Combining Primed Photoconversion and UV-Photoactivation for Aberration-Free, Live-Cell Compliant Multi-Color Single-Molecule Localization Microscopy Imaging

Abstract
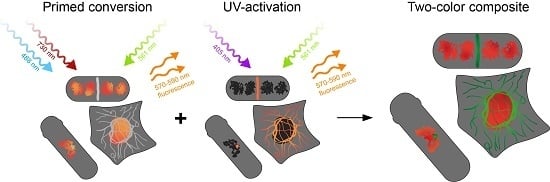
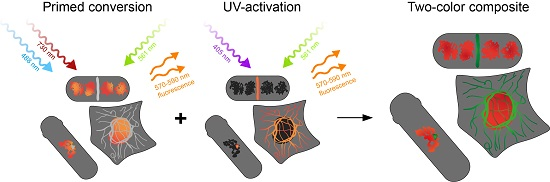
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
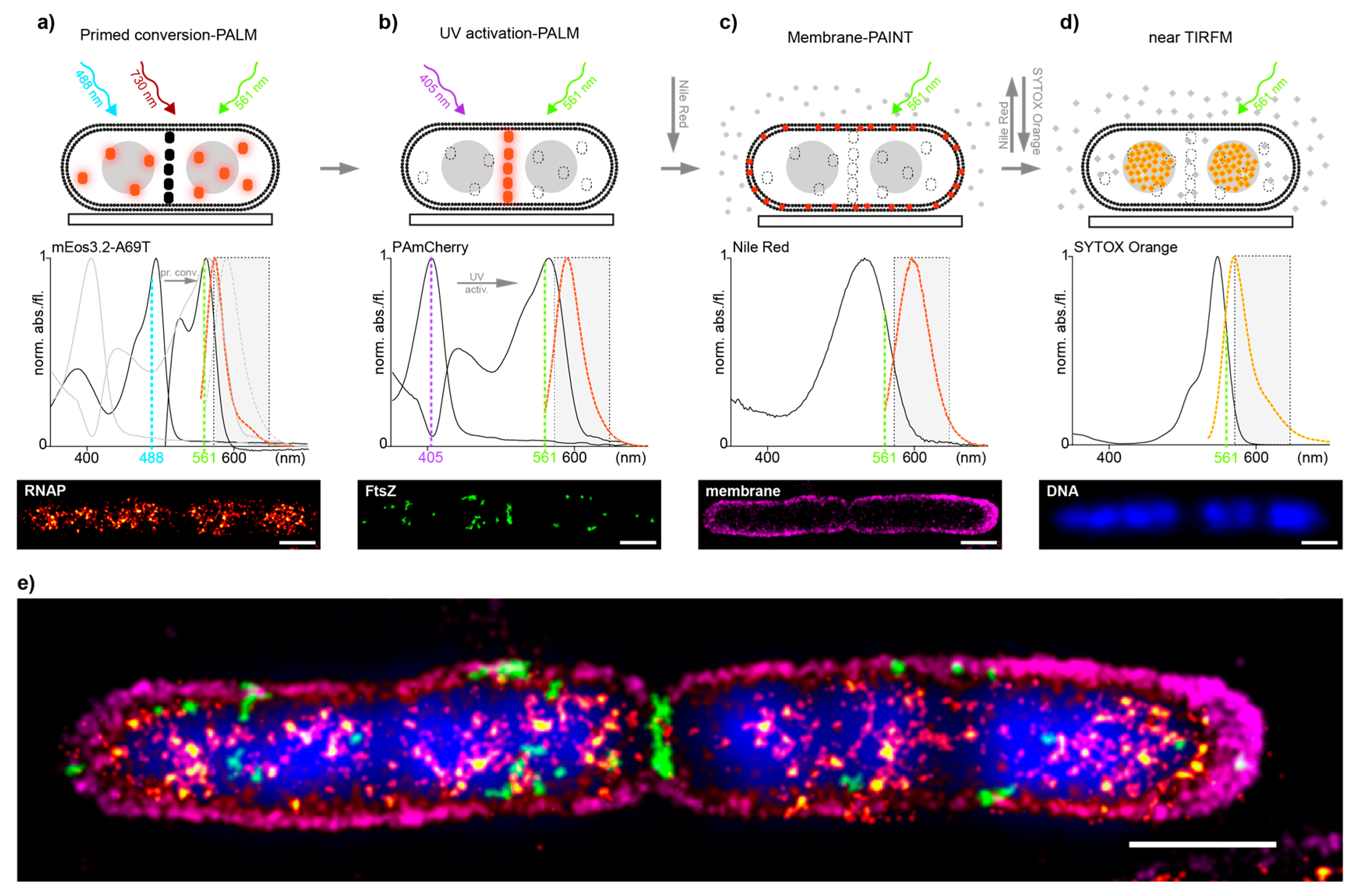
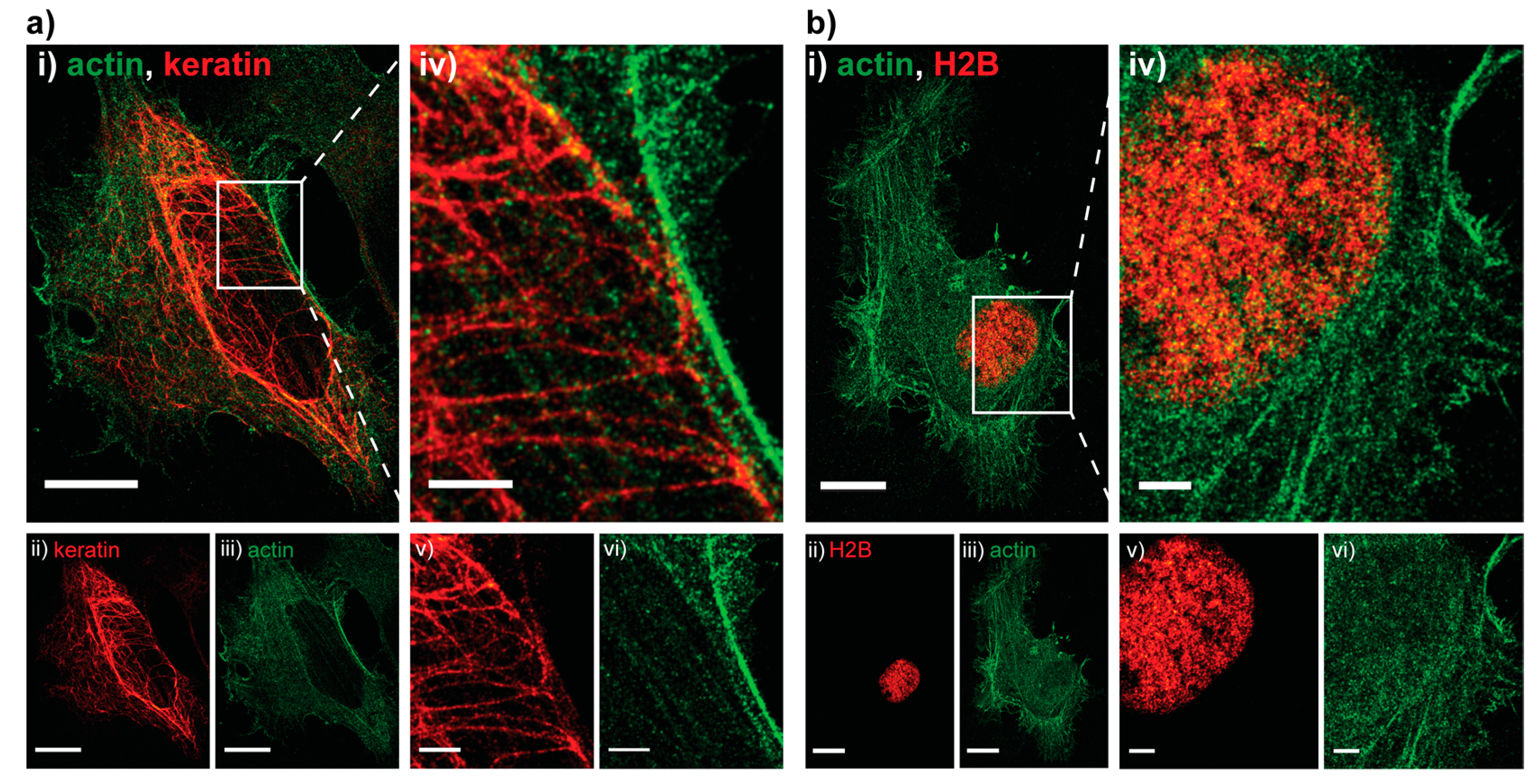
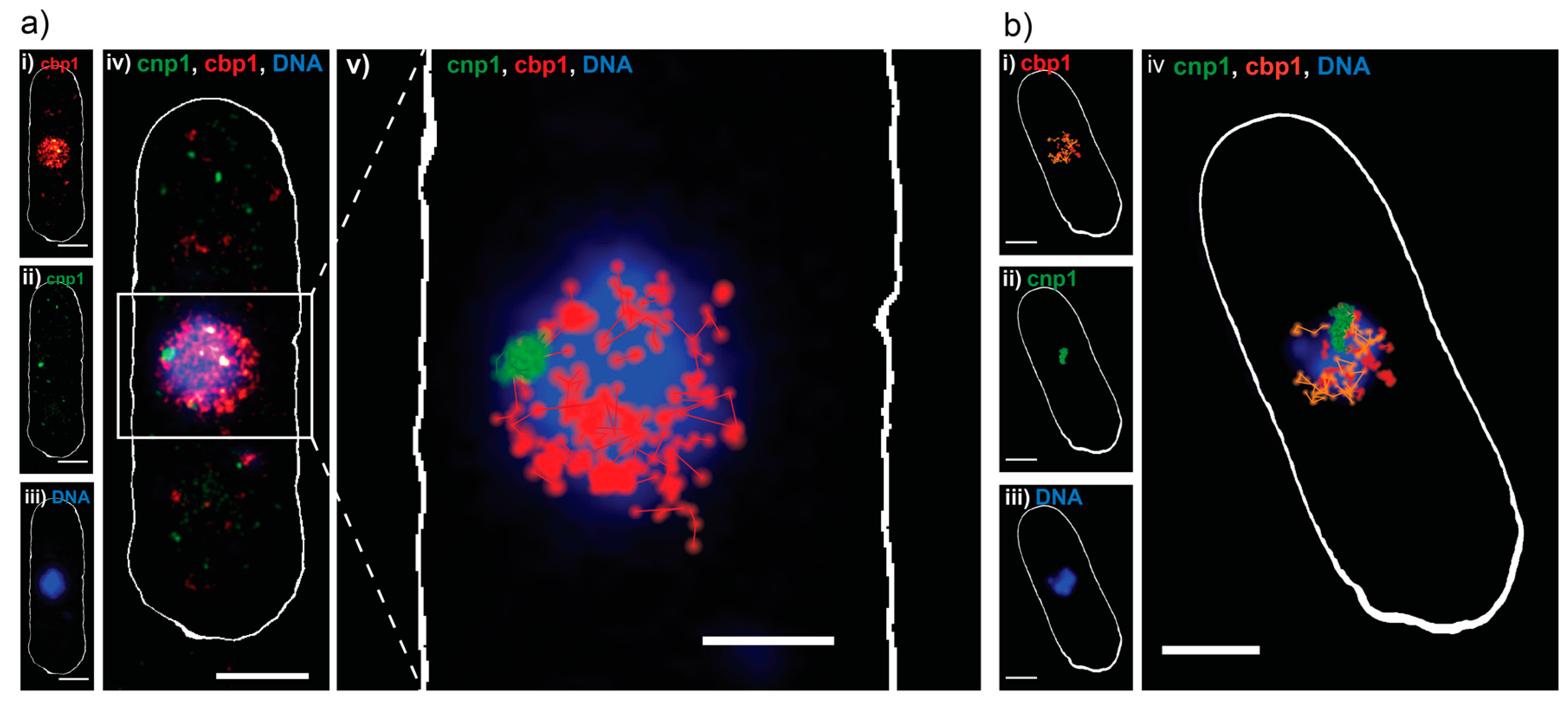
2. Results and Discussion
3. Materials and Methods
3.1. Imaging Procedures
3.1.1. Influence of High Intensity of 488 nm Light on PAmCherry and Dendra2
3.1.2. Quantitative Controls-RNAP Molecule Counting
3.1.3. E. coli Multi-Color Imaging
3.1.4. HeLa Dual-Color Imaging
3.1.5. S. pombe Multi-Color Imaging
3.1.6. Viability Controls
3.2. Data Analysis
3.2.1. Analysis of the Influence of High Intensity 488 nm Images
3.2.2. Super-Resolution Image Reconstruction of Bacterial and Mammalian Multi-Color Images
3.2.3. Single Particle Tracking in E. coli and S. pombe
4. Summary and Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| FP | Fluorescent protein |
| paFP | Photoactivatable fluorescent protein |
| PAINT | Points accumulation for imaging in nanoscale topography |
| PALM | Photoactivated localization microscopy |
| PC | Primed conversion |
| pcFP | Photoconvertible fluorescent protein |
| SMLM | Single molecule localization microscopy |
References
- Turkowyd, B.; Virant, D.; Endesfelder, U. From single molecules to life: Microscopy at the nanoscale. Anal. Bioanal. Chem. 2016, 408, 6885–6911. [Google Scholar] [CrossRef] [PubMed]
- Nicovich, P.R.; Owen, D.M.; Gaus, K. Turning single-molecule localization microscopy into a quantitative bioanalytical tool. Nat. Protoc. 2017, 12, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Sauer, M.; Heilemann, M. Single-molecule localization microscopy in eukaryotes. Chem. Rev. 2017, 117, 7478–7509. [Google Scholar] [CrossRef] [PubMed]
