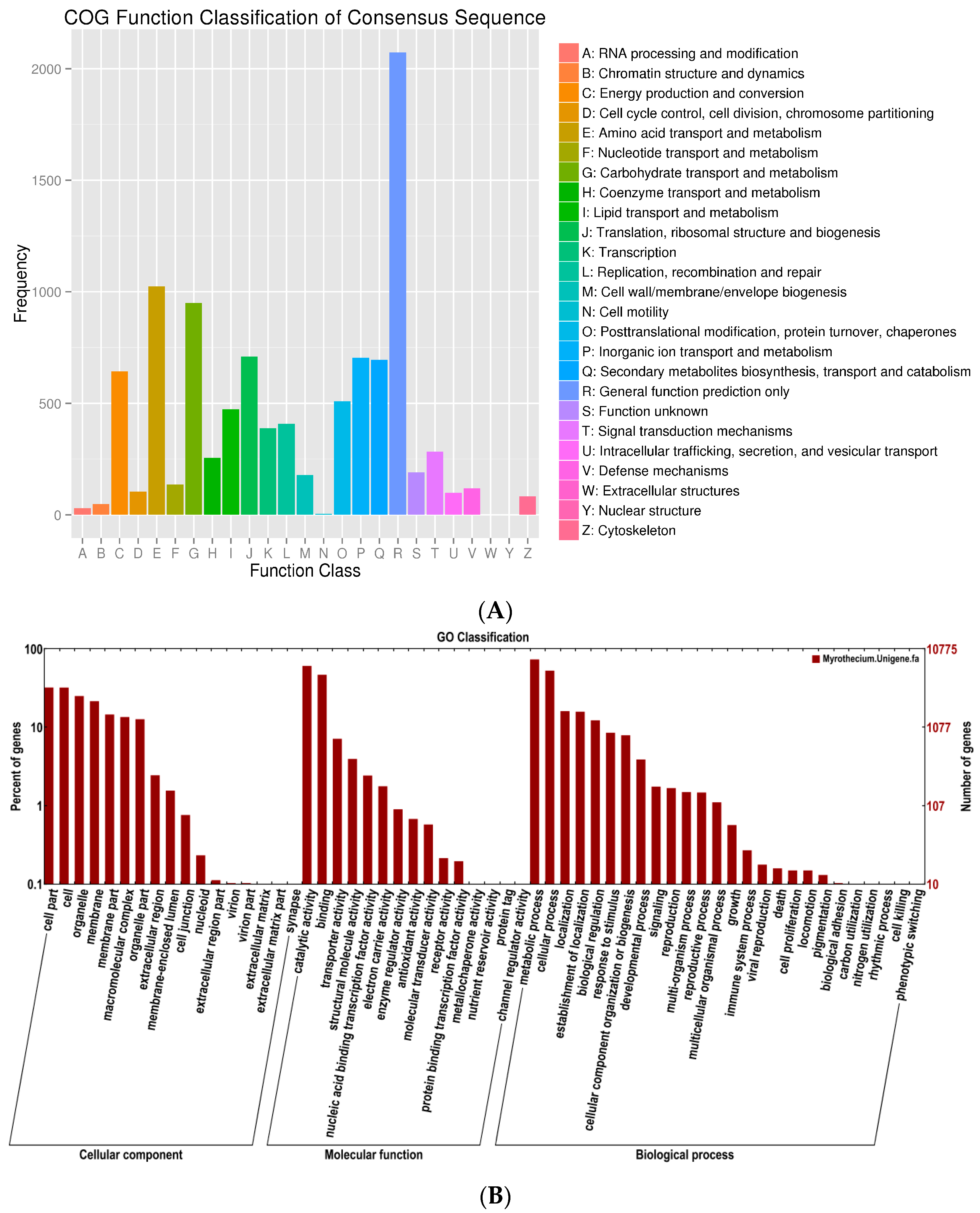
A total of 10,113 unigenes were annotated and grouped into 25 functional classifications (
Figure 2A). The most frequently identified classes were “general function” (2073, 20.5%), followed by transcription (1024, 10.1%), carbohydrate transport and metabolism (950, 9.4%), translation, ribosomal structure and biogenesis (709, 7.0%), inorganic ion transport and metabolism (704, 6.9%), secondary metabolites biosynthesis, transport, and catabolism (696, 6.8%), and post-translational modification, protein turnover, and chaperones (509, 5.0%). The nuclear structure (1, 0.001%) and extracellular structure (0, 0%) categories showed the fewest corresponding genes. Another 10,775 unigenes were annotated by GO database and classified into the biological process, cellular component, and molecular function categories (
Figure 2B). The high percentage of unigenes involved in the function of catalytic activity indicated the variety of the secondary metabolites produced by
