
Neuroinflammation and Oxidative Stress in Psychosis and Psychosis Risk

{kind=link}
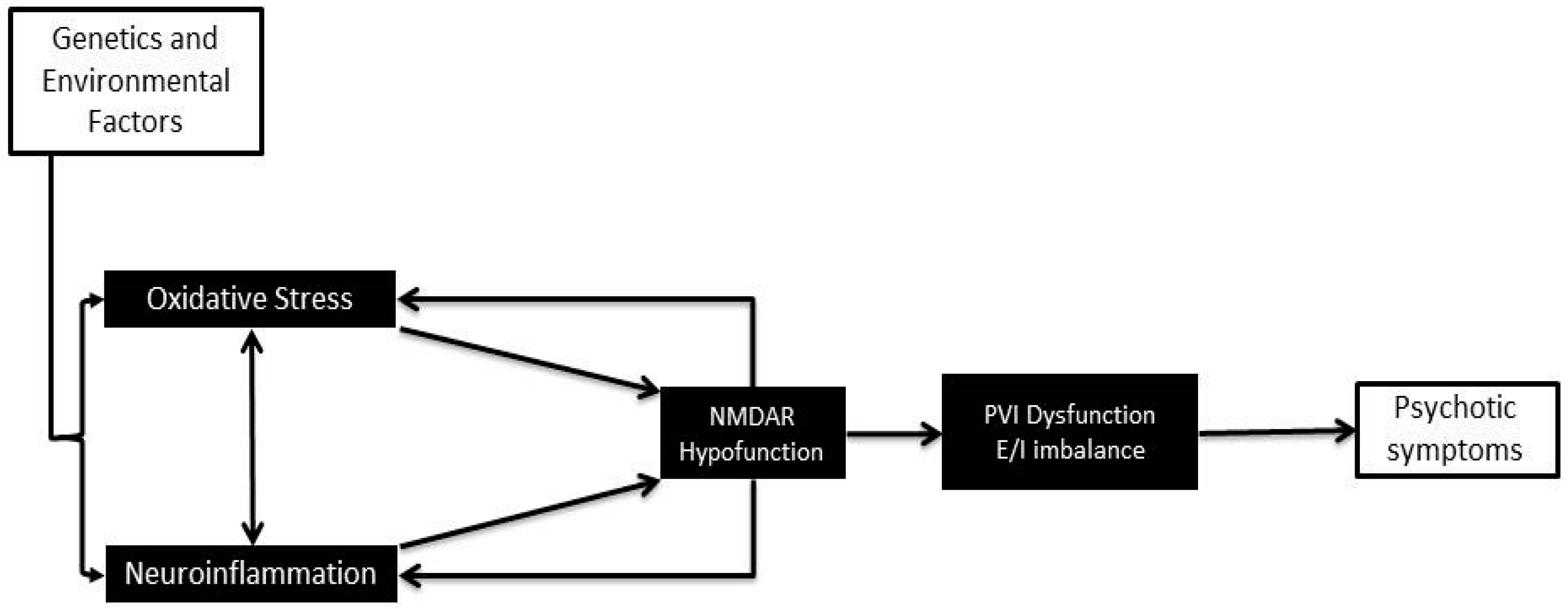
Abstract
:1. Introduction
2. NMDAR Hypofunction and Psychosis
3. Inflammation and Psychosis
3.1. Evidence from Preclinical Models
3.2. Clinical Studies
4. Oxidative Stress and Psychosis
4.1. Preclinical Studies
4.2. Clinical Studies
4.3. Connections between Inflammation and Oxidative Stress
5. Clinical Implications
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Riecher, A.; Maurer, K.; Loffler, W.; Fatkenheuer, B.; der Heiden, W.; Hafner, H. Schizophrenia—A disease of young single males? Preliminary results from an investigation on a representative cohort admitted to hospital for the first time. Eur. Arch. Psychiatry Neurol. Sci. 1989, 239, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Hafner, H.; Riecher-Rossler, A.; Hambrecht, M.; Maurer, K.; Meissner, S.; Schmidtke, A.; Fatkenheuer, B.; Loffler, W.; van der Heiden, W. Iraos: An instrument for the assessment of onset and early course of schizophrenia. Schizophr. Res. 1992, 6, 209–223. [Google Scholar] [CrossRef]
- Hafner, H.; Maurer, K.; Loffler, W.; an der Heiden, W.; Munk-Jorgensen, P.; Hambrecht, M.; Riecher-Rossler, A. The abc schizophrenia study: A preliminary overview of the results. Soc. Psychiatry Psychiatr. Epidemiol. 1998, 33, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Hafner, H.; Maurer, K.; Loffler, W.; Riecherrossler, A. The influence of age and sex on the onset and early course of schizophrenia. Br. J. Psychiatry 1993, 162, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Fusar-Poli, P.; Bonoldi, I.; Yung, A.R.; Borgwardt, S.; Kempton, M.J.; Valmaggia, L.; Barale, F.; Caverzasi, E.; McGuire, P. Predicting psychosis meta-analysis of transition outcomes in individuals at high clinical risk. Arch. Gen. Psychiatry 2012, 69, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Do, K.Q. Linking early-life nmdar hypofunction and oxidative stress in schizophrenia pathogenesis. Nat. Rev. Neurosci. 2016, 17, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Hoftman, G.D.; Datta, D.; Lewis, D.A. Layer 3 excitatory and inhibitory circuitry in the prefrontal cortex: Developmental trajectories and alterations in schizophrenia. Biol. Psychiatry 2016. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B., Jr.; Charney, D.S. Subanesthetic effects of the noncompetitive nmda antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Lancaster, E.; Martinez-Hernandez, E.; Rosenfeld, M.R.; Balice-Gordon, R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011, 10, 63–74. [Google Scholar] [CrossRef]
- Howes, O.D.; Kapur, S. The dopamine hypothesis of schizophrenia: Version III—The final common pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Grace, A.A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 2016, 17, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Modinos, G.; Allen, P.; Grace, A.A.; McGuire, P. Translating the mam model of psychosis to humans. Trends Neurosci. 2015, 38, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pinto-Duarte, A.; Sejnowski, T.J.; Behrens, M.M. How NOX2-containing NADPH oxidase affects cortical circuits in the NMDA receptor antagonist model of schizophrenia. Antioxid. Redox Signal. 2013, 18, 1444–1462. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Z.; Yang, S.F.; Xia, Y.; Johnson, K.M. Postnatal phencyclidine administration selectively reduces adult cortical parvalbumin-containing interneurons. Neuropsychopharmacology 2008, 33, 2442–2455. [Google Scholar] [CrossRef] [PubMed]