- Nienhaus, K.; Nienhaus, G.U. Fluorescent proteins for live-cell imaging with super-resolution. Chem. Soc. Rev. 2014, 43, 1088–1106. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, D.M.; Sengupta, P.; Lippincott-Schwartz, J.; Verkhusha, V.V. Photocontrollable fluorescent proteins for superresolution imaging. Annu. Rev. Biophys. 2014, 43, 303–329. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.B.; English, B.P.; Choi, H.; Muthusamy, A.K.; Mehl, B.P.; Dong, P.; Brown, T.A.; Lippincott-Schwartz, J.; Liu, Z.; Lionnet, T.; et al. Bright photoactivatable fluorophores for single-molecule imaging. Nat. Methods 2016, 13, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Gunewardene, M.S.; Subach, F.V.; Gould, T.J.; Penoncello, G.P.; Gudheti, M.V.; Verkhusha, V.V.; Hess, S.T. Superresolution imaging of multiple fluorescent proteins with highly overlapping emission spectra in living cells. Biophys. J. 2011, 101, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Gahlmann, A.; Moerner, W.E. Exploring bacterial cell biology with single-molecule tracking and super-resolution imaging. Nat. Rev. Microbiol. 2014, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, S.; Staufenbiel, M.; Lisse, D.; Richter, C.P.; Beutel, O.; Busch, K.B.; Hess, S.T.; Piehler, J. Triple-color super-resolution imaging of live cells: Resolving submicroscopic receptor organization in the plasma membrane. Angew. Chem. 2012, 51, 4868–4871. [Google Scholar] [CrossRef] [PubMed]
- Shroff, H.; Galbraith, C.G.; Galbraith, J.A.; White, H.; Gillette, J.; Olenych, S.; Davidson, M.W.; Betzig, E. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc. Natl. Acad. Sci. USA 2007, 104, 20308–20313. [Google Scholar] [CrossRef] [PubMed]