M. roridum. To investigate the biological function of unigenes further, a total of 4569 unigenes were assigned to the metabolic pathways described in the KEGG database, including metabolic pathways, biosynthesis of secondary metabolites, ubiquitin-mediated proteolyis, mitogen-activated protein kinase (MAPK) signaling pathway, and regulation of autophagy RNA transport.
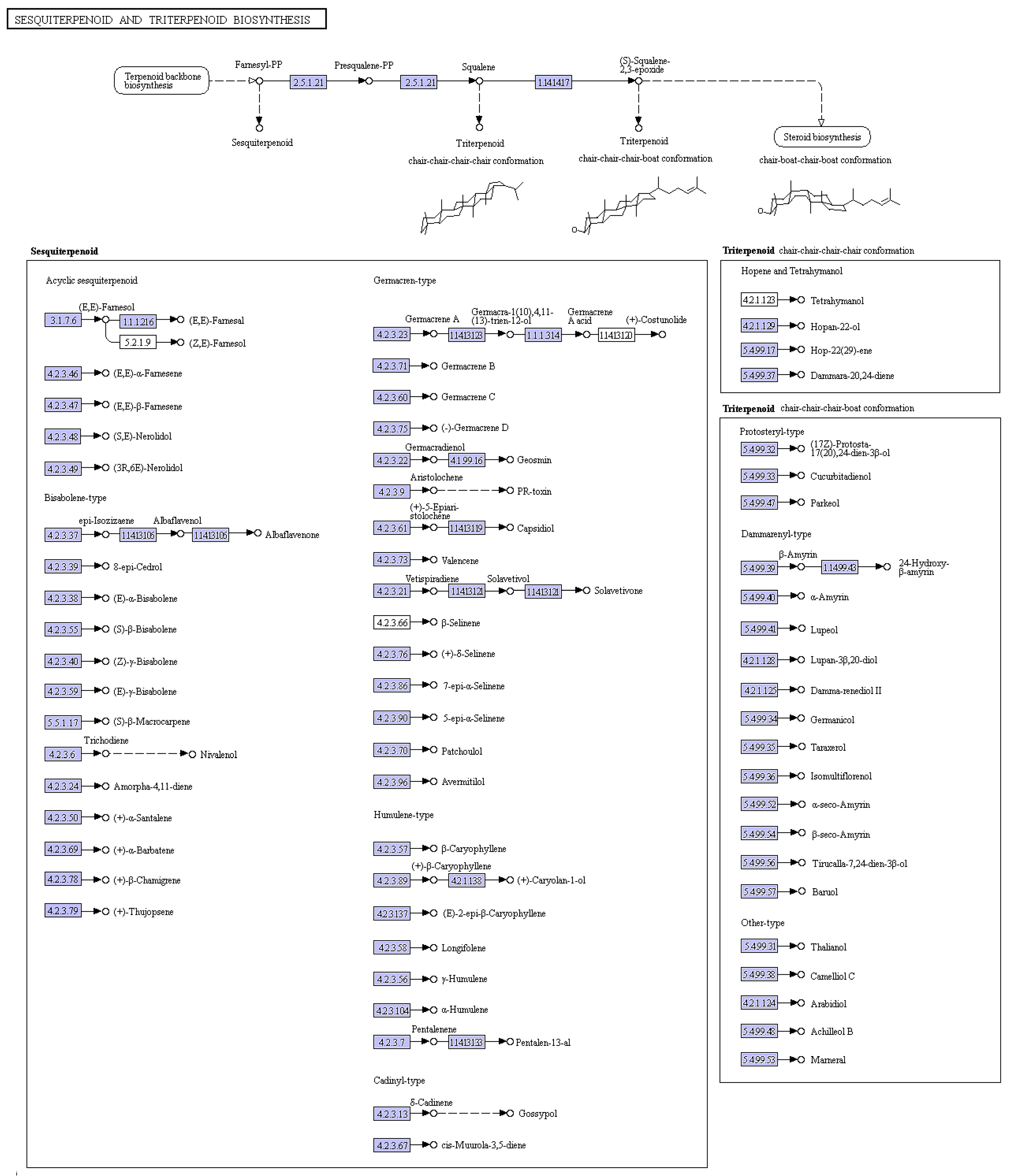
2.3. Candidate Genes Involved in Trichothecene Biosynthesis and Expression Analysis of Eight Candidate Genes by qPCR
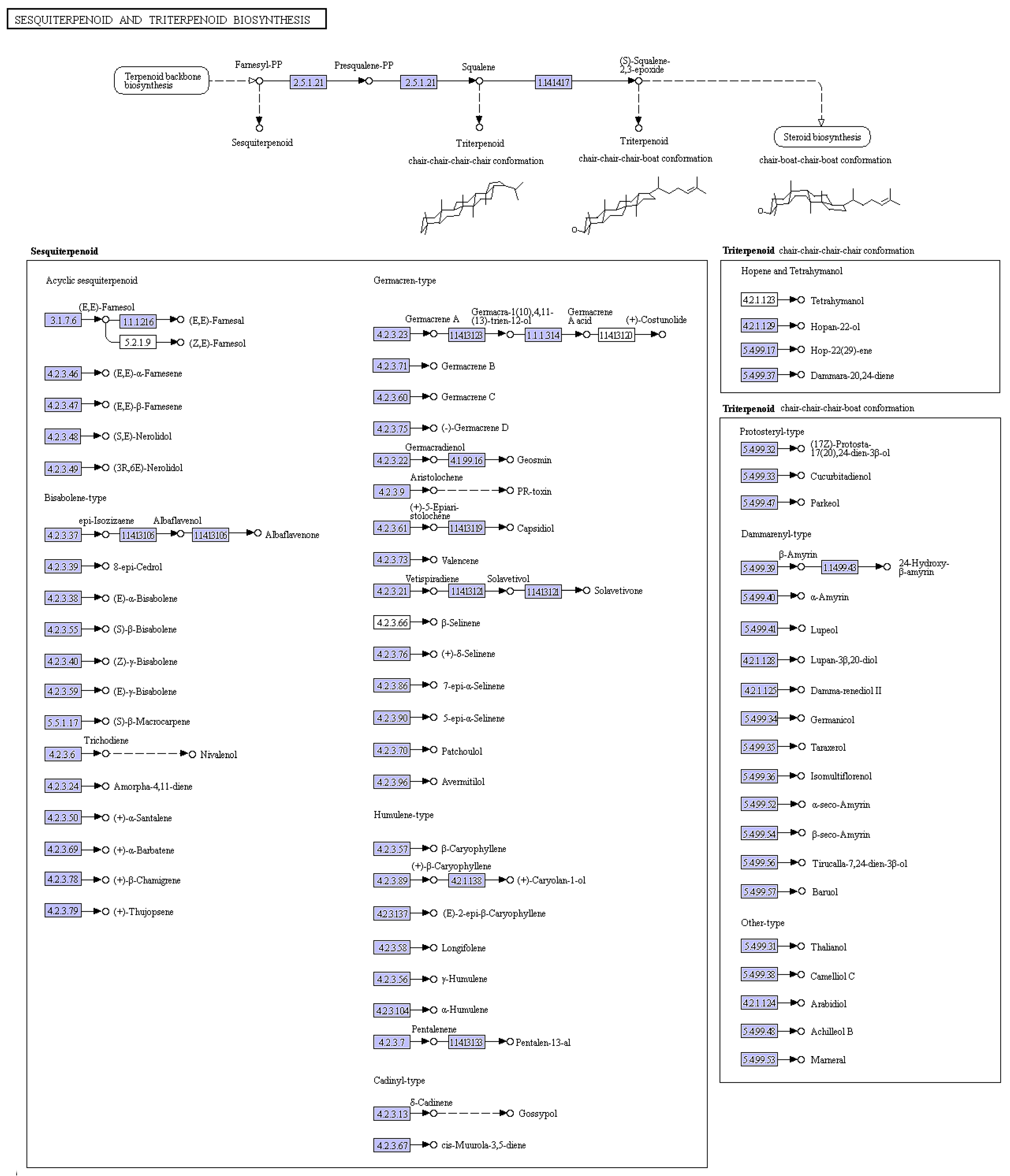
Trichothecene was postulated to play a very important role in the pathogenicity of
M. roridum [
4]. Thus the identification of genes involved in trichothecene biosynthesis establishes a foundation for the elucidation of the pathogenic mechanism of
M. roridum. Trichothecenes are sesquiterpene compounds, and the sesquiterpene and terpenoid backbone biosynthesis pathways in
M. roridum are shown in
Figure 3. Genes involved in trichothecene mycotoxin biosynthesis are summarized in
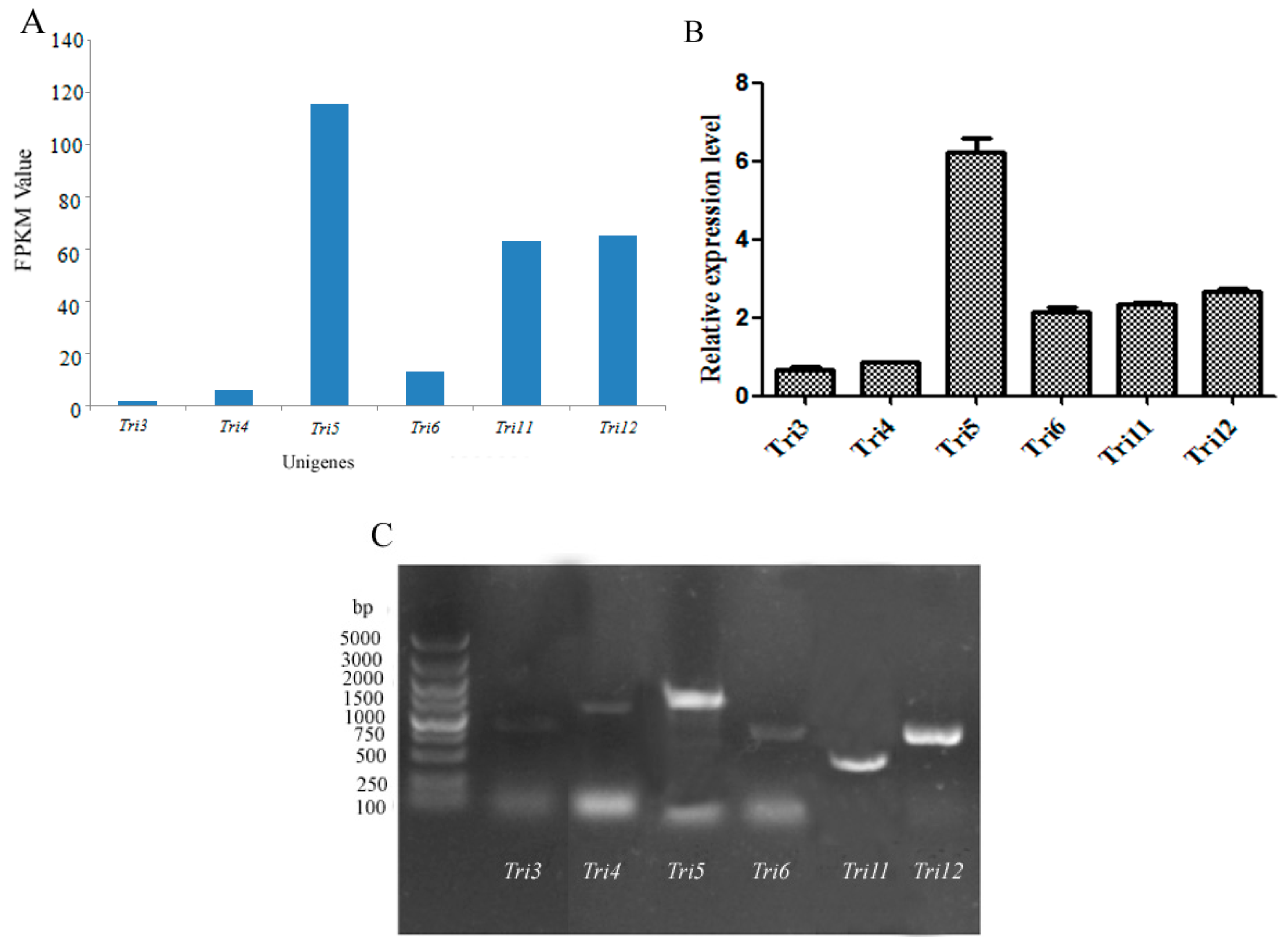
Table S1. The expression levels of unigenes, including
Tir3 (trichothecene 3-
O-acetyltransferase),
Tri4 (trichodiene oxygenase),
Tri5 (trichodiene synthase),
Tri6 (trichothecene biosynthesis transcription factor),