- Gulyas, A.I.; Megias, M.; Emri, Z.; Freund, T.F. Total number and ratio of excitatory and inhibitory synapses converging onto single interneurons of different types in the CA1 area of the rat hippocampus. J. Neurosci. 1999, 19, 10082–10097. [Google Scholar] [PubMed]
- Sullivan, E.M.; O’Donnell, P. Inhibitory interneurons, oxidative stress, and schizophrenia. Schizophr. Bull. 2012, 38, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Barr, C.E.; Mednick, S.A.; Munk-Jorgensen, P. Exposure to influenza epidemics during gestation and adult schizophrenia: A 40-year study. Arch. Gen. Psychiatry 1990, 47, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Mednick, S.A.; Machon, R.A.; Huttunen, M.O.; Bonett, D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch. Gen. Psychiatry 1988, 45, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Patterson, P.H. Maternal infection and schizophrenia: Implications for prevention. Schizophr. Bull. 2011, 37, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Feigenson, K.A.; Kusnecov, A.W.; Silverstein, S.M. Inflammation and the two-hit hypothesis of schizophrenia. Neurosci. Biobehav. Rev. 2014, 38, 72–93. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S. Prenatal infection as a risk factor for schizophrenia. Schizophr. Bull. 2006, 32, 200–202. [Google Scholar] [CrossRef] [PubMed]
- International Schizophrenia, C.; Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O'Donovan, M.C.; Sullivan, P.F.; Sklar, P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Ellman, L.M.; Deicken, R.F.; Vinogradov, S.; Kremen, W.S.; Poole, J.H.; Kern, D.M.; Tsai, W.Y.; Schaefer, C.A.; Brown, A.S. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr. Res. 2010, 121, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Steullet, P.; Cabungcal, J.; Monin, A.; Dwir, D.; O’Donnell, P.; Cuenod, M.; Do, K. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: A “central hub” in schizophrenia pathophysiology? Schizophr. Res. 2016, 176, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Piontkewitz, Y.; Arad, M.; Weiner, I. Abnormal trajectories of neurodevelopment and behavior following in utero insult in the rat. Biol. Psychiatry 2011, 70, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Feldon, J. To poly(I:C) or not to poly(I:C): Advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology 2012, 62, 1308–1321. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Feldon, J.; Schedlowski, M.; Yee, B.K. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci. Biobehav. Rev. 2005, 29, 913–947. [Google Scholar] [CrossRef] [PubMed]
- Bland, S.T.; Beckley, J.T.; Young, S.; Tsang, V.; Watkins, L.R.; Maier, S.F.; Bilbo, S.D. Enduring consequences of early-life infection on glial and neural cell genesis within cognitive regions of the brain. Brain Behav. Immun. 2010, 24, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Juckel, G.; Manitz, M.P.; Brune, M.; Friebe, A.; Heneka, M.T.; Wolf, R.J. Microglial activation in a neuroinflammational animal model of schizophrenia—A pilot study. Schizophr. Res. 2011, 131, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Ishibashi, H.; Hashimoto, K.; Nakanishi, H. Potentiation of the NMDA receptor-mediated responses through the activation of the glycine site by microglia secreting soluble factors. Glia 2006, 53, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Takaki, J.; Fujimori, K.; Miura, M.; Suzuki, T.; Sekino, Y.; Sato, K. l-glutamate released from activated microglia downregulates astrocytic l-glutamate transporter expression in neuroinflammation: The “collusion” hypothesis for increased extracellular l-glutamate concentration in neuroinflammation. J. Neuroin. 2012, 9, 275. [Google Scholar] [CrossRef] [PubMed]
