MicroRNA-210 Suppresses Junction Proteins and Disrupts Blood-Brain Barrier Integrity in Neonatal Rat Hypoxic-Ischemic Brain Injury

Abstract
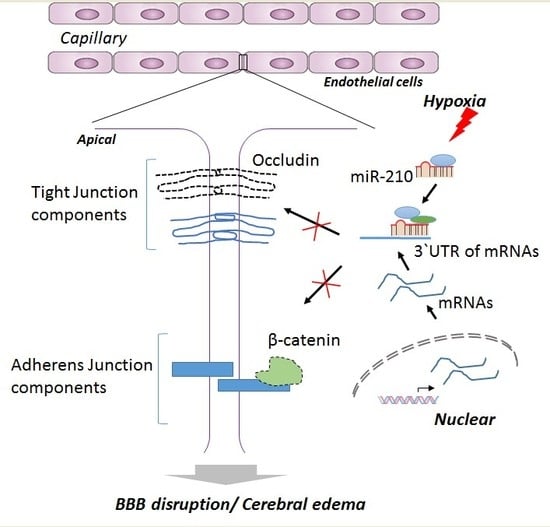
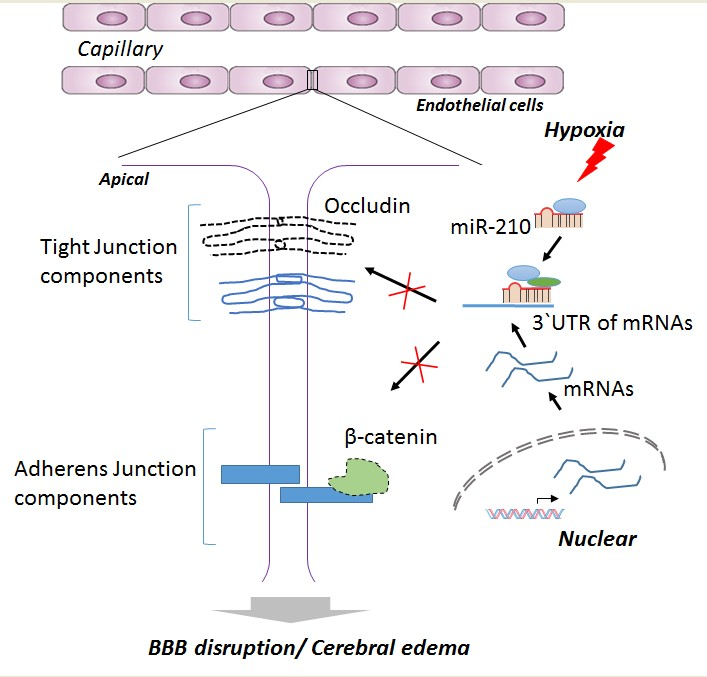
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
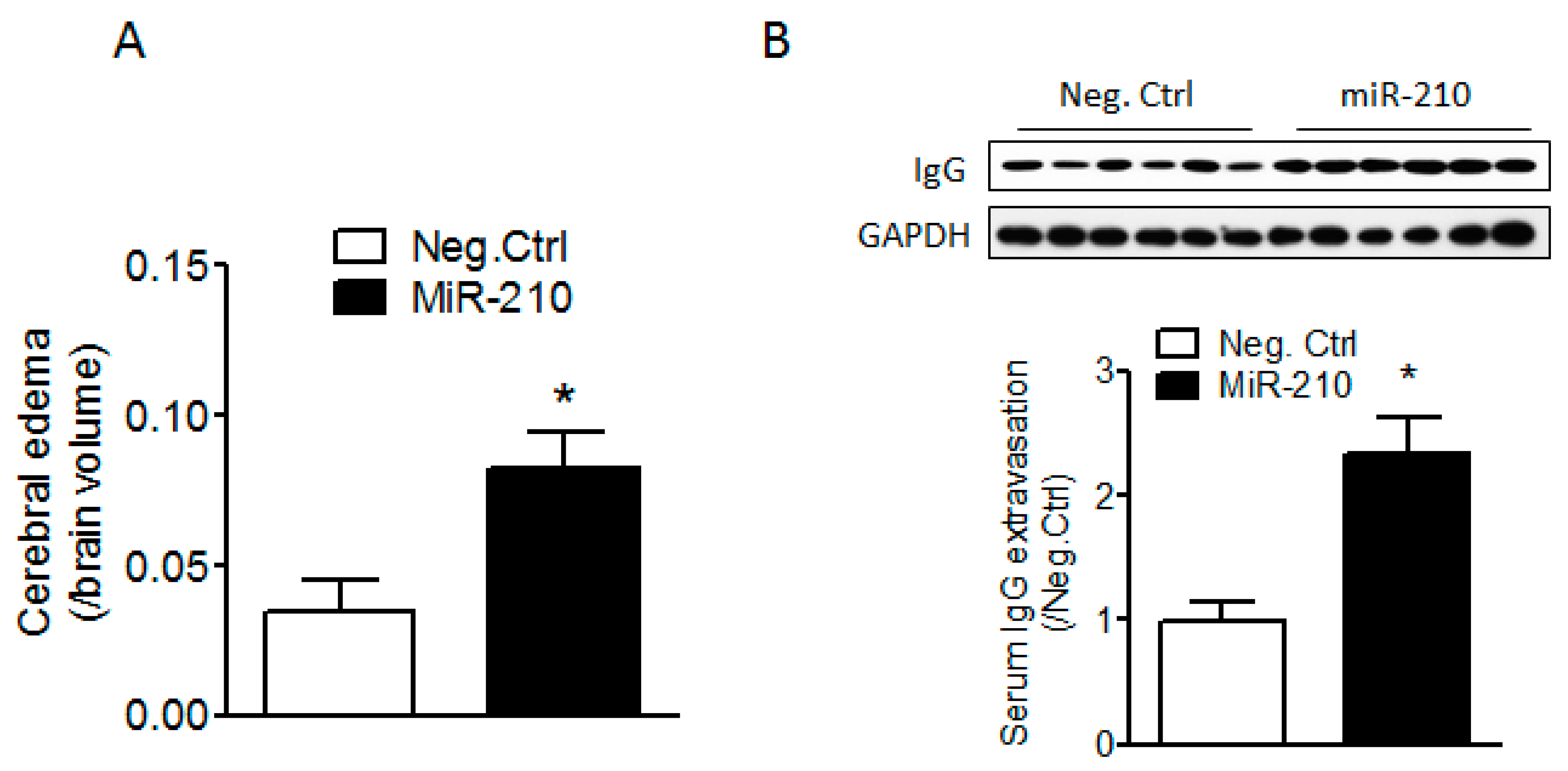