Tri12 (trichothecene efflux pump), and
Tri11 (trichothecene 15-
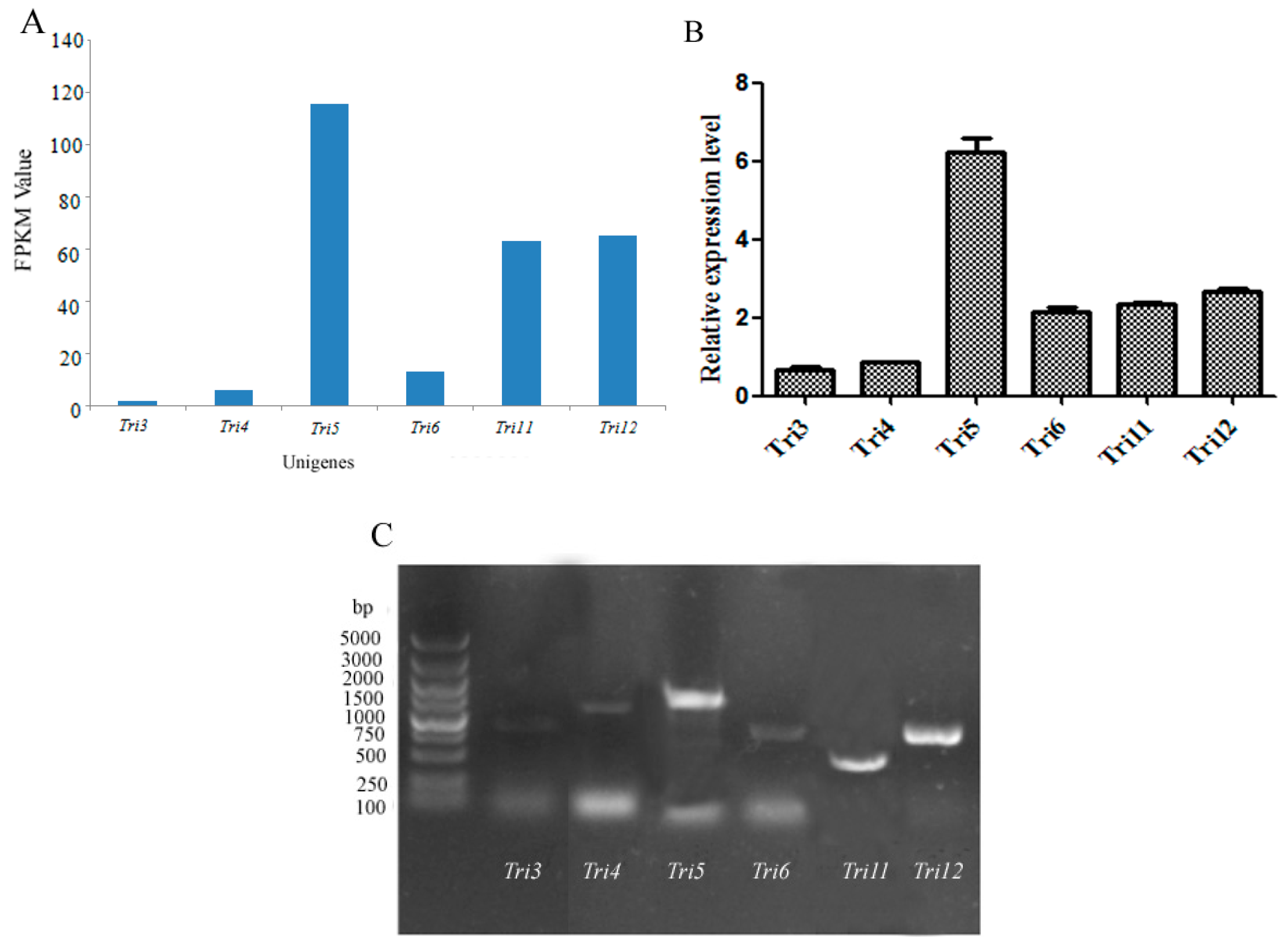
O-acetyltransferase) were predicted according to the FPKM (Fragments Per Kilobase of exon model per Million mapped reads) value and validated by qRT-PCR (Quantitative real-time polymerase chain reaction) using primers (
Table S2) according to the ORFs of the aforementioned genes (
Figure 4A,B).
Tri5 gene showed the highest expression level, followed by the genes
Tri12,
Tri6, and
Tri11, suggesting the important roles of these four genes in trichothecene biosynthesis, while
GAPDH was used as a reference gene. The expression levels identified by qRT-PCR were in accordance with the prediction. The qPCR products of
Tri3,
Tri4,
Tri5,
Tri6,
Tri11, and
Tri12 genes were detected by agarose gel (
Figure 4C). The sequencing results indicated that the fragments were desired genes involved in trichothecene biosynthesis.
2.4. Identification of Tri4, Tri5, and Tri11 Genes
Specific primers were designed to amplify the
Tri4,
Tri5, and
Tri11 genes with restriction enzyme sites (
NdeI and