- García-Bueno, B.; Bioque, M.; Mac-Dowell, K.S.; Barcones, M.F.; Martínez-Cengotitabengoa, M.; Pina-Camacho, L.; Rodríguez-Jiménez, R.; Sáiz, P.A.; Castro, C.; Lafuente, A. Pro-/anti-inflammatory dysregulation in patients with first episode of psychosis: Toward an integrative inflammatory hypothesis of schizophrenia. Schizophr. Bull. 2014, 40, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, F.; Stallings, C.; Origoni, A.; Schroeder, J.; Katsafanas, E.; Schweinfurth, L.; Savage, C.; Khushalani, S.; Yolken, R. Inflammatory markers in recent onset psychosis and chronic schizophrenia. Schizophr. Bull. 2016, 42, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.O.; Jeffries, C.D.; Addington, J.; Bearden, C.E.; Cadenhead, K.S.; Cannon, T.D.; Cornblatt, B.A.; Mathalon, D.H.; McGlashan, T.H.; Seidman, L.J.; et al. Towards a psychosis risk blood diagnostic for persons experiencing high-risk symptoms: Preliminary results from the napls project. Schizophr. Bull. 2015, 41, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.; Rapaport, M.; Miller, B. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cengotitabengoa, M.; Micó, J.A.; Arango, C.; Castro-Fornieles, J.; Graell, M.; Payá, B.; Leza, J.C.; Zorrilla, I.; Parellada, M.; López, M.P. Basal low antioxidant capacity correlates with cognitive deficits in early onset psychosis: A 2-year follow-up study. Schizophr. Res. 2014, 156, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Bosmans, E.; Ranjan, R.; Vandoolaeghe, E.; Meltzer, H.Y.; de Ley, M.; Berghmans, R.; Stans, G.; Desnyder, R. Lower plasma CC16, a natural anti-inflammatory protein, and increased plasma interleukin-1 receptor antagonist in schizophrenia: Effects of antipsychotic drugs. Schizophr. Res. 1996, 21, 39–50. [Google Scholar] [CrossRef]
- Maes, M.; Bosmans, E.; Kenis, G.; de Jong, R.; Smith, R.S.; Meltzer, H.Y. In vivo immunomodulatory effects of clozapine in schizophrenia. Schizophr. Res. 1997, 26, 221–225. [Google Scholar] [CrossRef]
- Carter, C.S.; Bullmore, E.T.; Harrison, P. Is there a flame in the brain in psychosis? Biol. Psychiatry 2014, 75, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Kenk, M.; Selvanathan, T.; Rao, N.; Suridjan, I.; Rusjan, P.; Remington, G.; Meyer, J.H.; Wilson, A.A.; Houle, S.; Mizrahi, R. Imaging neuroinflammation in gray and white matter in schizophrenia: An in vivo pet study with [18F]-FEPPA. Schizophr. Bull. 2015, 41, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, O.; Kubicki, M.; Shenton, M.E. In vivo imaging of neuroinflammation in schizophrenia. Schizophr. Res. 2016, 173, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Hafizi, S.; Tseng, H.H.; Rao, N.; Selvanathan, T.; Kenk, M.; Bazinet, R.P.; Suridjan, I.; Wilson, A.A.; Meyer, J.H.; Remington, G.; et al. Imaging microglial activation in untreated first-episode psychosis: A pet study with [18F]FEPPA. Am. J. Psychiatry 2017, 174, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, J.; Wang, Y.; Ambinder, E.; Ward, R.; Minn, I.; Vranesic, M.; Kim, P.; Ford, C.; Higgs, C.; Hayes, L. In vivo markers of inflammatory response in recent-onset schizophrenia: A combined study using [11C]DPA-713 PET and analysis of CSF and plasma. Transl. Psychiatry 2016, 6, e777. [Google Scholar] [CrossRef] [PubMed]