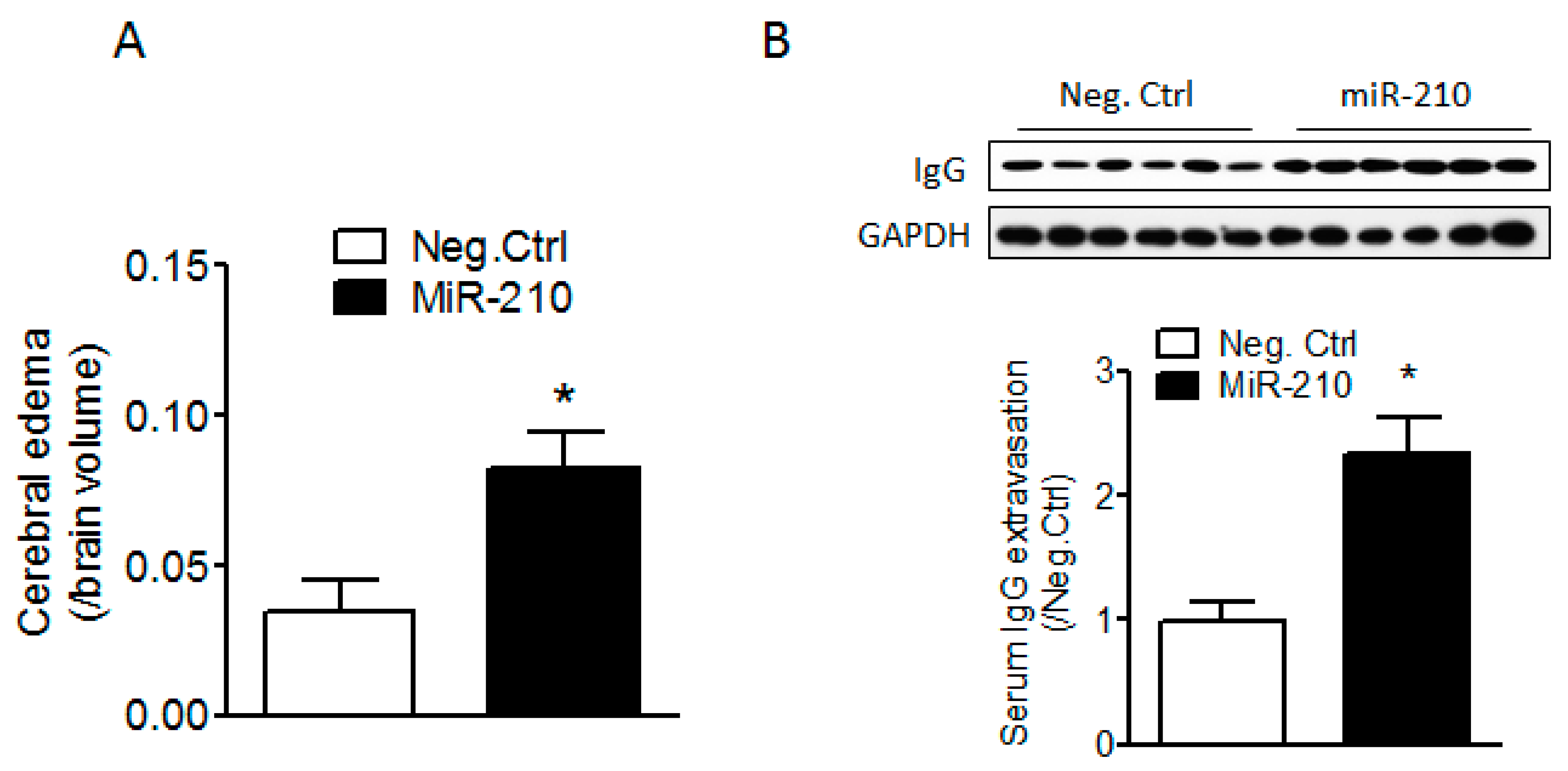
2.1. miR-210 Mimic Exacerbates Cerebral Edema and Immunoglobulin G (IgG) Leakage Following Neonatal HI Insult
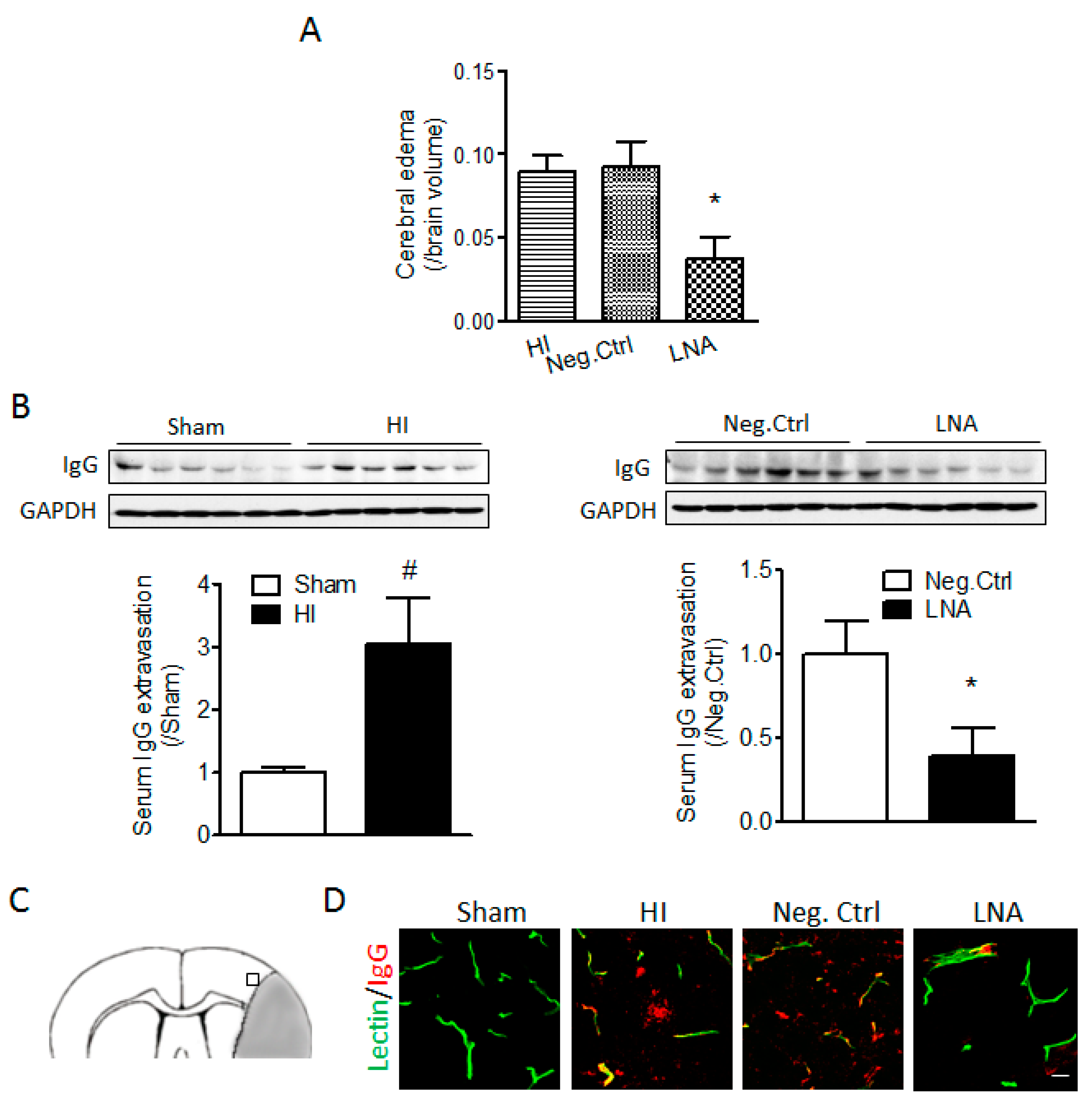