XhoI), then the amplified fragment was inserted into vector pET28a and expressed in
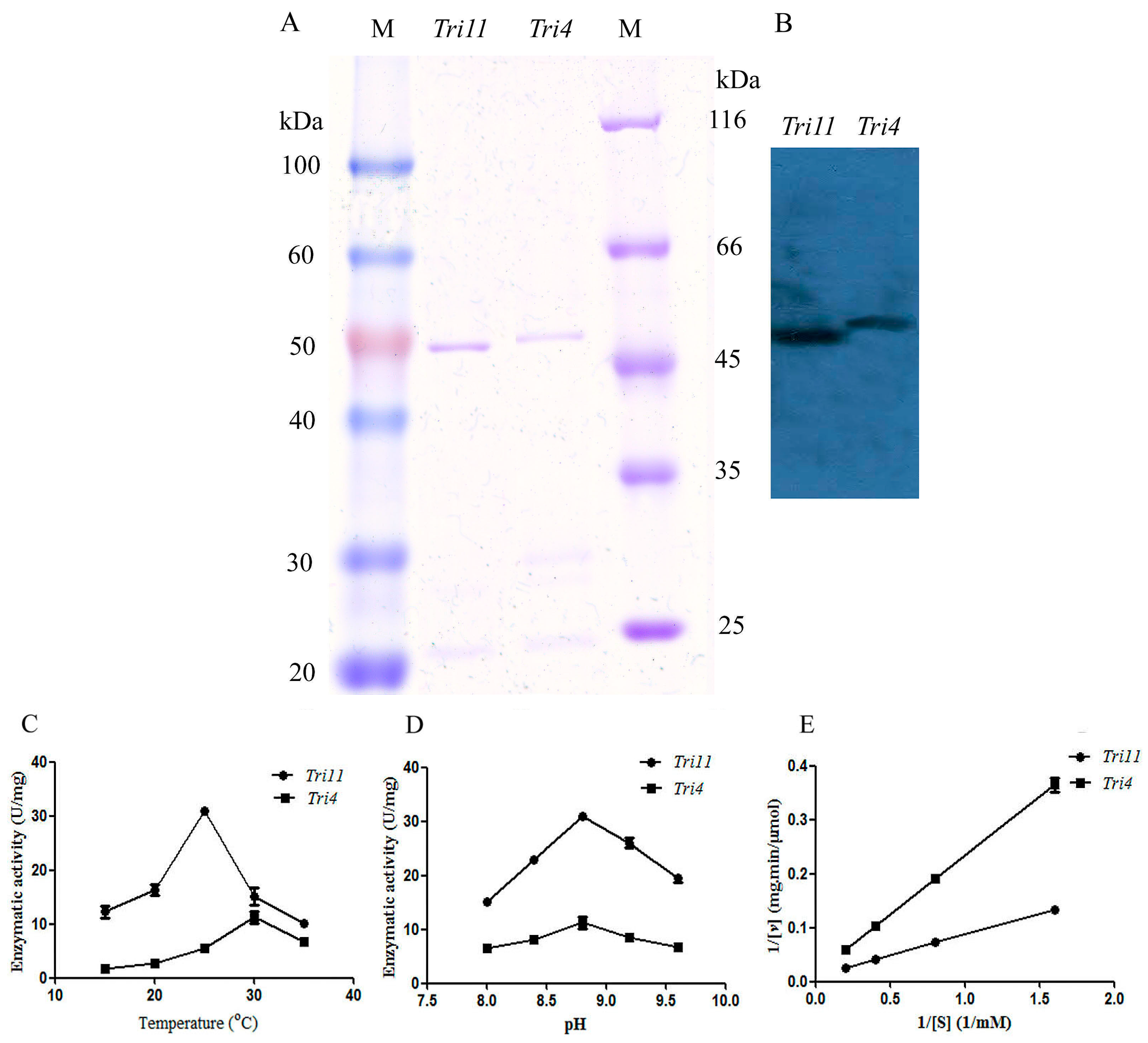
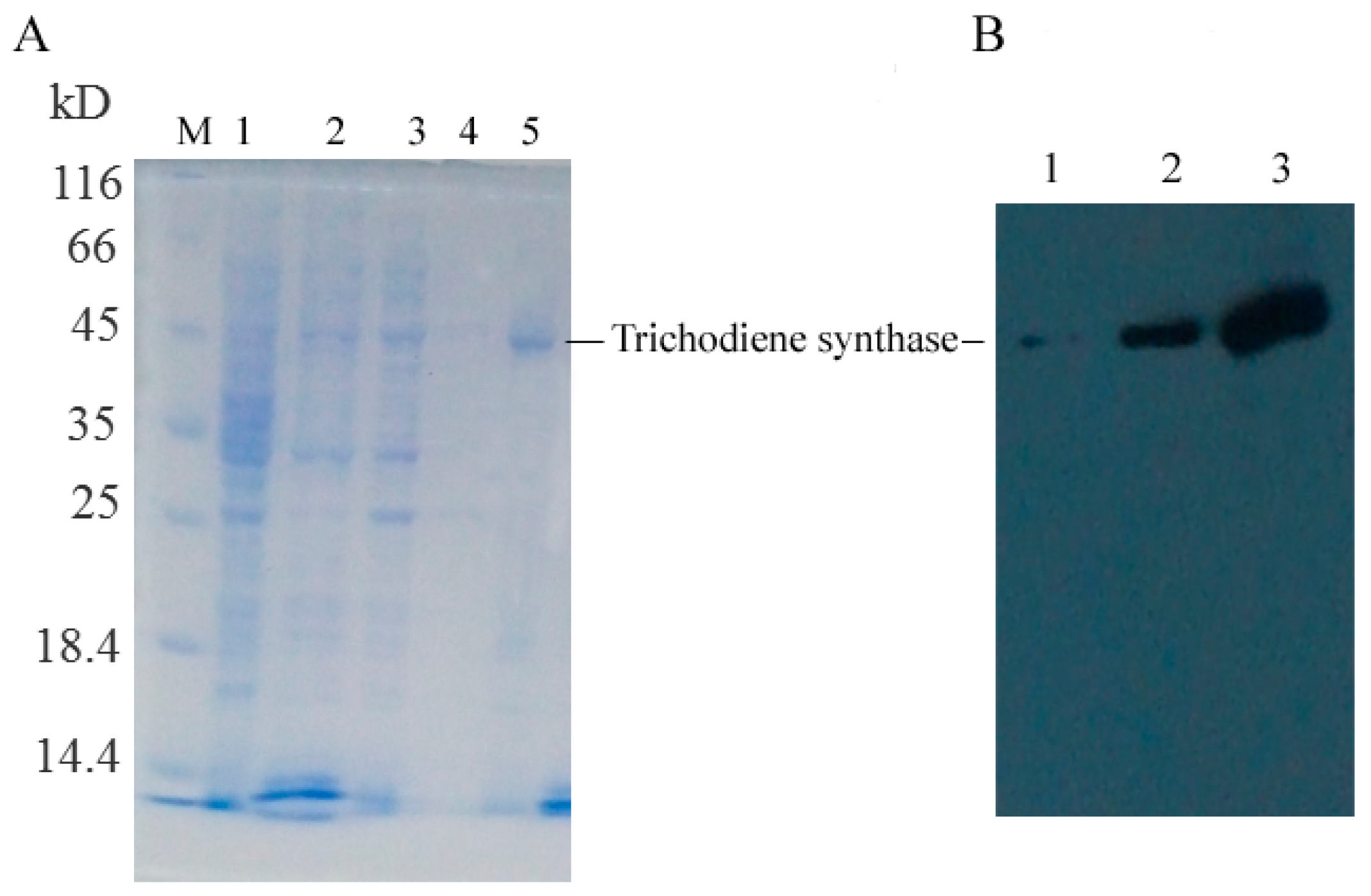
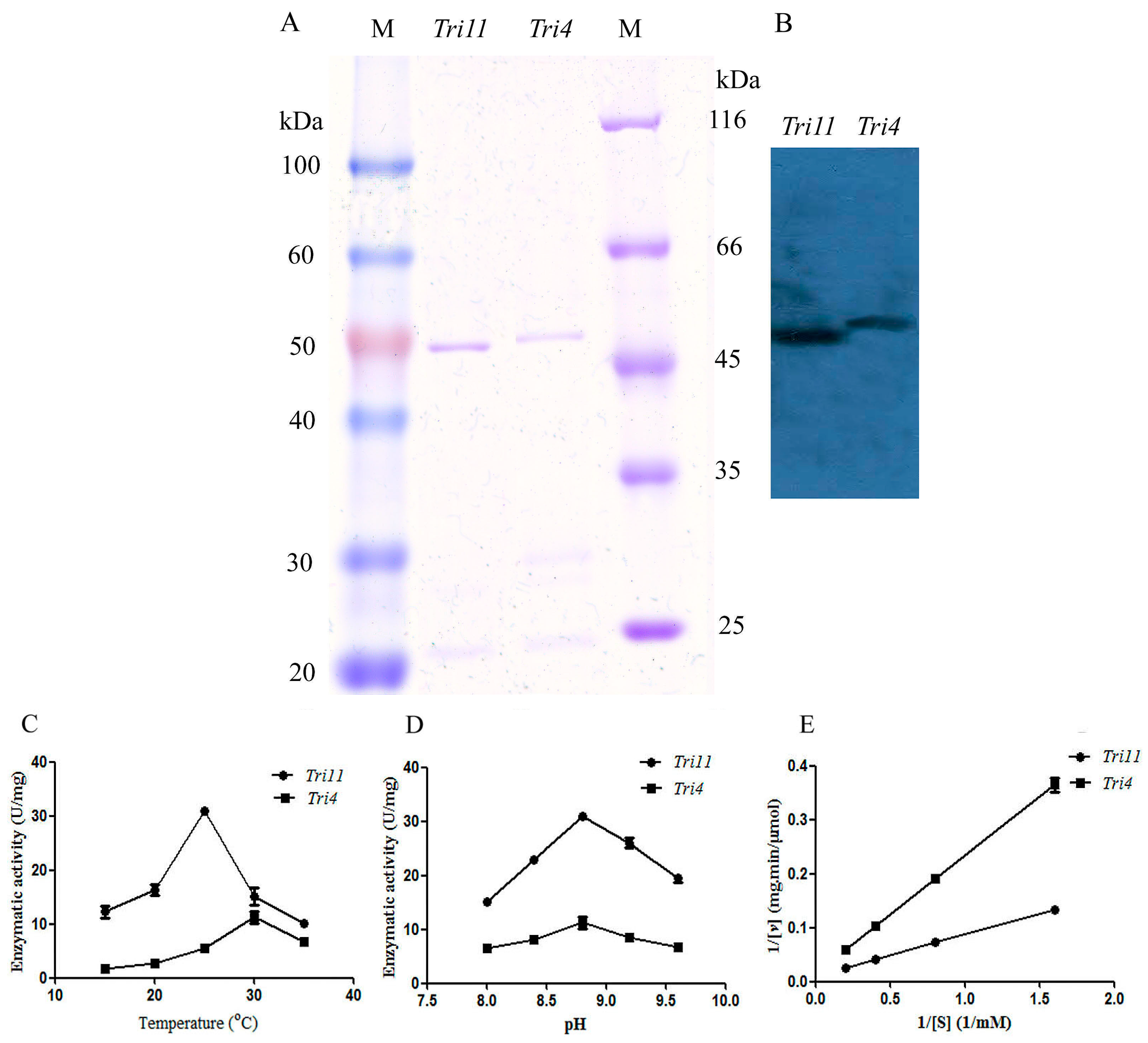
Escherichia coli BL21 (DE3) with a molecular weight of 52.0, 45.0, and 50.0 kDa, respectively, which was in accordance with the theoretical value. The trichodiene synthase was purified by Ni affinity chromatography with a purity of 91.9% (
Figure 5A). The Western blot analysis result using anti-His monoclonal antibody confirmed the successful expression and purification of the
Tri5 gene (
Figure 5B). Trichodiene synthase from
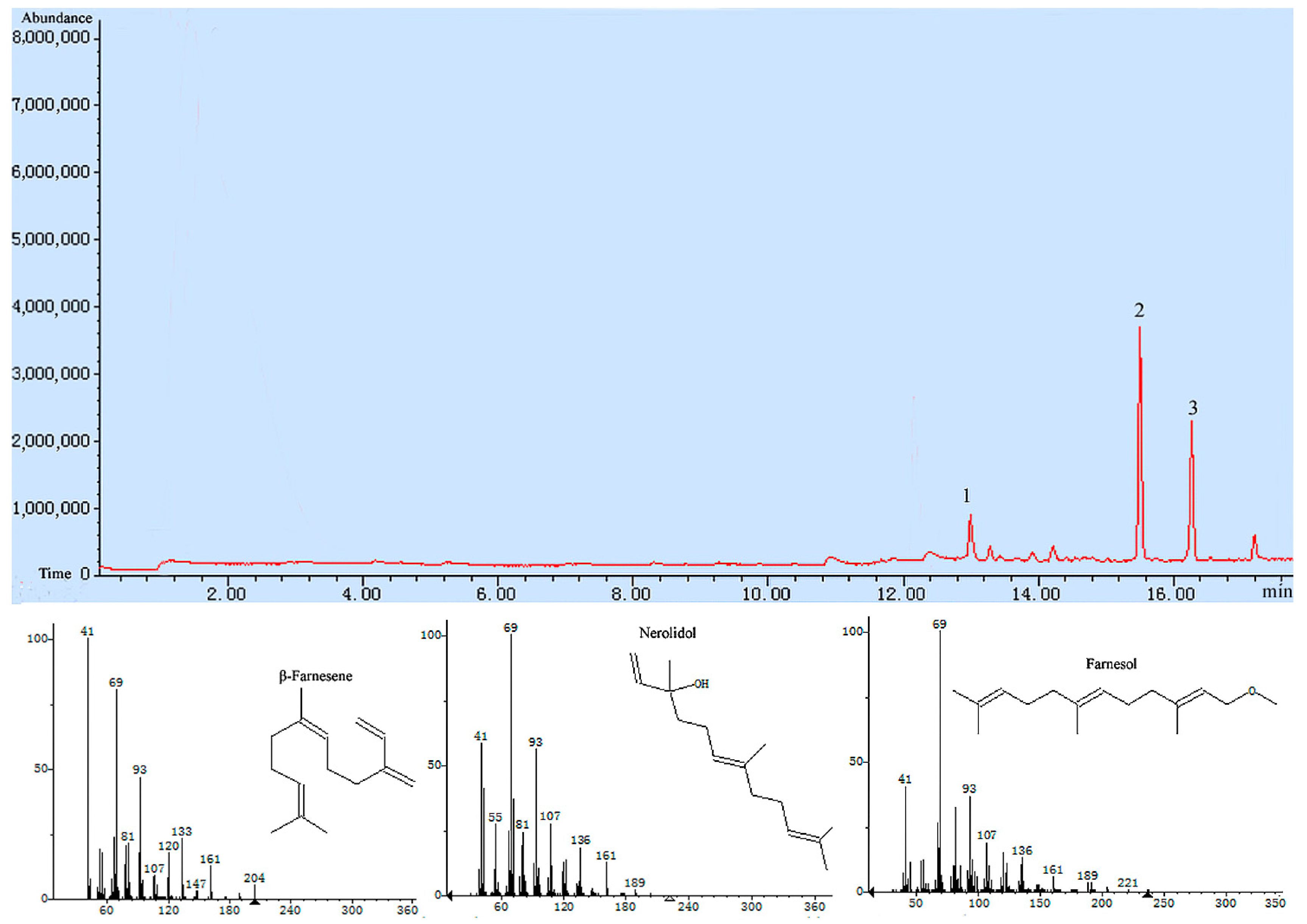
M. roridum encoded by
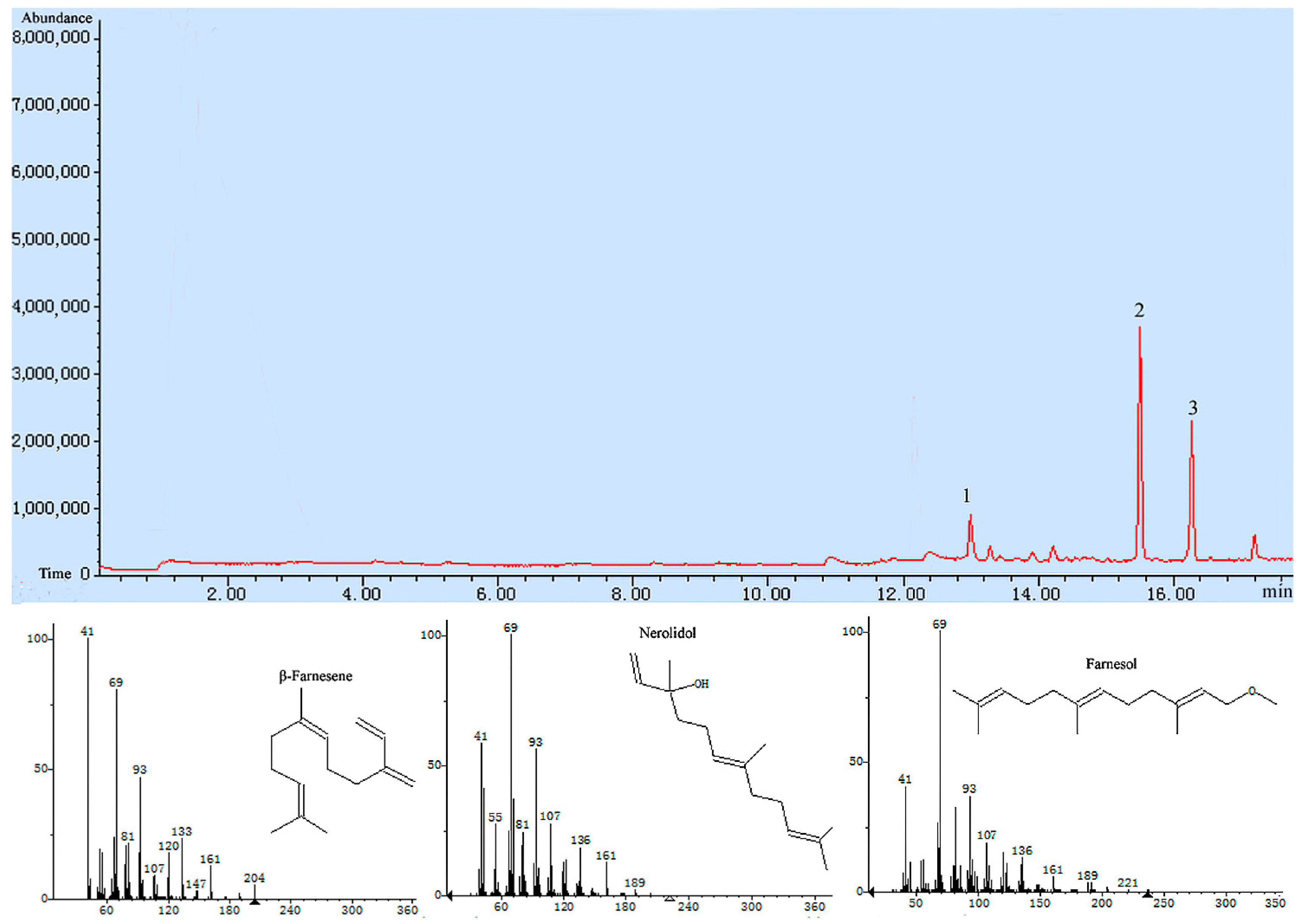
Tri5 gene were added to the substrate FPP, and the catalyzation products were detected by GC-MS (Gas Chromatograph-Mass Spectrometer), and the sesquiterpenoids including β-farnesene, nerolidol and farnesol were detected, nerolidol was detected as a main product, which accounted for 68.2% of all the catalyation products (
Figure 6). The result further demonstrated that the trichodiene synthase encoded by
Tri5 was a sesquiterpene synthase.