- Doorduin, J.; de Vries, E.F.; Willemsen, A.T.; de Groot, J.C.; Dierckx, R.A.; Klein, H.C. Neuroinflammation in schizophrenia-related psychosis: A pet study. J. Nucl. Med. 2009, 50, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Van Berckel, B.N.; Bossong, M.G.; Boellaard, R.; Kloet, R.; Schuitemaker, A.; Caspers, E.; Luurtsema, G.; Windhorst, A.D.; Cahn, W.; Lammertsma, A.A. Microglia activation in recent-onset schizophrenia: A quantitative (r)-[11C] PK11195 positron emission tomography study. Biol. Psychiatry 2008, 64, 820–822. [Google Scholar] [CrossRef] [PubMed]
- Takano, A.; Arakawa, R.; Ito, H.; Tateno, A.; Takahashi, H.; Matsumoto, R.; Okubo, Y.; Suhara, T. Peripheral benzodiazepine receptors in patients with chronic schizophrenia: A pet study with [11C]DAA1106. Int. J. Neuropsychopharmacol. 2010, 13, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.E.; Hinz, R.; Drake, R.J.; Gregory, C.J.; Conen, S.; Matthews, J.C.; Anton-Rodriguez, J.M.; Gerhard, A.; Talbot, P.S. In vivo imaging of brain microglial activity in antipsychotic-free and medicated schizophrenia: A [11C](r)-PK11195 positron emission tomography study. Mol. Psychiatry 2016, 21, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Van der Doef, T.F.; de Witte, L.D.; Sutterland, A.L.; Jobse, E.; Yaqub, M.; Boellaard, R.; de Haan, L.; Eriksson, J.; Lammertsma, A.A.; Kahn, R.S. In vivo (r)-[11C] PK11195 pet imaging of 18 kDa translocator protein in recent onset psychosis. NPJ Schizophr. 2016, 2, 16031. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, P.S.; Selvaraj, S.; Veronese, M.; Rizzo, G.; Bertoldo, A.; Owen, D.R.; Bloomfield, M.A.; Bonoldi, I.; Kalk, N.; Turkheimer, F. Microglial activity in people at ULTRA high risk of psychosis and in schizophrenia: An [11C] PBR28 pet brain imaging study. Am. J. Psychiatry 2015, 173, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Narendran, R.; Frankle, W.G. Comment on analyses and conclusions of “microglial activity in people at ULTRA high risk of psychosis and in schizophrenia: An [11C] PBR28 pet brain imaging study”. Am. J. Psychiatry 2016, 173, 536–537. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Flatow, J.; Buckley, P.; Miller, B.J. Meta-analysis of oxidative stress in schizophrenia. Biol. Psychiatry 2013, 74, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Machado, A.K.; Pan, A.Y.; da Silva, T.M.; Duong, A.; Andreazza, A.C. Upstream pathways controlling mitochondrial function in major psychosis a focus on bipolar disorder. Can. J. Psychiatry 2016, 61, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.T.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.P.; Rodriguiz, R.M.; Mork, A.; Wetsel, W.C. Monoaminergic dysregulation in glutathione-deficient mice: Possible relevance to schizophrenia? Neuroscience 2005, 132, 1055–1072. [Google Scholar] [CrossRef] [PubMed]
- Steullet, P.; Neijt, H.C.; Cuenod, M.; Do, K.Q. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: Relevance to schizophrenia. Neuroscience 2006, 137, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Morishita, H.; Cabungcal, J.H.; Chen, Y.; Do, K.Q.; Hensch, T.K. Prolonged period of cortical plasticity upon redox dysregulation in fast-spiking interneurons. Biol. Psychiatry 2015, 78, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.H.; Steullet, P.; Kraftsik, R.; Cuenod, M.; Do, K.Q. Early-life insults impair parvalbumin interneurons via oxidative stress: Reversal by N-acetylcysteine. Biol. Psychiatry 2013, 73, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Steullet, P.; Cabungcal, J.H.; Kulak, A.; Kraftsik, R.; Chen, Y.; Dalton, T.P.; Cuenod, M.; Do, K.Q. Redox dysregulation affects the ventral but not dorsal hippocampus: Impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J. Neurosci. 