Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
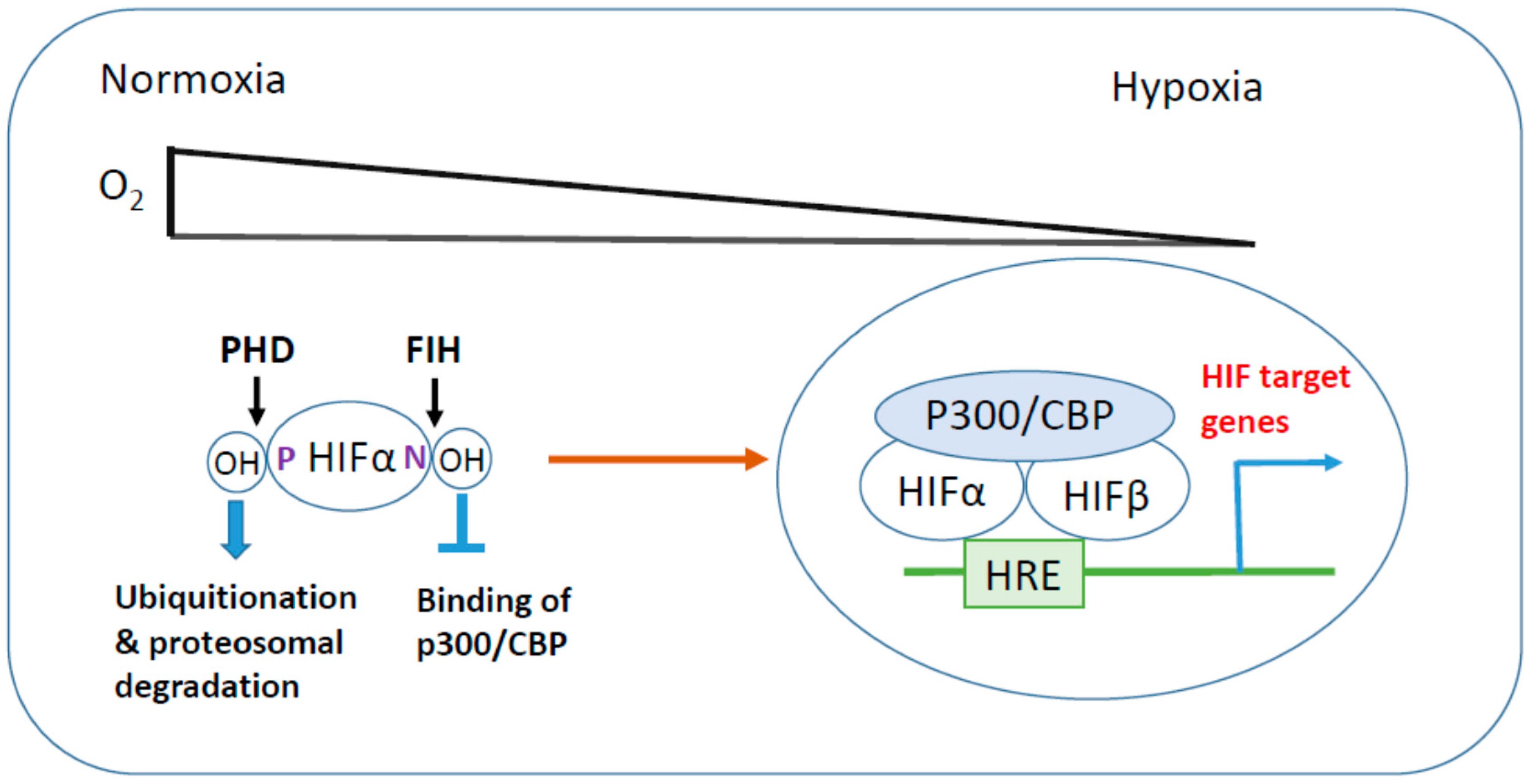
1.1. Hypoxia-Inducible Factor (HIF)
1.1.1. HIF Regulation in Hypoxia
1.1.2. HIF Expression in Tumors
1.2. Mechanism of Tumor Adaptation to Hypoxia, and Its Implication in Drug Resistance
1.2.1. Hypoxia-Induced Autophagy
1.2.2. Regulation of Cell Death under Hypoxia
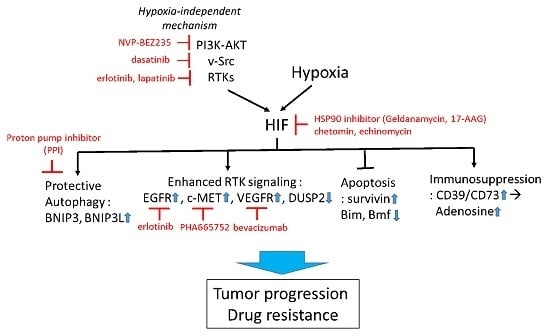
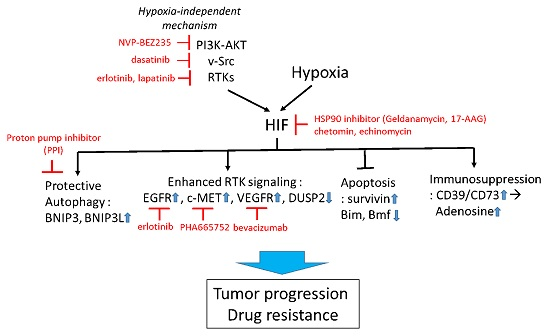
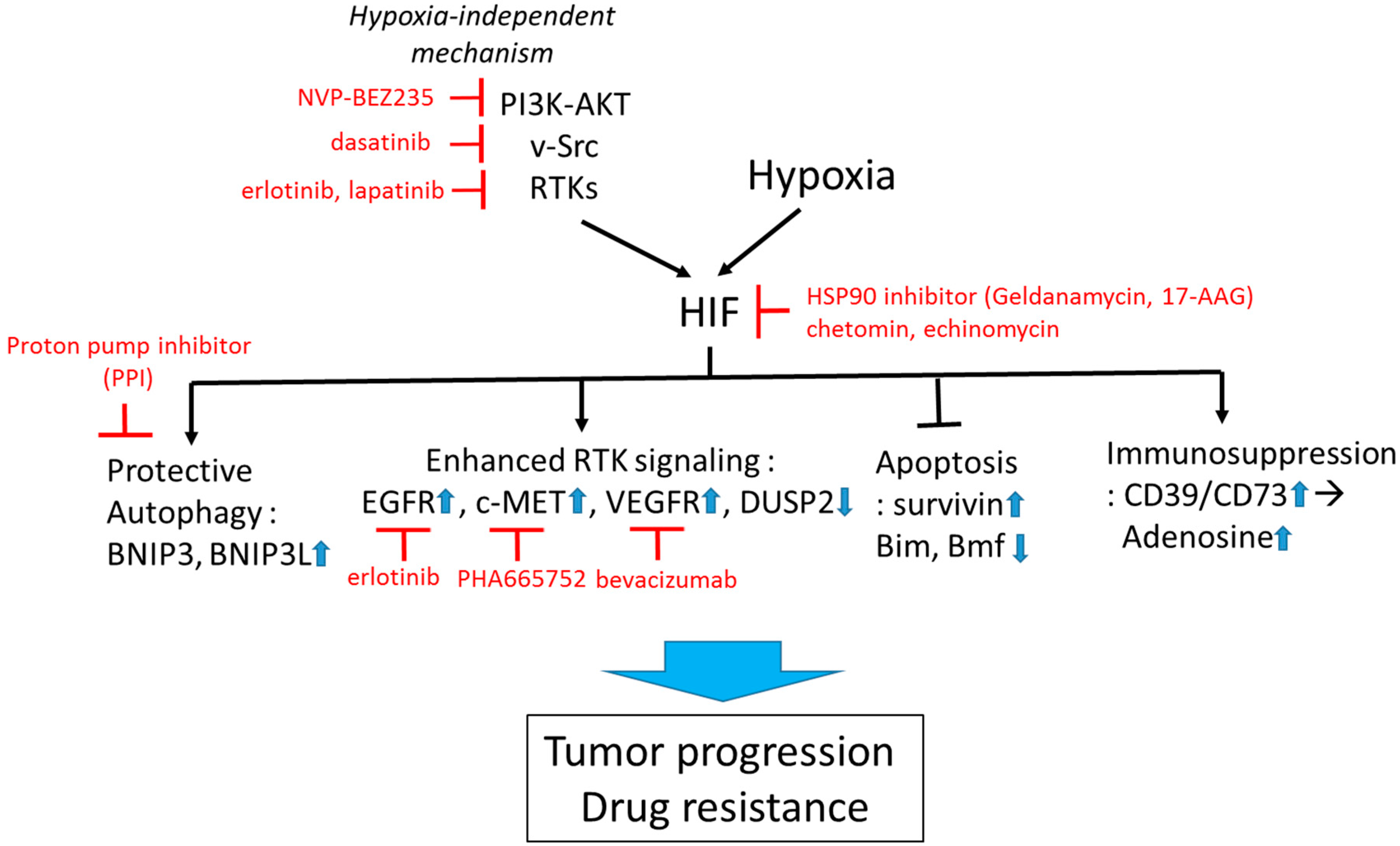
1.3. Strategies to Improve Therapy
1.3.1. Targeting HIF-1 Directly
1.3.2. Targeting HIF-1 Signaling
1.3.3. Targeting Hypoxia-Induced Autophagy
1.3.4. Targeting Hypoxia-HIF to Improve Immunotherapy
2. Summary
Acknowledgments
Conflicts of Interest
References
- Brown, L.M.; Cowen, R.L.; Debray, C.; Eustace, A.; Erler, J.T.; Sheppard, F.C.; Parker, C.A.; Stratford, I.J.; Williams, K.J. Reversing hypoxic cell chemoresistance in vitro using genetic and small molecule approaches targeting hypoxia inducible factor-1. Mol. Pharmacol. 2006, 69, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Hypoxia and drug resistance. Cancer Metastasis Rev. 1994, 13, 139–168. [Google Scholar] [CrossRef] [PubMed]
- Tatum, J.L.; Kelloff, G.J.; Gillies, R.J.; Arbeit, J.M.; Brown, J.M.; Chao, K.S.; Chapman, J.D.; Eckelman, W.C.; Fyles, A.W.; Giaccia, A.J.; et al. Hypoxia: Importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy. Inte. J. Radiat. Biol. 2006, 82, 699–757. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, M.S.; Jurgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Horstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol.Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Hahnfeldt, P.; Hlatky, L. Cancer: Looking outside the genome. Nat. Rev. Mol. Cell Biol. 2000, 1, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, M.C.; Chiche, J.; Pouyssegur, J. Hypoxia and cancer. J. Mol. Med. 2007, 85, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; de Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Irigoyen, M.; Garcia-Ruiz, J.C.; Berra, E. The hypoxia signalling pathway in haematological malignancies. Oncotarget 2017, 8, 36832–36844. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Thall, P.F.; Yi, C.A.; Borthakur, G.; Coveler, A.; Bueso-Ramos, C.; Benito, J.; Konoplev, S.; Gu, Y.; Ravandi, F.; et al. Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid leukemia and acute lymphoblastic leukemia. Haematologica 2015, 100, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.O.; Mortensen, B.T.; Hodgkiss, R.J.; Iversen, P.O.; Christensen, I.J.; Helledie, N.; Larsen, J.K. Increased cellular hypoxia and reduced proliferation of both normal and leukaemic cells during progression of acute myeloid leukaemia in rats. Cell Prolif. 2000, 33, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Agani, F.; Passaniti, A.; Semenza, G.L. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: Involvement of HIF-1 in tumor progression. Cancer Res. 1997, 57, 5328–5335. [Google Scholar] [PubMed]
- Feldser, D.; Agani, F.; Iyer, N.V.; Pak, B.; Ferreira, G.; Semenza, G.L. Reciprocal positive regulation of hypoxia-inducible factor 1α and insulin-like growth factor 2. Cancer Res. 1999, 59, 3915–3918. [Google Scholar] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Takeuchi, T.; Kuwano, H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann. Surg. Oncol. 2009, 16, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Karpathiou, G.; Sivridis, E.; Koukourakis, M.I.; Mikroulis, D.; Bouros, D.; Froudarakis, M.E.; Giatromanolaki, A. Light-chain 3A autophagic activity and prognostic significance in non-small cell lung carcinomas. Chest 2011, 140, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Sivridis, E.; Koukourakis, M.I.; Mendrinos, S.E.; Karpouzis, A.; Fiska, A.; Kouskoukis, C.; Giatromanolaki, A. Beclin-1 and LC3A expression in cutaneous malignant melanomas: A biphasic survival pattern for beclin-1. Melanoma Res. 2011, 21, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Thomas, S.; Golden, E.B.; Hofman, F.M.; Chen, T.C.; Petasis, N.A.; Schonthal, A.H.; Louie, S.G. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett. 2012, 326, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta 2010, 1806, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, S.M.; Moretti, L.; Varki, V.; Lu, B. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: Implications for future therapeutic approaches. Drug Resist. Updates 2010, 13, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, Y.; Liu, Q.; Wang, J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med. Oncol. 2011, 28, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in philadelphia chromosome-positive cells, including primary cml stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting er stress-induced autophagy overcomes braf inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.W.; Su, Y.; Zhu, H.; Cao, J.; Ding, W.J.; Zhao, Y.C.; He, Q.J.; Yang, B. HIF-1α-dependent autophagy protects Hela cells from fenretinide (4-HPR)-induced apoptosis in hypoxia. Pharmacol. Res. 2010, 62, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol.Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Sakamoto, K. Ampk: A key sensor of fuel and energy status in skeletal muscle. Physiology 2006, 21, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef] [PubMed]
- Kilic, M.; Kasperczyk, H.; Fulda, S.; Debatin, K.M. Role of hypoxia inducible factor-1α in modulation of apoptosis resistance. Oncogene 2007, 26, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar] [PubMed]