The trichodiene oxygenase encoded by
Tri4 from
M. roridum (TRO-MR) and isotrichodermin C-15 hydroxylase encoded by
Tri11 from
M. roridum (ITH-MR) were purified by Ni affinity chromatography with purities of 92.5% and 95.7%, respectively, which was also demonstrated by Western blot analysis (
Figure 7A,B). The substrate thioanisole was used to investigate the enzymatic properties of enzymes encoded by
Tri4 and
Tri11. Trichodiene oxygenase showed the highest enzymatic activity of 11.26 ± 1.53 U/mg towards thioanisole at 30 °C, whereas isotrichodermin C-15 hydroxylase showed the highest enzymatic activity of 31.0 ± 0.6 U/mg towards thioanisole at 25 °C (
Figure 7C). The optimal reaction pH values of trichodiene oxygenase and isotrichodermin C-15 hydroxylase from
M. roridum with thioanisole were 8.8 (
Figure 7D). The enzymatic kinetics of TRO-MR and ITH-MR towards thioanisole was investigated by using different substrates (
Figure 7E). ITH-MR and TRO-MR showed
Vmax and
Km values of 94.34 μmol/mg∙min, 7.19 mM, and 71.43 µmol/mg∙min, 15.53 mM, respectively. The results indicated that ITH-MR showed higher substrate-binding affinity than TRO-MR.