2010, 30, 2547–2558. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.H.; Counotte, D.S.; Lewis, E.M.; Tejeda, H.A.; Piantadosi, P.; Pollock, C.; Calhoon, G.G.; Sullivan, E.M.; Presgraves, E.; Kil, J.; et al. Juvenile antioxidant treatment prevents adult deficits in a developmental model of schizophrenia. Neuron 2014, 83, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Otte, D.M.; Sommersberg, B.; Kudin, A.; Guerrero, C.; Albayram, O.; Filiou, M.D.; Frisch, P.; Yilmaz, O.; Drews, E.; Turck, C.W.; et al. N-acetyl cysteine treatment rescues cognitive deficits induced by mitochondrial dysfunction in g72/g30 transgenic mice. Neuropsychopharmacology 2011, 36, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.H.; Steullet, P.; Morishita, H.; Kraftsik, R.; Cuenod, M.; Hensch, T.K.; Do, K.Q. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 9130–9135. [Google Scholar] [CrossRef] [PubMed]
- Papadia, S.; Soriano, F.X.; Leveille, F.; Martel, M.A.; Dakin, K.A.; Hansen, H.H.; Kaindl, A.; Sifringer, M.; Fowler, J.; Stefovska, V.; et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 2008, 11, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Bell, K.F.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.E. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 2015, 6, 6761. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Choi, Y.B.; Takahashi, H.; Zhang, D.; Li, W.; Godzik, A.; Bankston, L.A. Cysteine regulation of protein function—as exemplified by NMDA-receptor modulation. Trends Neurosci. 2002, 25, 474–480. [Google Scholar] [CrossRef]
- Behrens, M.M.; Ali, S.S.; Dao, D.N.; Lucero, J.; Shekhtman, G.; Quick, K.L.; Dugan, L.L. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 2007, 318, 1645–1647. [Google Scholar] [CrossRef] [PubMed]
- Gawryluk, J.W.; Wang, J.F.; Andreazza, A.C.; Shao, L.; Young, L.T. Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int. J. Neuropsychopharmacol. 2011, 14, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Do, K.Q.; Trabesinger, A.H.; Kirsten-Kruger, M.; Lauer, C.J.; Dydak, U.; Hell, D.; Holsboer, F.; Boesiger, P.; Cuenod, M. Schizophrenia: Glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci. 2000, 12, 3721–3728. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Q.; Kosten, T.R.; Zhang, X.Y. Free radicals, antioxidant defense systems, and schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 46, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Micó, J.A.; Rojas-Corrales, M.O.; Gibert-Rahola, J.; Parellada, M.; Moreno, D.; Fraguas, D.; Graell, M.; Gil, J.; Irazusta, J.; Castro-Fornieles, J. Reduced antioxidant defense in early onset first-episode psychosis: A case-control study. BMC Psychiatry 2011, 11, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraguas, D.; Gonzalez-Pinto, A.; Micó, J.A.; Reig, S.; Parellada, M.; Martínez-Cengotitabengoa, M.; Castro-Fornieles, J.; Rapado-Castro, M.; Baeza, I.; Janssen, J. Decreased glutathione levels predict loss of brain volume in children and adolescents with first-episode psychosis in a two-year longitudinal study. Schizophr. Res. 2012, 137, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.K.; Reddy, R.D.; van Kammen, D.P. Human plasma glutathione peroxidase and symptom severity in schizophrenia. Biol. Psychiatry 1999, 45, 1512–1515. [Google Scholar] [CrossRef]