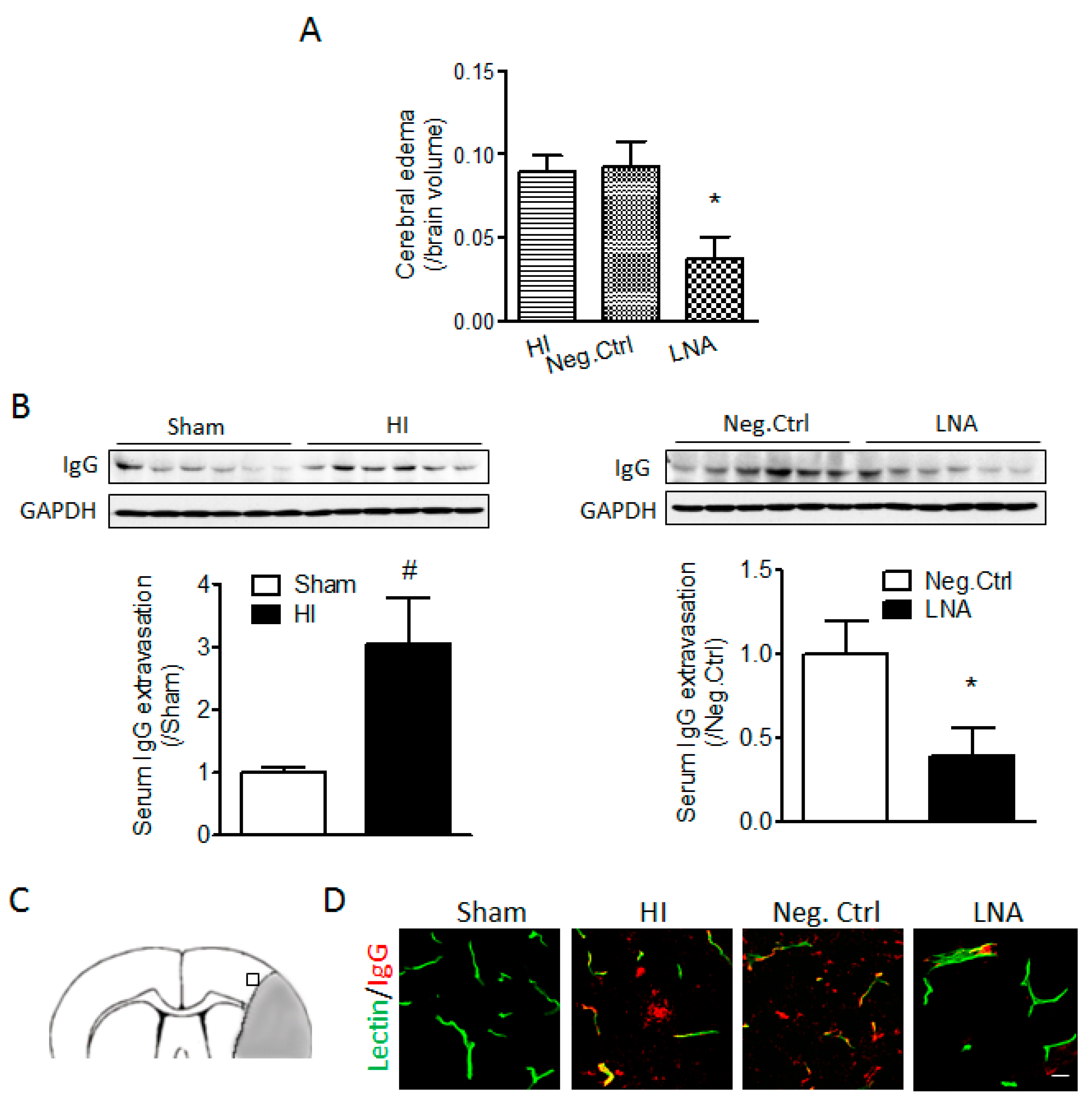
2.2. miR-210 Inhibition Reduces Cerebral Edema and Serum IgG Leakage Following Neonatal HI