2.5. Development and Characterization of cDNA-Derived SSR Markers
Different unigenes in M. roridum were identified by MISA analysis, and 2200 unigenes containing SSR (Simple Sequence Repeat) were identified, among which, 402 sequences contained more than one SSR. Moreover, 202 SSRs were present in the compound formation. The distribution and frequency of the mono-, di-, tri-, tetra-, and hexa-nucleotide repeats were analyzed. The most abundant repeat motifs were mononucleotides (1115, 50.68%), followed by trinucleotides (592, 26.91%), dinucleotides (216, 9.82%), and tetra-nucleotides (48, 2.18%). The most frequent was A/T (904, 41.09%), followed by C/G (340, 15.45%), AGC/CTG (143, 6.5%), AAAC/GTTT (129, 5.86%), AG/CT (128, 5.82%), AAG/CTT (125, 5.68%), AGG/CTT (91, 4.14%), and ACG/CGT (72, 3.27%).
2.6. Phylogenetic Analysis of Genes Involved in Trichothecene Mycotoxin Biosynthesis
Tri5 gene, encoding trichodiene synthase in
M. roridum, played a very important role in trichothecene biosynthesis. Therefore, phylogenetic analysis was conducted using an alignment of all known predicted protein sequences in the NCBI database (
Figure S1A), and the result demonstrated that the highest similarity of trichodiene synthase from
M. roridum (c10283) with that from
Fusarium sporotrichioides was only 26%, indicating significant differences between the trichodiene synthase from
M. roridum and the trichodiene synthases from other fungal species.
Tri4 was annotated as the trichodiene oxygenase from
F. sporotrichioides in the Nr database. It shared a similarity of 32% with the cytochrome P450 protein from
Eutypa lata, and nine kinds of trichodiene oxygenases were found in
M. roridum, exhibiting low similarities (
Figure S1B). The alignment results demonstrated the diversity and novelty of trichodiene oxygenases from
M. roridum.
2.7. Construction of Protein–Protein Interaction Network
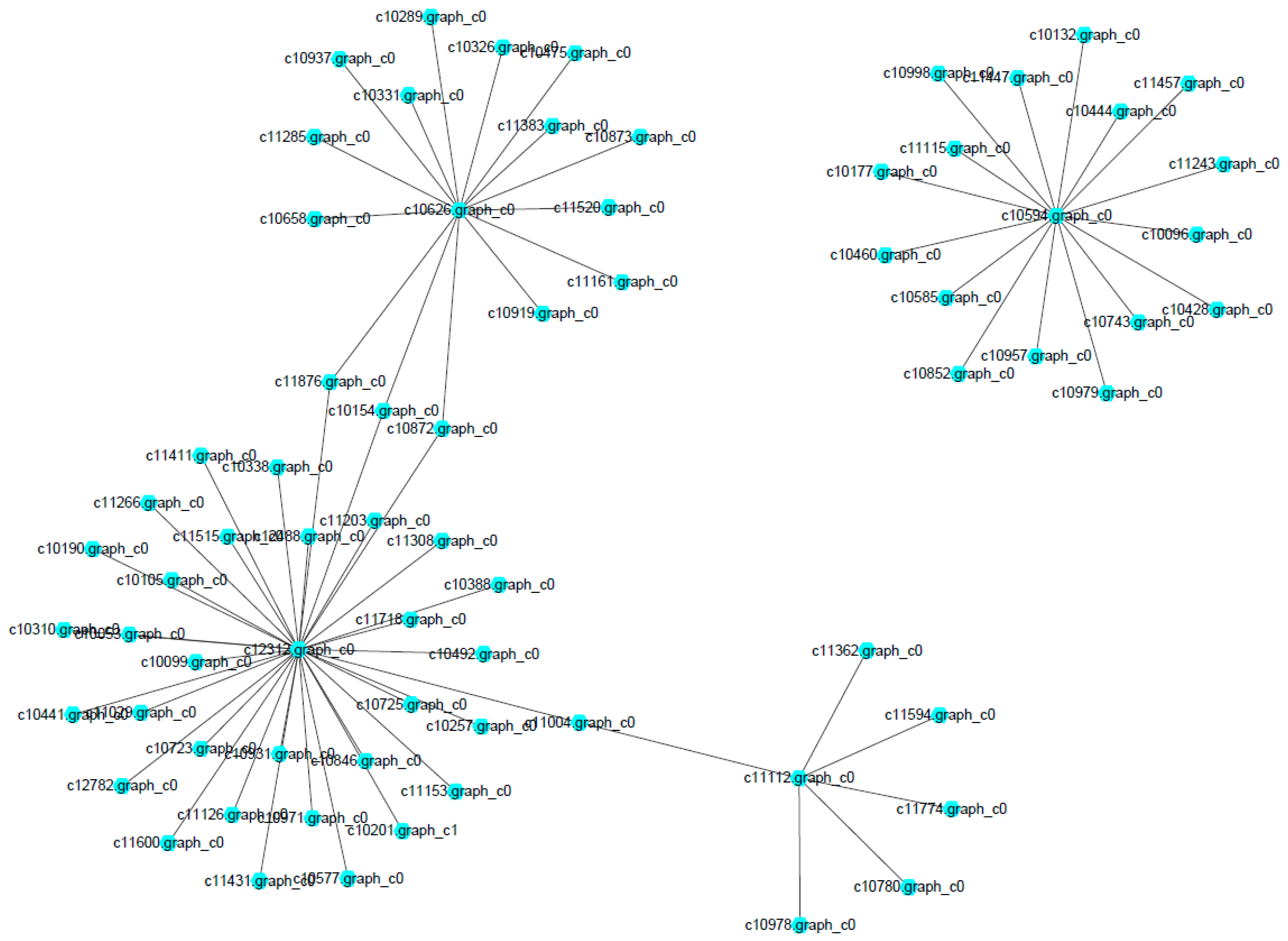
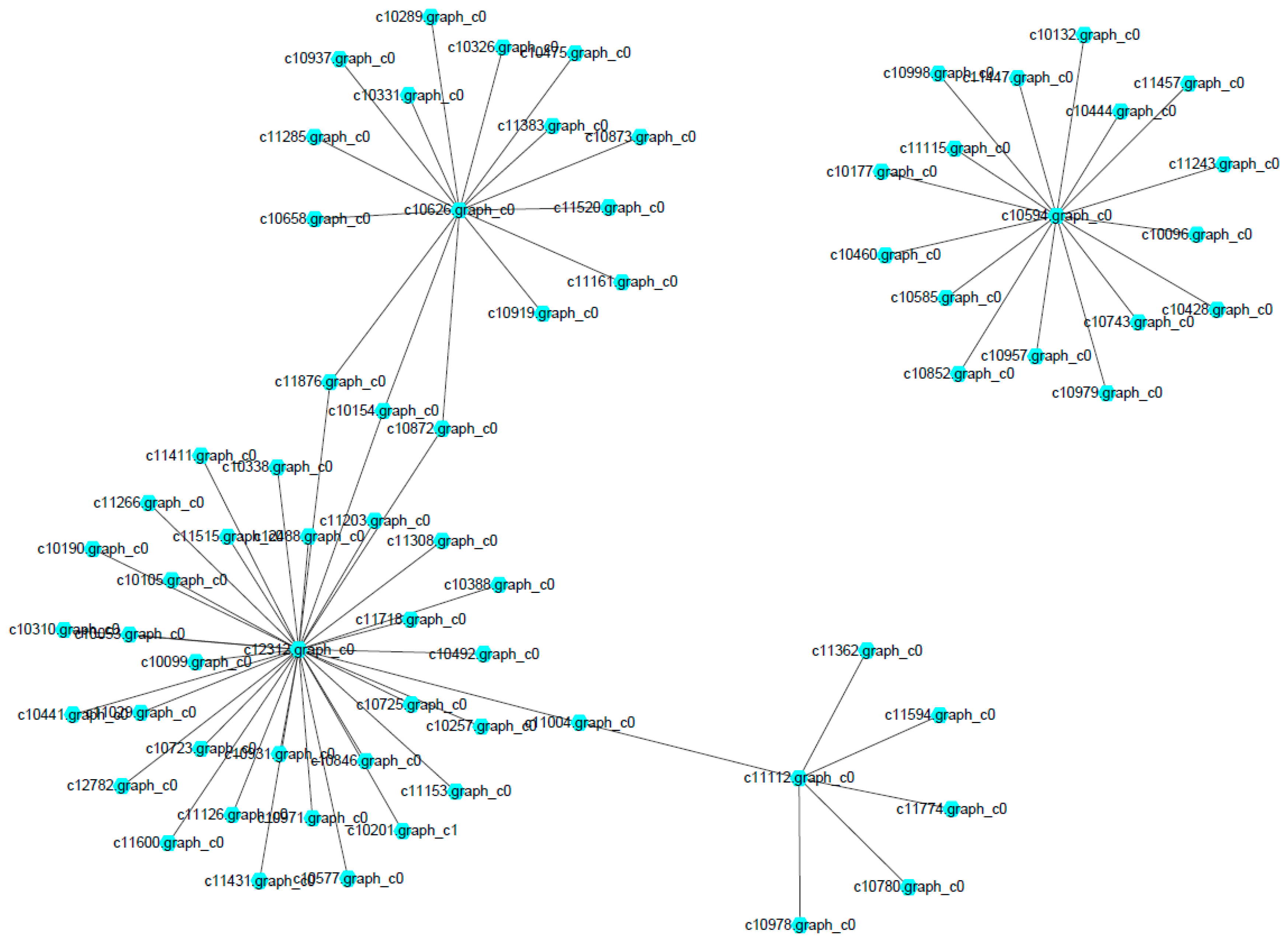
The network of protein–protein interaction involved in the MAPK signaling pathway and ribosome biogenesis in
M. roridum was constructed (
Figure 8). A total of 54 and 17 unigenes were involved in the MAPK signaling pathway and ribosome biogenesis, respectively. The two pathways played very important roles in the growth and virulence of
M. roridum. Unigene c10594 encoding U3 small nucleolar RNA-associated protein 7 is the central unigene for the network of ribosome biogenesis in
M. roridum. The Unigene 10626 encoding CDC42-GTP-binding protein of the Ras superfamily, unigene c12312 encoding ubiquitin, and unigene c11112 encoding transport protein SEC23 are the central unigenes for the network of MAPK signaling pathway in
M. roridum. 3. Discussion
M. roridum is a plant pathogenic fungus that causes great loss to economical crops. The pathogenic mechanism remains unclear. Fungi of
Collecotrichum,
Nemania,
Xylaria,
Phomopsis, and
Alternaria isolated from leaves of
Ageratina adenophora showed strong pathogenicity towards 11 kinds of native plants and four types of economical crops [
14].
Phyllosticta capitalensis, a kind of plant endophyte, displayed pathogenicity towards various plant families [
15]. The transcriptome analysis is an important approach to reveal the molecular pathology of plant pathogenic fungi.
Rhizoctonia solani is an important plant pathogen, which cause disease in many crops as well as ornamental plants and forest trees. The transcriptome analysis of
R. solani and the identification of pathogenicity-related genes revealed that genes encoding cellulose, pectin and lignin degrading enzymes and genes related to the MAPK pathway played important roles in the pathogenicity of
R. solani [
13].
In our previous work [
16], three trichothecene compounds were obtained from the fermentation liquid of
M. roridum. In this study, de novo transcriptome analysis of
M. roridum isolated from
Pogostemon cablin was performed to investigate the pathogenic mechanism and genes associated with trichothecene mycotoxin biosynthesis. A total of 4569 unigenes were assigned to 104 KEGG pathways, and 202 SSRs were identified. Our
M. roridum transcriptome results revealed that 47 unigenes, which encoded key enzymes in the trichothecene biosynthesis, including
Tri3,
Tri4,
Tri5,
Tri6,
Tri11, and
Tri12, were postulated according to the biosynthesis pathway of trichothecene mycotoxins in
Fusarium spp. qPCR analysis was employed to validate the expression levels of these unigenes. PCR and sequencing were performed to confirm the results of the transcriptome analysis. qRT-PCR analysis results revealed that
Tri5 gene showed the highest expressed level,
Tri12,
Tri6 genes took the second and the third positions, respectively. These results demonstrated the important role of
Tri5 gene in the biosynthesis of trichothecene mycotoxins. The phylogenetic analysis of genes
Tri4 and
Tri5 revealed that significant differences between the trichothecene-biosynthesis-related unigenes from
M. roridum and those from other fungal species existed. The identification of