- Lavoie, S.; Murray, M.M.; Deppen, P.; Knyazeva, M.G.; Berk, M.; Boulat, O.; Bovet, P.; Bush, A.I.; Conus, P.; Copolov, D.; et al. Glutathione precursor, N-acetyl-cysteine, improves mismatch negativity in schizophrenia patients. Neuropsychopharmacology 2008, 33, 2187–2199. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, D.; Hashimoto, K. Magnetic resonance spectroscopy study of the antioxidant defense system in schizophrenia. Antioxid Redox Signal. 2011, 15, 2057–2065. [Google Scholar] [CrossRef] [PubMed]
- Tosic, M.; Ott, J.; Barral, S.; Bovet, P.; Deppen, P.; Gheorghita, F.; Matthey, M.L.; Parnas, J.; Preisig, M.; Saraga, M.; et al. Schizophrenia and oxidative stress: Glutamate cysteine ligase modifier as a susceptibility gene. Am. J. Hum. Genet. 2006, 79, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Serritella, A.V.; Sawa, A.; Sedlak, T.W. Implications for reactive oxygen species in schizophrenia pathogenesis. Schizophr. Res. 2016, 176, 52–71. [Google Scholar] [CrossRef] [PubMed]
- Lett, T.A.; Chakavarty, M.M.; Felsky, D.; Brandl, E.J.; Tiwari, A.K.; Gonçalves, V.F.; Rajji, T.K.; Daskalakis, Z.J.; Meltzer, H.Y.; Lieberman, J.A. The genome-wide supported microrna-137 variant predicts phenotypic heterogeneity within schizophrenia. Mol. Psychiatry 2013, 18, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-κB. G Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Bitanihirwe, B.K.; Woo, T.U. Oxidative stress in schizophrenia: An integrated approach. Neurosci. Biobehav. Rev. 2011, 35, 878–893. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Qu, J.; Shen, X. NF-κB/p65 antagonizes NRF2-are pathway by depriving CBP from NRF2 and facilitating recruitment of HDAC3 to MAFK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Lante, F.; Meunier, J.; Guiramand, J.; de Jesus Ferreira, M.C.; Cambonie, G.; Aimar, R.; Cohen-Solal, C.; Maurice, T.; Vignes, M.; Barbanel, G. Late N-acetylcysteine treatment prevents the deficits induced in the offspring of dams exposed to an immune stress during gestation. Hippocampus 2008, 18, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Lante, F.; Meunier, J.; Guiramand, J.; Maurice, T.; Cavalier, M.; de Jesus Ferreira, M.C.; Aimar, R.; Cohen-Solal, C.; Vignes, M.; Barbanel, G. Neurodevelopmental damage after prenatal infection: Role of oxidative stress in the fetal brain. Free Radic. Biol. Med. 2007, 42, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.; Conus, P.; Eide, P.; Mass, R.; Karow, A.; Moritz, S.; Golks, D.; Naber, D. Impact of present and past antipsychotic side effects on attitude toward typical antipsychotic treatment and adherence. Eur. Psychiatry 2004, 19, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Milev, P.; Ho, B.C.; Arndt, S.; Andreasen, N.C. Predictive values of neurocognition and negative symptoms on functional outcome in schizophrenia: A longitudinal first-episode study with 7-year follow-up. Am. J. Psychiatry 2005, 162, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Valenstein, M.; Ganoczy, D.; McCarthy, J.F.; Kim, H.M.; Lee, T.A.; Blow, F.C. Antipsychotic adherence over time among patients receiving treatment for schizophrenia: A retrospective review. J. Clin. Psychiatry 2006, 67, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- García, S.; Martínez-Cengotitabengoa, M.; López-Zurbano, S.; Zorrilla, I.; López, P.; Vieta, E.; González-Pinto, A. Adherence to antipsychotic medication in bipolar disorder and schizophrenic patients: A systematic review. J. Clin. Psychopharmacol. 2016, 36, 355. [Google Scholar] [CrossRef] [PubMed]