- Nahmani, M.; Lanahan, C.; DeRosier, D.; Turrigiano, G.G. High-numerical-aperture cryogenic light microscopy for increased precision of superresolution reconstructions. Proc. Natl. Acad. Sci. USA. 2017, 114, 3832–3836. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, W.P.; Georgieva, L.; Helbling, P.M.; Sonay, A.Y.; Truong, T.V.; Haffner, M.; Pantazis, P. In vivo single-cell labeling by confined primed conversion. Nat. Methods 2015, 12, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Klementieva, N.V.; Lukyanov, K.A.; Markina, N.M.; Lukyanov, S.A.; Zagaynova, E.V.; Mishin, A.S. Green-to-red primed conversion of dendra2 using blue and red lasers. Chem. Commun. 2016, 52, 13144–13146. [Google Scholar] [CrossRef] [PubMed]
- Turkowyd, B.; Balinovic, A.; Virant, D.; Golz Carnero, H.G.; Caldana, F.; Endesfelder, M.; Bourgeois, D.; Endesfelder, U. A general mechanism of photoconversion of green-to-red fluorescent proteins based on blue and infrared light reduces phototoxicity in live-cell single-molecule imaging. Angew. Chem. 2017. [Google Scholar] [CrossRef]
- Mohr, M.A.; Argast, P.; Pantazis, P. Labeling cellular structures in vivo using confined primed conversion of photoconvertible fluorescent proteins. Nat. Protoc. 2016, 11, 2419–2431. [Google Scholar] [CrossRef] [PubMed]
- Subach, F.V.; Patterson, G.H.; Manley, S.; Gillette, J.M.; Lippincott-Schwartz, J.; Verkhusha, V.V. Photoactivatable mcherry for high-resolution two-color fluorescence microscopy. Nat. Methods 2009, 6, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Moffitt, J.R.; Dempsey, G.T.; Xie, X.S.; Zhuang, X. Characterization and development of photoactivatable fluorescent proteins for single-molecule–based superresolution imaging. Proc. Natl. Acad. Sci. USA 2014, 111, 8452–8457. [Google Scholar] [CrossRef] [PubMed]
- Wäldchen, S.; Lehmann, J.; Klein, T.; van de Linde, S.; Sauer, M. Light-induced cell damage in live-cell super-resolution microscopy. Sci. Rep. 2015, 5, 15348. [Google Scholar] [CrossRef] [PubMed]