Tri5 gene from
M. roridum confirmed that the function of
Tri5-encoding protein as a sesquiterpene synthase, meanwhile, trichothecene mytoxins are known as a kind of sesquiterpenoids, thus demonstrating the important role of
Tri5 gene in the biosynthesis of trichothecene mytoxins. The identification of
Tri4 and
Tri11 genes from
M. roridum demonstrated the function of
Tri4 and
Tri11 as monooxygenase, which played an important role in the biosynthesis of trichothecene mytoxins. The transcriptome analysis of
M. roridum and identification of the genes related to trichothecene biosynthesis establishes a foundation for the elucidation of molecular plant pathology of the
M. roridum.Eleven unigenes, which were found in the transcriptome of
M. roridum, were annotated as
Tri4 gene encoding trichodiene oxygenase. Different
Tri4 unigenes showed significant differences between each other, indicating that different kinds of trichodiene oxygenases in
M. roridum contributed to the biosynthesis of different secondary metabolites. Epiroridin acid, epiroridin E, and mytoxin B were isolated from the fermentation liquid of
M. roridum [
16] (
Figure S2), and trichodiene oxygenase was assumed to play an important role in the transformation of epiroridin E into mytoxin B. Only one unigene encoding trichodiene synthase was annotated in the
M. roridum transcriptome, and this trichodiene synthase showed the highest similarity of 26% with that from the
F. sporotrichioides, suggesting that unigene c3810 encodes a novel trichodiene synthase. The three trichothecene compounds, epiroridin E, epiroridin acid, and mytoxin B, isolated from
M. roridum showed different structures with known trichothecene mycotoxins, such as T-2 and DON toxin, indicating the possible special biosynthesis mechanism of trichothecens in
M. roridum. Moreover, the amino acid sequence of regulatory protein encoded by the
Tri6 gene from
M. roridum showed the highest similarity of 65% with that from
F. sporotrichioides, implying the possibly different regulatory mechanism of trichothecene biosynthesis in
M. roridum.The
stel2 gene involved in the MAPK signal pathway in opportunistic pathogenic fungus
Cryptococcus neoformans was reportedly capable of regulating the expression of virulence-related genes, including capsule and melanin-related genes [
17,
18], regulating the virulence of
C. neoformans. MAPK signal pathway was also reported to be involved in the process of phenotype switching and mycelium formation, which played an important role in the invasive infection of
Candida albicans [
19]. Thus, the MAPK signal pathway, especially the transport protein SEC23, is assumed to have played an important role in the pathogenicity of
M. roridum. 4. Materials and Methods
4.1. cDNA Library Construction and RNA Sequencing
The strain
M. roridum A553 (Accession No. KJ813720) was isolated from the medicinal plant
Pogostemon cablin, a kind of Guangdong medicinal plant collected from Xinyi City, Guangdong province.
M. roridum was inoculated on a PDA medium and incubated at 30 °C for 3, 5, 7, and 9 days. The total RNAs of
M. roridum at different growth stages were determined using the RNA extracting kit (Umagen, Guangzhou, China). The quantity, purity, and integrity of RNA were checked on a 1.5% (
w/
v) agarose gel and the Nanodrop-2000 spectrophotometer (GE, Fairfield, CT, USA). HPLC (High Performance Liquid Chromatography) Agilent 2100 (Agilent, Santa Clara, CA, USA) was used to detect the RIN value of the total RNA. High-quality samples (RNA ≥ 6.0) were selected for high-throughput sequencing. Then, the different RNAs were mixed with the same amount. Total RNAs in an amount of 5.0 µg were resuspended in RNase free-water and stored at −80 °C until use. The extracted RNA samples were used for the complementary DNA (cDNA) synthesis. Poly(A) mRNA was isolated using oligo-dT beads (Qiagen, Germantown, MD, USA). All mRNAs were broken into short fragments (200 nt) by adding a fragmentation buffer. First-strand cDNA was generated using random hexamer-primed reverse transcription, followed by the synthesis of the second-strand cDNA using RNase H and DNA polymerase I. The cDNA fragments were purified using a QIAquick PCR extraction kit (Qiagen, Germantown, MD, USA). These purified fragments were then washed with Elution buffer for end reparation poly(A) addition and ligated to sequencing adapters. Following the agarose gel electrophoresis and extraction of cDNA from gels, the cDNA fragments (200 ± 25 bp) were purified and enriched by PCR to construct the final cDNA library. The cDNA library was sequenced on the Illumina sequencing platform (Illumina HiSeq™ 2000, Illumina, San Diego, CA, USA) using the single-end paired-end technology in a single run. The original image processes of sequencing, base-calling, and quality value calculation were performed by the Illumina GA Pipeline (version 1.6, Illumina, San Diego, CA, USA), in which 90-bp paired-end reads were obtained [
20,
21].