- McGorry, P.D.; Killackey, E.; Yung, A. Early intervention in psychosis: Concepts, evidence and future directions. World Psychiatry 2008, 7, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Addington, J.; Cadenhead, K.S.; Cornblatt, B.A.; Mathalon, D.H.; McGlashan, T.H.; Perkins, D.O.; Seidman, L.J.; Tsuang, M.T.; Walker, E.F.; Woods, S.W.; et al. North american prodrome longitudinal study (NAPLS 2): Overview and recruitment. Schizophr. Res. 2012, 142, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Tsuang, M.T.; Van Os, J.; Tandon, R.; Barch, D.M.; Bustillo, J.; Gaebel, W.; Gur, R.E.; Heckers, S.; Malaspina, D.; Owen, M.J.; et al. Attenuated psychosis syndrome in DSM-5. Schizophr. Res. 2013, 150, 31–35. [Google Scholar] [CrossRef] [PubMed]
- McGlashan, T.H.; Zipursky, R.B.; Perkins, D.; Addington, J.; Miller, T.; Woods, S.W.; Hawkins, K.A.; Hoffman, R.E.; Preda, A.; Epstein, I.; et al. Randomized, double-blind trial of olanzapine versus placebo in patients prodromally symptomatic for psychosis. Am. J. Psychiatry 2006, 163, 790–799. [Google Scholar] [CrossRef] [PubMed]
- McGorry, P.D.; Nelson, B.; Phillips, L.J.; Yuen, H.P.; Francey, S.M.; Thampi, A.; Berger, G.E.; Amminger, G.P.; Simmons, M.B.; Kelly, D.; et al. Randomized controlled trial of interventions for young people at ULTRA-high risk of psychosis: Twelve-month outcome. J. Clin. Psychiatry 2013, 74, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Phillips, L.J.; McGorry, P.D.; Yuen, H.P.; Ward, J.; Donovan, K.; Kelly, D.; Francey, S.M.; Yung, A.R. Medium term follow-up of a randomized controlled trial of interventions for young people at ULTRA high risk of psychosis. Schizophr. Res. 2007, 96, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Addington, J.; Epstein, I.; Liu, L.; French, P.; Boydell, K.M.; Zipursky, R.B. A randomized controlled trial of cognitive behavioral therapy for individuals at clinical high risk of psychosis. Schizophr. Res. 2011, 125, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Bechdolf, A.; Wagner, M.; Ruhrmann, S.; Harrigan, S.; Putzfeld, V.; Pukrop, R.; Brockhaus-Dumke, A.; Berning, J.; Janssen, B.; Decker, P.; et al. Preventing progression to first-episode psychosis in early initial prodromal states. Br. J. Psychiatry 2012, 200, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.P.; French, P.; Parker, S.; Roberts, M.; Stevens, H.; Bentall, R.P.; Lewis, S.W. Three-year follow-up of a randomized controlled trial of cognitive therapy for the prevention of psychosis in people at ultrahigh risk. Schizophr. Bull. 2007, 33, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.P.; French, P.; Stewart, S.L.; Birchwood, M.; Fowler, D.; Gumley, A.I.; Jones, P.B.; Bentall, R.P.; Lewis, S.W.; Murray, G.K. Early detection and intervention evaluation for people at risk of psychosis: Multisite randomised controlled trial. BMJ 2012, 344. [Google Scholar] [CrossRef] [PubMed]