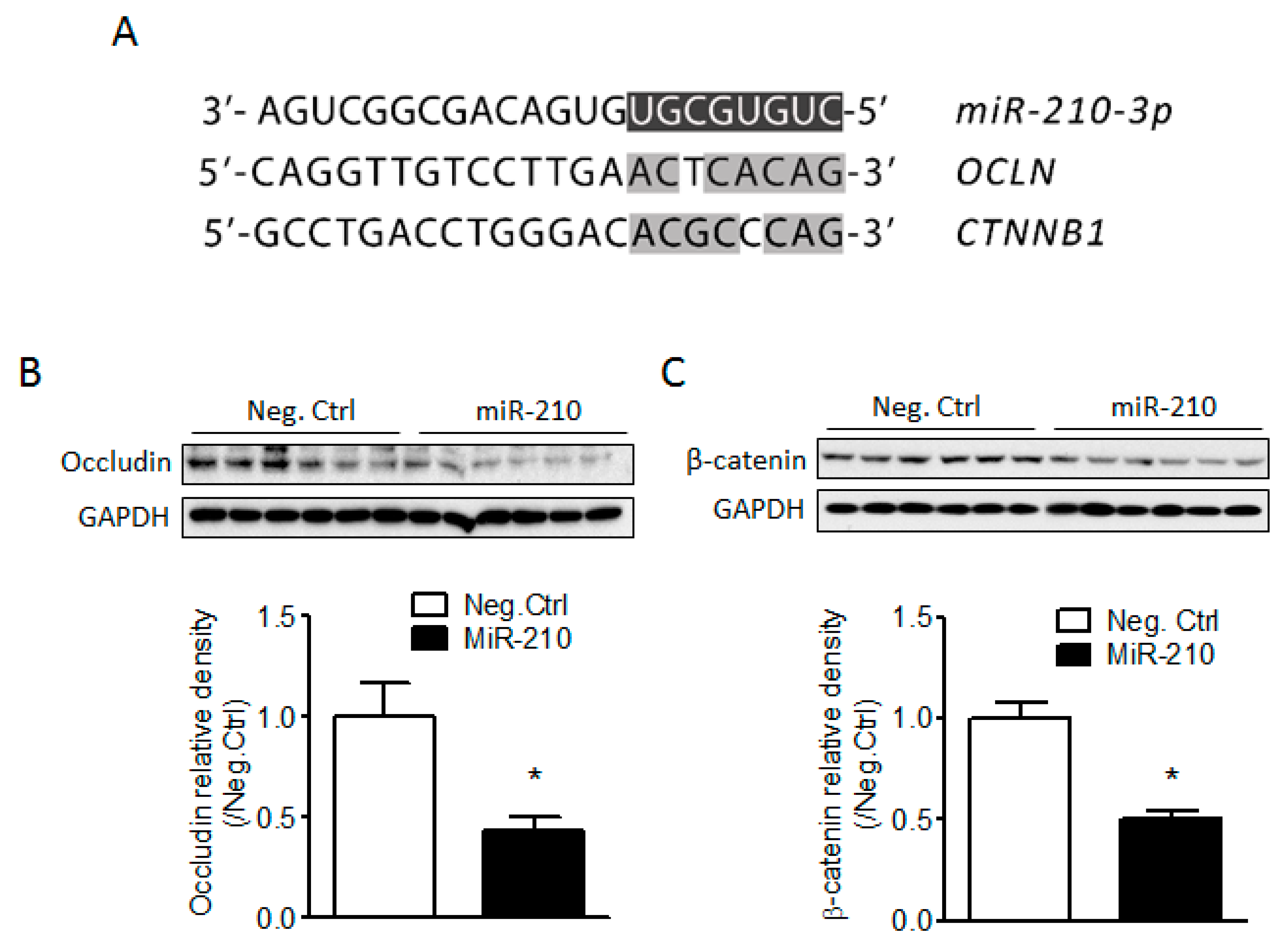
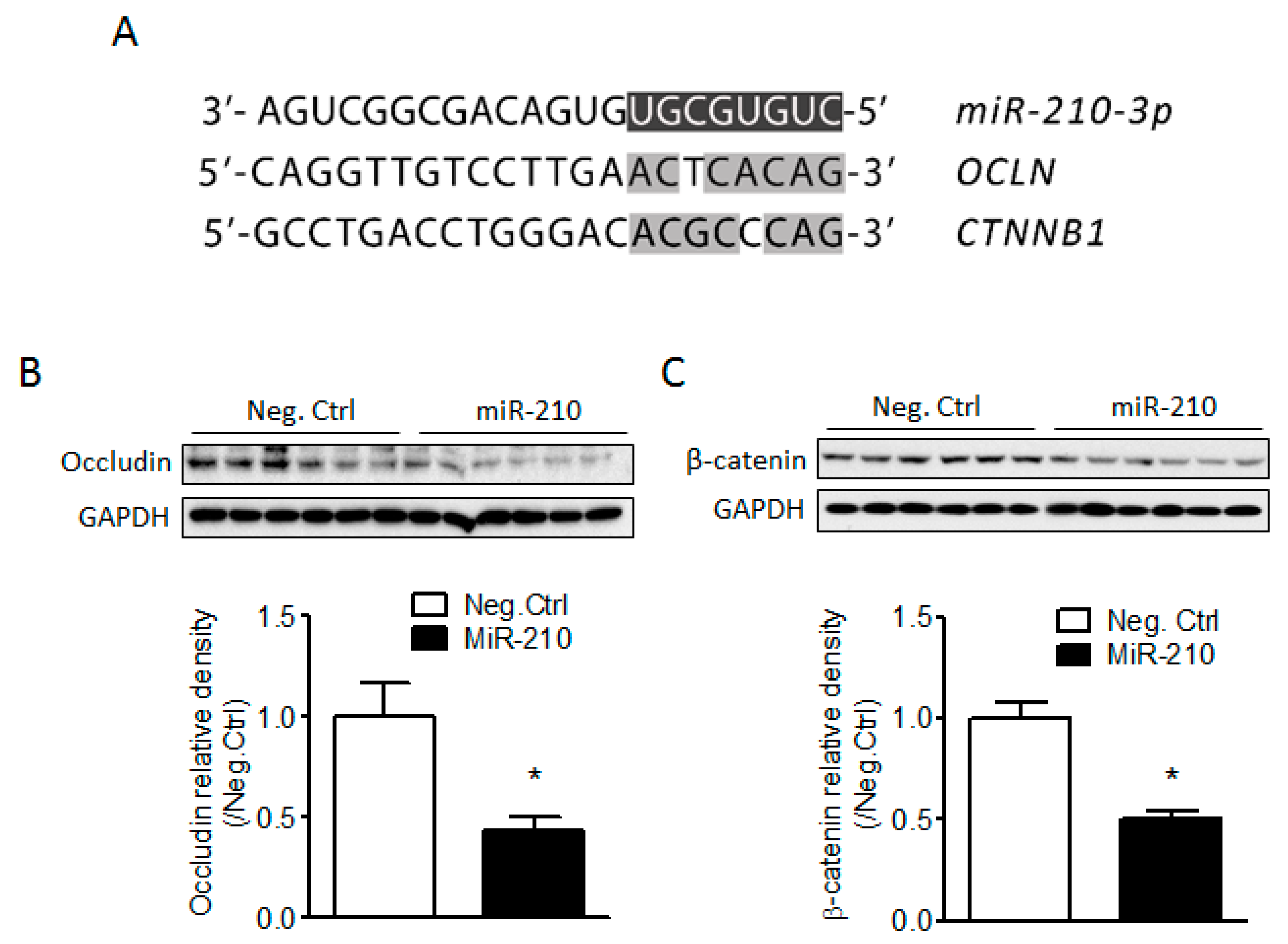
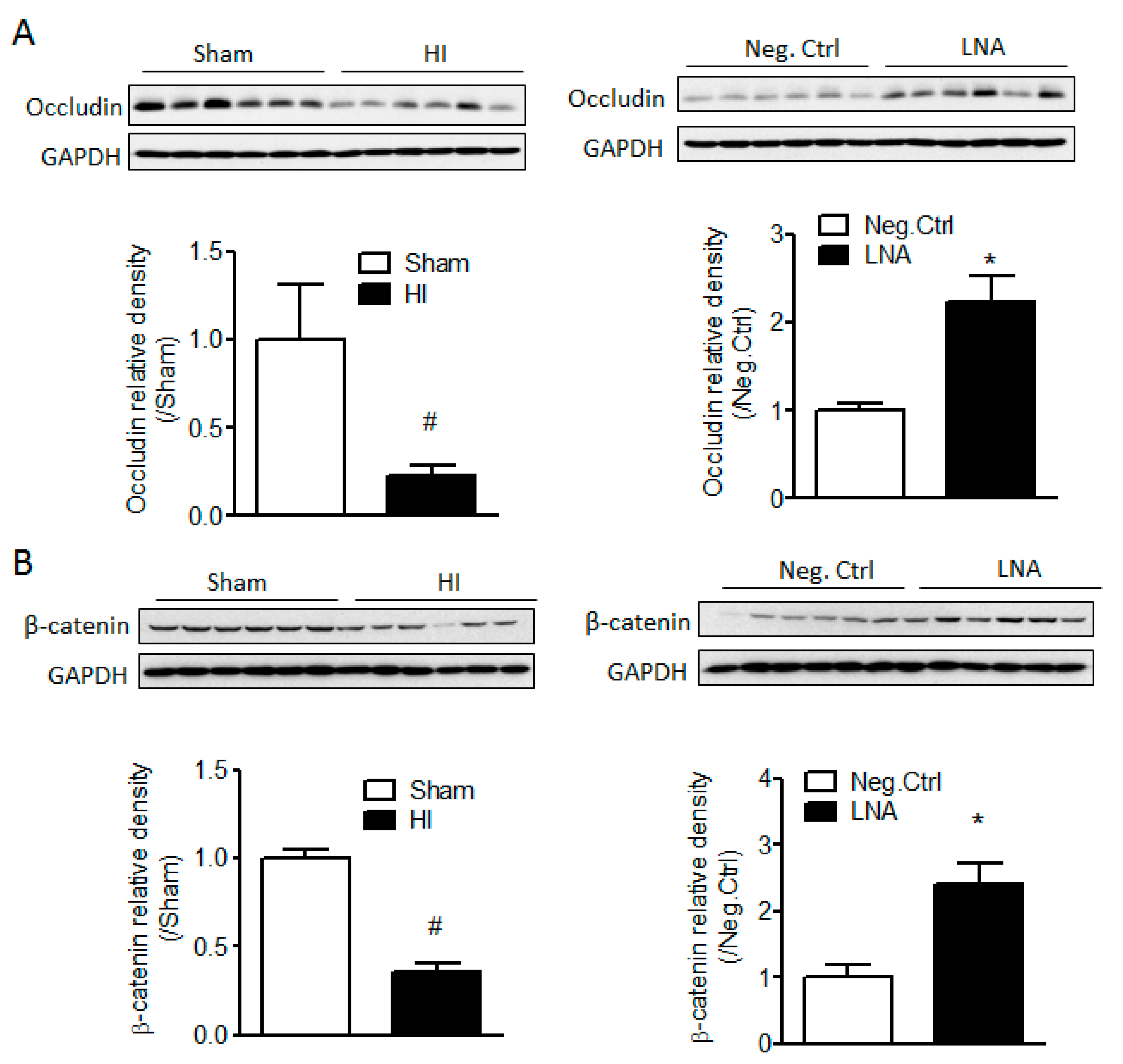
2.3. miR-210 Downregulates the Expression of Junction Proteins Occludin and β-Catenin