4.2. Data Processing, Assembly, and Annotation
Transcriptome sequencing was conducted using the Illumina HiSeq™ 2000 sequencing platform. To assemble the entire transcriptomes of the different samples better, a paired-end (PE)100 sequencing strategy was used. All sequences were examined to ensure their accuracy. A Perl program was written to select clean reads by removing low-quality sequences (more than 50% bases with quality lower than 20 in one sequence and Q30 less than 80% were identified), reads with more than 5% N bases (bases unknown), and reads containing adaptor sequences. Subsequently, the clean reads were assembled using the Trinity software (version 1.4, Campton, NH, USA) to construct unique consensus sequences. Adaptor and low-quality sequences were trimmed. Short sequences (<50 bp) were removed using a custom Perl program (version 5.14, USA). The resulting high-quality sequences were deposited in the National Center for Biotechnology Information (NCBI) database and de novo assembled into contigs and transcripts. To reduce data redundancy, transcripts with a minimum length of 200 bp were assembled and clustered using the CLC NGS Cell software (version 1.3, Illumina, San Diego, CA, USA) under default parameters. The longest sequences in each cluster were reserved and designated as unigenes. Searches were performed using local BLASTX programs (NCBI, NIH, Bethesda, MD, USA) against sequences in the NCBI non-redundant (nr) protein database and the SWISS-PROT database (the
e value cut-off was 1 × 10
−5) [
22]. Unigenes were tentatively identified according to top hits against known sequences. The resulting unigenes were used as references for the determination of GO and COG terms and were analyzed further using the KEGG database.
4.3. KEGG Pathway Analysis and Predicted CDS
Pathway assignments were made according to KEGG mapping. Enzyme commission numbers were assigned to unique sequences that had the best BLASTX scores with cutoff e values of 1.00 × 10−5, as determined from our KEGG database search. The KEGG database is capable of analyzing gene products during the metabolism process and related gene function in the cellular processes. The sequences were mapped to the KEGG biochemical pathways according to the Enzyme Commission (EC) distribution within the pathway database.
Then, we used blast results information to extract CDS (Coding Domain Sequences) from Unigene sequences and translated them into peptide sequences. Moreover, blast results information was also used to train ESTScan [
23]. The CDS of unigenes that had no hit in BLAST were predicted by ESTScan and then translated into peptide sequences.
4.4. Quantitative Real-Time Polymerase Chain Reaction Analysis
To verify the quality of the sequences assembled in this study, unigenes related to the biosynthesis of trichothecene were validated by quantitative real-time polymerase chain reaction (qRT-PCR). qRT-PCR was performed using the Mastercycler ep realplex System (Eppendorf, Westbury, NY, USA) with 2SYBR Green mix (Fermentas, Harrington, DE, USA) according to the manufacturer’s instructions. First-strand cDNA was synthesized from 1 μg of total RNA with reverse transcriptase (Takara, Tokyo, Japan) and oligo (dT) 15 primer, and the resulting products were used as templates for qRT-PCR. The specific primers used for qRT-PCR are listed in
Table S2. The qRT-PCR thermal cycling condition for all reactions was 95 °C for 1 min 50 s, followed by 40 cycles at 95 °C for 10 s and 55 °C for 33 s. All reactions were conducted in triplicates, and the results were expressed as relative expression levels to the
GAPDH gene. The
Ct values obtained were used as the original data to calculate the relative expression level of different genes to histone gene by the 2
−ΔΔCt method [
24,
25]. Each sample was analyzed in triplicate.
4.5. Identification of Tri4, Tri5, and Tri11 Genes
The
Tri4,
Tri5, and
Tri11 genes were amplified with restriction enzymes
NdeI and
XhoI using primers listed in
Table S3, then inserted into the expression vector pET28a and transformed with
E. coli, and the recombinant vector pET28a-
Tri4, pET28a-
Tri5, and pET28a-
Tri11 were expressed in
E. coli BL21 (DE3) after being induced using 1 mM of IPTG for 3.0 h. A supernatant of 200 mL fermentation liquid containing pET28a-
Tri5 after sonication was loaded onto the Ni affinity chromatography. The target protein was eluted with 20 mM Tris–HCl (pH 8.0) containing different concentrations of imidazole. Different protein samples were identified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to the NC membrane. After being blocked by 5% non-fat milk, the membrane was incubated with mouse anti-His monoclonal antibody (Earthox, Millbrae, CA, USA) and goat anti mouse IgG antibody (Promega, Madison, WI, USA), and target bands were finally visualized using an ECL kit (Fermentas, Harrington, DE, USA) following the manufacturer’s instructions.
4.6. Enzymatic Activity Assay of Enzymes Encoded by Tri4, Tri5, and Tri11
Briefly, farnesyl pyrophosphate (FPP) substrate with a concentration of 46 µM and 50 µL Tri5 encoding-protein with a concentration of 0.6 mg/mL were added to a buffer containing 25 mM Tris-HCl (pH 7.0), 100 mM MgSO4, 10% glycerol, and 5 mM DTT. A 200 µL aliquot of the mixture was immediately sealed and incubated at 37 °C for 1 h. A solid phase extraction column (Anpel, Shanghai, China) was used to absorb sesquiterpenes produced by catalysis of Tri5 encoding-protein at 65 °C for 0.5 h. The absorbed products were loaded onto a HP 6890/5975C GC-MS apparatus (Agilent, Santa Clara, CA, USA). The GC-MS running procedure is as following: the injection port temperature is 250 °C, the initial temperature is 80 °C, increased to 180 °C with a gradient of 5 °C/min, then increased to 250 °C with a gradient of 20 °C/min. The flow rate of helium is 1.0 mL/min. The structure and abundance of the produced sesquiterpenes were analyzed.
NADPH with content of 0.40 µmol and 0.04 mg enzymes were added into substrate thioanisole with a concentration of 1.0 µmol, 50 mM Tris-HCl (pH 8.8) was added to total volume of 200 µL. The mixture was incubated at 25 °C, the increase or the decrease in absorbance at 340 nm were monitored. 1 U was defined as 1 µmol NADPH was consumed in one minute. The optimal reaction temperature and reaction pH for trichodiene oxygenase encoded by Tri4 and isotrichodermin C-15 hydroxylase encoded by Tri11 were investigated under the reaction temperatures of 15 °C, 20 °C, 25 °C, 30 °C, 35 °C and reaction pH values of 8.0, 8.4, 8.8, 9.2, and 9.6. Substrate thioanisole with different concentrations of 5.0 mM, 2.5 mM, 1.25 mM, and 0.625 mM were used to investigate the enzymatic kinetics of enzymes encoded by Tri4 and Tri11 genes.
4.7. Phylogenetic Analysis of Genes Related to Trichothecene Mycotoxin Biosynthesis
A phylogenetic tree was constructed with MEGA version 5.0 (Arizona State University, Tempe, AZ, USA) using the neighbor-joining method [
26]. NCBI and the transcriptome of
M. roridum were searched for different trichodiene synthases and trichodiene oxygenases. The functional interaction networks of proteins were integrated using the STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) database with the confidence parameter set at 0.15 threshold. The expression clusters of the
CsWRKY genes from each cultivar were analyzed using Cluster (
http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm). A diagram was drawn using Tree View (
http://jtreeview.sourceforge.net/).
4.8. Identification of SSRs
The MIcroSAtellite identification tool (MISA,
http://pgrc.inpk-gatersleben.de/misa/) was used to identify SSRs [
27]. The parameters were adjusted to identify perfect dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide motifs with a minimum of 6, 5, 5, 4, and 4 repetitions, respectively. Primer pairs were designed using the Primer 3 software and selected according to the following criteria: (1) primers with SSRs were eliminated; (2) primers aligned with unigene sequences were allowed three mismatches at the 5′ site and one mismatch at the 3′ site; (3) primers that aligned to more than one unigene were eliminated; and (4) SSRs were identified using the ssr_finder (
http://www.fresnostate.edu/ssrfinder/). We kept the products whose results from ssr_finder and MISA were the same.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}