- van der Gaag, M.; Nieman, D.H.; Rietdijk, J.; Dragt, S.; Ising, H.K.; Klaassen, R.M.; Koeter, M.; Cuijpers, P.; Wunderink, L.; Linszen, D.H. Cognitive behavioral therapy for subjects at ultrahigh risk for developing psychosis: A randomized controlled clinical trial. Schizophr. Bull. 2012, 38, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- McGorry, P.D.; Nelson, B.; Markulev, C.; Yuen, H.P.; Schäfer, M.R.; Mossaheb, N.; Schlögelhofer, M.; Smesny, S.; Hickie, I.B.; Berger, G.E. Effect of ω-3 polyunsaturated fatty acids in young people at ultrahigh risk for psychotic disorders: The neurapro randomized clinical trial. JAMA Psychiatry 2017, 74, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J.; Andrieux, A.; Bartzokis, G.; Cadenhead, K.; Dazzan, P.; Fusar-Poli, P.; Gallinat, J.; Giedd, J.; Grayson, D.R.; Heinrichs, M.; et al. Altering the course of schizophrenia: Progress and perspectives. Nat. Rev. Drug Discov. 2016, 15, 485–515. [Google Scholar] [CrossRef] [PubMed]
- Amminger, G.P.; Schafer, M.R.; Schlogelhofer, M.; Klier, C.M.; McGorry, P.D. Longer-term outcome in the prevention of psychotic disorders by the vienna omega-3 study. Nat. Commun. 2015, 6, 7934. [Google Scholar] [CrossRef] [PubMed]
- Schlogelhofer, M.; Amminger, G.P.; Schaefer, M.R.; Fusar-Poli, P.; Smesny, S.; McGorry, P.; Berger, G.; Mossaheb, N. Polyunsaturated fatty acids in emerging psychosis: A safer alternative? Early Interv. Psychiatry 2014, 8, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Bondi, C.O.; Taha, A.Y.; Tock, J.L.; Totah, N.K.; Cheon, Y.; Torres, G.E.; Rapoport, S.I.; Moghaddam, B. Adolescent behavior and dopamine availability are uniquely sensitive to dietary ω-3 fatty acid deficiency. Biol. Psychiatry 2014, 75, 38–46. [Google Scholar] [CrossRef] [PubMed]
- English, J.A.; Harauma, A.; Focking, M.; Wynne, K.; Scaife, C.; Cagney, G.; Moriguchi, T.; Cotter, D.R. Omega-3 fatty acid deficiency disrupts endocytosis, neuritogenesis, and mitochondrial protein pathways in the mouse hippocampus. Front. Genet. 2013, 4, 208. [Google Scholar] [CrossRef] [PubMed]
- Amminger, G.P.; Schafer, M.R.; Papageorgiou, K.; Klier, C.M.; Cotton, S.M.; Harrigan, S.M.; Mackinnon, A.; McGorry, P.D.; Berger, G.E. Long-chain ω-3 fatty acids for indicated prevention of psychotic disorders: A randomized, placebo-controlled trial. Arch. Gen. Psychiatry 2010, 67, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [PubMed]
- Fillman, S.G.; Weickert, T.W.; Lenroot, R.K.; Catts, S.V.; Bruggemann, J.M.; Catts, V.S.; Weickert, C.S. Elevated peripheral cytokines characterize a subgroup of people with schizophrenia displaying poor verbal fluency and reduced broca’s area volume. Mol. Psychiatry 2016, 21, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Klosterkötte, J. The clinical staging and the endophenotype approach as an integrative future perspective for psychiatry. World Psychiatry 2008, 7, 159–160. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barron, H.; Hafizi, S.; Andreazza, A.C.; Mizrahi, R. Neuroinflammation and Oxidative Stress in Psychosis and Psychosis Risk. Int. J. Mol. Sci. 2017, 18, 651. https://doi.org/10.3390/ijms18030651
Barron H, Hafizi S, Andreazza AC, Mizrahi R. Neuroinflammation and Oxidative Stress in Psychosis and Psychosis Risk. International Journal of Molecular Sciences. 2017; 18(3):651. https://doi.org/10.3390/ijms18030651
Chicago/Turabian StyleBarron, Henry, Sina Hafizi, Ana C. Andreazza, and Romina Mizrahi. 2017. "Neuroinflammation and Oxidative Stress in Psychosis and Psychosis Risk" International Journal of Molecular Sciences 18, no. 3: 651. https://doi.org/10.3390/ijms18030651
APA StyleBarron, H., Hafizi, S., Andreazza, A. C., & Mizrahi, R. (2017). Neuroinflammation and Oxidative Stress in Psychosis and Psychosis Risk. International Journal of Molecular Sciences, 18(3), 651. https://doi.org/10.3390/ijms18030651