- Patterson, G.H.; Lippincott-Schwartz, J. A photoactivatable gfp for selective photolabeling of proteins and cells. Science 2002, 297, 1873–1877. [Google Scholar] [CrossRef] [PubMed]
- Subach, F.V.; Patterson, G.H.; Renz, M.; Lippincott-Schwartz, J.; Verkhusha, V.V. Bright monomeric photoactivatable red fluorescent protein for two-color super-resolution sptpalm of live cells. J. Am. Chem. Soc. 2010, 132, 6481–6491. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, U.; Finan, K.; Holden, S.J.; Cook, P.R.; Kapanidis, A.N.; Heilemann, M. Multiscale spatial organization of rna polymerase in escherichia coli. Biophys. J. 2013, 105, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Stracy, M.; Lesterlin, C.; Garza de Leon, F.; Uphoff, S.; Zawadzki, P.; Kapanidis, A.N. Live-cell superresolution microscopy reveals the organization of rna polymerase in the bacterial nucleoid. Proc. Natl. Acad. Sci. USA 2015, 112, E4390–E4399. [Google Scholar] [CrossRef] [PubMed]
- Spahn, C.; Cella-Zannacchi, F.; Endesfelder, U.; Heilemann, M. Correlative super-resolution imaging of rna polymerase distribution and dynamics, bacterial membrane and chromosomal structure inescherichia coli. Methods Appl. Fluoresc. 2015, 3, 014005. [Google Scholar] [CrossRef]
- Sharonov, A.; Hochstrasser, R.M. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl. Acad. Sci. USA 2006, 103, 18911–18916. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.R.; Fujimoto, B.S.; Chiu, D.T. Quantitative microscopy based on single-molecule fluorescence. Curr. Opin. Chem. Biol. 2017, 39, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Yu, J. Investigating intracellular dynamics of ftsz cytoskeleton with photoactivation single-molecule tracking. Biophys. J. 2008, 95, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Laplante, C.; Huang, F.; Tebbs, I.R.; Bewersdorf, J.; Pollard, T.D. Molecular organization of cytokinesis nodes and contractile rings by super-resolution fluorescence microscopy of live fission yeast. Proc. Natl. Acad. Sci. USA 2016, 113, E5876–E5885. [Google Scholar] [CrossRef] [PubMed]