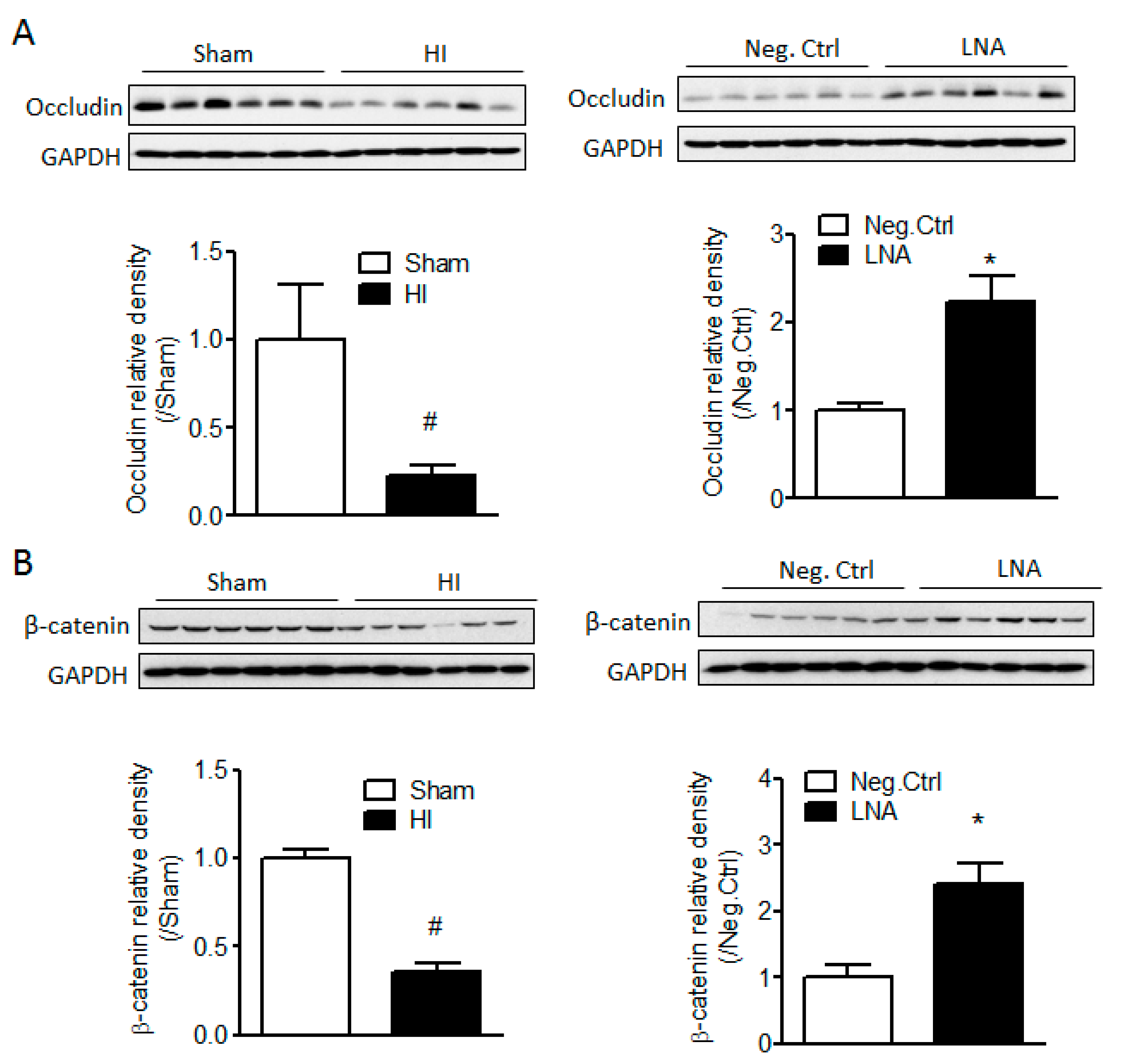
2.4. miR-210-LNA Treatment Protects Junction Proteins Occludin and β-Catenin from Neonatal HI Insult