- Daigaku, Y.; Etheridge, T.J.; Nakazawa, Y.; Nakayama, M.; Watson, A.T.; Miyabe, I.; Ogi, T.; Osborne, M.A.; Carr, A.M. Pcna ubiquitylation ensures timely completion of unperturbed DNA replication in fission yeast. PLoS Genet. 2017, 13, e1006789. [Google Scholar] [CrossRef] [PubMed]
- Zaratiegui, M.; Vaughn, M.W.; Irvine, D.V.; Goto, D.; Watt, S.; Bahler, J.; Arcangioli, B.; Martienssen, R.A. Cenp-b preserves genome integrity at replication forks paused by retrotransposon ltr. Nature 2011, 469, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, T.; Barrowman, J.; Grewal, S.I. Chromosome domain architecture and dynamic organization of the fission yeast genome. FEBS Lett. 2015, 589, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, U.; Malkusch, S.; Fricke, F.; Heilemann, M. A simple method to estimate the average localization precision of a single-molecule localization microscopy experiment. Histochem. Cell Biol. 2014, 141, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Etheridge, T.J.; Boulineau, R.L.; Herbert, A.; Watson, A.T.; Daigaku, Y.; Tucker, J.; George, S.; Jonsson, P.; Palayret, M.; Lando, D.; et al. Quantification of DNA-associated proteins inside eukaryotic cells using single-molecule localization microscopy. Nucleic Acids Res. 2014, 42, e146. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Wolter, S.; Loschberger, A.; Holm, T.; Aufmkolk, S.; Dabauvalle, M.C.; van de Linde, S.; Sauer, M. Rapidstorm: Accurate, fast open-source software for localization microscopy. Nat. Methods 2012, 9, 1040–1041. [Google Scholar] [CrossRef] [PubMed]
- Malkusch, S.; Heilemann, M. Extracting quantitative information from single-molecule super-resolution imaging data with lama—Localization microscopy analyzer. Sci. Rep. 2016, 6, 34486. [Google Scholar] [CrossRef] [PubMed]
- Ester, M.; Kriegel, H.P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise; Kdd: Portland, OH, USA, 1996; pp. 226–231. [Google Scholar]
- Legant, W.R.; Shao, L.; Grimm, J.B.; Brown, T.A.; Milkie, D.E.; Avants, B.B.; Lavis, L.D.; Betzig, E. High-density three-dimensional localization microscopy across large volumes. Nat. Methods 2016, 13, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. Halotag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, A.; Baker, B.; Sun, L.; Howard, A.; Buswell, J.; Maurel, D.; Masharina, A.; Johnsson, K.; Noren, C.J.; et al. Development of snap-tag fluorogenic probes for wash-free fluorescence imaging. Chembiochem 2011, 12, 2217–2226. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Virant, D.; Turkowyd, B.; Balinovic, A.; Endesfelder, U. Combining Primed Photoconversion and UV-Photoactivation for Aberration-Free, Live-Cell Compliant Multi-Color Single-Molecule Localization Microscopy Imaging. Int. J. Mol. Sci. 2017, 18, 1524. https://doi.org/10.3390/ijms18071524
Virant D, Turkowyd B, Balinovic A, Endesfelder U. Combining Primed Photoconversion and UV-Photoactivation for Aberration-Free, Live-Cell Compliant Multi-Color Single-Molecule Localization Microscopy Imaging. International Journal of Molecular Sciences. 2017; 18(7):1524. https://doi.org/10.3390/ijms18071524
Chicago/Turabian StyleVirant, David, Bartosz Turkowyd, Alexander Balinovic, and Ulrike Endesfelder. 2017. "Combining Primed Photoconversion and UV-Photoactivation for Aberration-Free, Live-Cell Compliant Multi-Color Single-Molecule Localization Microscopy Imaging" International Journal of Molecular Sciences 18, no. 7: 1524. https://doi.org/10.3390/ijms18071524