3. Discussion
4. Materials and Methods
4.1. Hypoxic-Ischemia Model in Rat Pups
4.2. Intracerebroventricular Injection (i.c.v.)
4.3. Brain Infarct Staining and Cerebral Edema Assay
4.4. Western Blotting
4.5. Labeling of Cerebral Microvessels In Vivo and Immunofluorescence Staining
4.6. Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fatemi, A.; Wilson, M.A.; Johnston, M.V. Hypoxic-ischemic encephalopathy in the term infant. Clin. Perinatol. 2009, 36, 835–858. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Kozuki, N.; Blencowe, H.; Vos, T.; Bahalim, A.; Darmstadt, G.L.; Niermeyer, S.; Ellis, M.; Robertson, N.J.; Cousens, S.; et al. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr. Res. 2013, 74, 50–72. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, R.C. Hypoxic-ischemic encephalopathy. Am. J. Perinatol. 2000, 17, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Koenigsberger, M.R. Advances in neonatal neurology: 1950–2000. Rev. Neurol. 2000, 31, 202–211. [Google Scholar] [PubMed]
- Zanelli, G.; Petrarca, M.; Cappa, P.; Castelli, E.; Berthoz, A. Reorientation ability of adults and healthy children submitted to whole body horizontal rotations. Cogn. Process. 2009, 10, S346–S350. [Google Scholar] [CrossRef] [PubMed]
- Shalak, L.; Perlman, J.M. Hypoxic-ischemic brain injury in the term infant-current concepts. Early Hum. Dev. 2004, 80, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Baburamani, A.A.; Ek, C.J.; Walker, D.W.; Castillo-Melendez, M. Vulnerability of the developing brain to hypoxic-ischemic damage: Contribution of the cerebral vasculature to injury and repair? Front. Physiol. 2012, 3, 424. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.C.; Nesic, O.; Perez-Polo, J.R. Perspectives on neonatal hypoxia/ischemia-induced edema formation. Neurochem. Res. 2010, 35, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Boichot, C.; Walker, P.M.; Durand, C.; Grimaldi, M.; Chapuis, S.; Gouyon, J.B.; Brunotte, F. Term neonate prognoses after perinatal asphyxia: Contributions of MR imaging, MR spectroscopy, relaxation times, and apparent diffusion coefficients. Radiology 2006, 239, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Sargent, M.A.; Poskitt, K.J.; Roland, E.H.; Hill, A.; Hendson, G. Cerebellar vermian atrophy after neonatal hypoxic-ischemic encephalopathy. Am. J. Neuroradiol. 2004, 25, 1008–1015. [Google Scholar] [PubMed]
- Soul, J.S.; Robertson, R.L.; Tzika, A.A.; du Plessis, A.J.; Volpe, J.J. Time course of changes in diffusion-weighted magnetic resonance imaging in a case of neonatal encephalopathy with defined onset and duration of hypoxic-ischemic insult. Pediatrics 2001, 108, 1211–1214. [Google Scholar] [CrossRef] [PubMed]
- Qiao, M.; Latta, P.; Meng, S.; Tomanek, B.; Tuor, U.I. Development of acute edema following cerebral hypoxia-ischemia in neonatal compared with juvenile rats using magnetic resonance imaging. Pediatr. Res. 2004, 55, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.C.; Chang, Y.C.; Lin, Y.C.; Sze, C.I.; Huang, C.C.; Ho, C.J. Cerebral microvascular damage occurs early after hypoxia-ischemia via nnos activation in the neonatal brain. J. Cereb. Blood Flow Metab. 2014, 34, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Ek, C.J.; D’Angelo, B.; Baburamani, A.A.; Lehner, C.; Leverin, A.L.; Smith, P.L.; Nilsson, H.; Svedin, P.; Hagberg, H.; Mallard, C. Brain barrier properties and cerebral blood flow in neonatal mice exposed to cerebral hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, H.; Mu, H.; Zhu, W.; Jiang, X.; Hu, X.; Shi, Y.; Leak, R.K.; Dong, Q.; Chen, J.; et al. ω-3 polyunsaturated fatty acids mitigate blood-brain barrier disruption after hypoxic-ischemic brain injury. Neurobiol. Dis. 2016, 91, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mittal, R.; Khanna, H.D.; Basu, S. Free radical injury and blood-brain barrier permeability in hypoxic-ischemic encephalopathy. Pediatrics 2008, 122, e722–e727. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E. Endothelial adherens junctions: Implications in the control of vascular permeability and angiogenesis. J. Clin. Investig. 1996, 98, 1949–1953. [Google Scholar] [CrossRef] [PubMed]
- Cristante, E.; McArthur, S.; Mauro, C.; Maggioli, E.; Romero, I.A.; Wylezinska-Arridge, M.; Couraud, P.O.; Lopez-Tremoleda, J.; Christian, H.C.; Weksler, B.B.; et al. Identification of an essential endogenous regulator of blood-brain barrier integrity, and its pathological and therapeutic implications. Proc. Natl. Acad. Sci. USA 2013, 110, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Marin, N.; Zamorano, P.; Carrasco, R.; Mujica, P.; Gonzalez, F.G.; Quezada, C.; Meininger, C.J.; Boric, M.P.; Duran, W.N.; Sanchez, F.A. S-nitrosation of β-catenin and p120 catenin: A novel regulatory mechanism in endothelial hyperpermeability. Circ. Res. 2012, 111, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Irmady, K.; Jackman, K.A.; Padow, V.A.; Shahani, N.; Martin, L.A.; Cerchietti, L.; Unsicker, K.; Iadecola, C.; Hempstead, B.L. Mir-592 regulates the induction and cell death-promoting activity of p75NTR in neuronal ischemic injury. J. Neurosci. 2014, 34, 3419–3428. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.M.; Xu, L.; Giffard, R.G. Inhibition of microRNA-181 reduces forebrain ischemia-induced neuronal loss. J. Cereb. Blood Flow Metab. 2013, 33, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Impey, S.; Goodman, R.H. Role reversal: The regulation of neuronal gene expression by micrornas. Curr. Opin. Neurobiol. 2005, 15, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S.; Krichevsky, A.M. The elegance of the microRNAs: A neuronal perspective. Neuron 2005, 47, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Nallamshetty, S.; Chan, S.Y.; Loscalzo, J. Hypoxia: A master regulator of microRNA biogenesis and activity. Free Radic. Biol. Med. 2013, 64, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Fasanaro, P.; D’Alessandra, Y.; di Stefano, V.; Melchionna, R.; Romani, S.; Pompilio, G.; Capogrossi, M.C.; Martelli, F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J. Biol. Chem. 2008, 283, 15878–15883. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; He, X.; Wang, Y.; Tang, Y.; Zheng, C.; Cai, H.; Liu, J.; Wang, Y.; Fu, Y.; Yang, G.Y. MicroRNA-210 overexpression induces angiogenesis and neurogenesis in the normal adult mouse brain. Gene Ther. 2014, 21, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Dasgupta, C.; Li, Y.; Bajwa, N.M.; Xiong, F.; Harding, B.; Hartman, R.; Zhang, L. Inhibition of microRNA-210 provides neuroprotection in hypoxic-ischemic brain injury in neonatal rats. Neurobiol. Dis. 2016, 89, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.A.; Morton, M.T.; Tsao-Wu, G.; Savalos, R.A.; Davidson, C.; Sharp, F.R. A semiautomated method for measuring brain infarct volume. J. Cereb. Blood Flow Metab. 1990, 10, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Blecharz, K.; Kahles, T.; Schwarz, T.; Kraft, P.; Gobel, K.; Meuth, S.G.; Burek, M.; Thum, T.; Stoll, G.; et al. Glucocorticoid insensitivity at the hypoxic blood-brain barrier can be reversed by inhibition of the proteasome. Stroke 2011, 42, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A. Hypoxic-ischemic injury in neonatal brain: Involvement of a novel neuronal molecule in neuronal cell death and potential target for neuroprotection. Int. J. Dev. Neurosci. 2008, 26, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Zonouzi, M.; Scafidi, J.; Li, P.; McEllin, B.; Edwards, J.; Dupree, J.L.; Harvey, L.; Sun, D.; Hubner, C.A.; Cull-Candy, S.G.; et al. GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat. Neurosci. 2015, 18, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, K.; Fukuda, A.; Togari, H.; Wada, Y.; Nishino, H. Vulnerability to cerebral hypoxic-ischemic insult in neonatal but not in adult rats is in parallel with disruption of the blood-brain barrier. Stroke 1997, 28, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Brain endothelial cell-cell junctions: How to “open” the blood brain barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Michinaga, S.; Koyama, Y. Pathogenesis of brain edema and investigation into anti-edema drugs. Int. J. Mol. Sci. 2015, 16, 9949–9975. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A. Ischemic brain edema. Prog. Cardiovasc. Dis. 1999, 42, 209–216. [Google Scholar] [CrossRef]
- Todd, N.V.; Picozzi, P.; Crockard, A.; Russell, R.W. Duration of ischemia influences the development and resolution of ischemic brain edema. Stroke 1986, 17, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Kent, T.A.; Chen, M.; Tarasov, K.V.; Gerzanich, V. Brain oedema in focal ischaemia: Molecular pathophysiology and theoretical implications. Lancet Neurol. 2007, 6, 258–268. [Google Scholar] [CrossRef]
- Ferrari, D.C.; Nesic, O.B.; Perez-Polo, J.R. Oxygen resuscitation does not ameliorate neonatal hypoxia/ischemia-induced cerebral edema. J. Neurosci. Res. 2010, 88, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, K.E.; Witt, K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Furuse, M.; Sasaki, H.; Schulzke, J.D.; Fromm, M.; Takano, H.; Noda, T.; Tsukita, S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 2000, 11, 4131–4142. [Google Scholar] [CrossRef] [PubMed]
- Steed, E.; Balda, M.S.; Matter, K. Dynamics and functions of tight junctions. Trends Cell Biol. 2010, 20, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Sawant, D.A.; Tharakan, B.; Hunter, F.A.; Smythe, W.R.; Childs, E.W. Role of β-catenin in regulating microvascular endothelial cell hyperpermeability. J. Trauma Acute Care Surg. 2011, 70, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Cattelino, A.; Liebner, S.; Gallini, R.; Zanetti, A.; Balconi, G.; Corsi, A.; Bianco, P.; Wolburg, H.; Moore, R.; Oreda, B.; et al. The conditional inactivation of the β-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J. Cell Biol. 2003, 162, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.K.; Sun, Y.; Zhao, Y.D.; Potula, H.H.; Frey, R.S.; Vogel, S.M.; Malik, A.B.; Zhao, Y.Y. FoxM1 regulates re-annealing of endothelial adherens junctions through transcriptional control of β-catenin expression. J. Exp. Med. 2010, 207, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.A.; Zhang, X.; Predescu, D.; Huang, X.; Machado, R.F.; Gothert, J.R.; Malik, A.B.; Valyi-Nagy, T.; Zhao, Y.Y. Endothelial β-catenin signaling is required for maintaining adult blood-brain barrier integrity and central nervous system homeostasis. Circulation 2016, 133, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Huntley, M.A.; Bien-Ly, N.; Daneman, R.; Watts, R.J. Dissecting gene expression at the blood-brain barrier. Front. Neurosci. 2014, 8, 355. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Lieberman, J.; Lal, A. Desperately seeking microrna targets. Nat. Struct. Mol. Biol. 2010, 17, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Forster, C.; Silwedel, C.; Golenhofen, N.; Burek, M.; Kietz, S.; Mankertz, J.; Drenckhahn, D. Occludin as direct target for glucocorticoid-induced improvement of blood-brain barrier properties in a murine in vitro system. J. Physiol. 2005, 565, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Forster, C.; Waschke, J.; Burek, M.; Leers, J.; Drenckhahn, D. Glucocorticoid effects on mouse microvascular endothelial barrier permeability are brain specific. J. Physiol. 2006, 573, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Singer, K.L.; Stevenson, B.R.; Woo, P.L.; Firestone, G.L. Relationship of serine/threonine phosphorylation/dephosphorylation signaling to glucocorticoid regulation of tight junction permeability and ZO-1 distribution in nontransformed mammary epithelial cells. J. Biol. Chem. 1994, 269, 16108–16115. [Google Scholar] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Q.; Dasgupta, C.; Li, Y.; Huang, L.; Zhang, L. MicroRNA-210 Suppresses Junction Proteins and Disrupts Blood-Brain Barrier Integrity in Neonatal Rat Hypoxic-Ischemic Brain Injury. Int. J. Mol. Sci. 2017, 18, 1356. https://doi.org/10.3390/ijms18071356
Ma Q, Dasgupta C, Li Y, Huang L, Zhang L. MicroRNA-210 Suppresses Junction Proteins and Disrupts Blood-Brain Barrier Integrity in Neonatal Rat Hypoxic-Ischemic Brain Injury. International Journal of Molecular Sciences. 2017; 18(7):1356. https://doi.org/10.3390/ijms18071356
Chicago/Turabian StyleMa, Qingyi, Chiranjib Dasgupta, Yong Li, Lei Huang, and Lubo Zhang. 2017. "MicroRNA-210 Suppresses Junction Proteins and Disrupts Blood-Brain Barrier Integrity in Neonatal Rat Hypoxic-Ischemic Brain Injury" International Journal of Molecular Sciences 18, no. 7: 1356. https://doi.org/10.3390/ijms18071356