- Boyd, J.M.; Malstrom, S.; Subramanian, T.; Venkatesh, L.K.; Schaeper, U.; Elangovan, B.; D′Sa-Eipper, C.; Chinnadurai, G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 1994, 79, 341–351. [Google Scholar] [CrossRef]
- Vande Velde, C.; Cizeau, J.; Dubik, D.; Alimonti, J.; Brown, T.; Israels, S.; Hakem, R.; Greenberg, A.H. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol. 2000, 20, 5454–5468. [Google Scholar] [CrossRef] [PubMed]
- Formigli, L.; Papucci, L.; Tani, A.; Schiavone, N.; Tempestini, A.; Orlandini, G.E.; Capaccioli, S.; Orlandini, S.Z. Aponecrosis: Morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J. Cell. Physiol. 2000, 182, 41–49. [Google Scholar] [CrossRef]
- Papandreou, I.; Krishna, C.; Kaper, F.; Cai, D.; Giaccia, A.J.; Denko, N.C. Anoxia is necessary for tumor cell toxicity caused by a low-oxygen environment. Cancer Res. 2005, 65, 3171–3178. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.M.; Denko, N.C.; Dorie, M.J.; Abraham, R.T.; Giaccia, A.J. Hypoxia links atr and p53 through replication arrest. Mol. Cell. Biol. 2002, 22, 1834–1843. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.M.; Giaccia, A.J. The role of p53 in hypoxia-induced apoptosis. Biochem. Biophys. Res. Commun. 2005, 331, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Tomida, A.; Tsuruo, T. Dephosphorylated hypoxia-inducible factor 1α as a mediator of p53-dependent apoptosis during hypoxia. Oncogene 2001, 20, 5779–5788. [Google Scholar] [CrossRef] [PubMed]
- An, W.G.; Kanekal, M.; Simon, M.C.; Maltepe, E.; Blagosklonny, M.V.; Neckers, L.M. Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature 1998, 392, 405–408. [Google Scholar] [PubMed]
- Blagosklonny, M.V.; An, W.G.; Romanova, L.Y.; Trepel, J.; Fojo, T.; Neckers, L. p53 inhibits hypoxia-inducible factor-stimulated transcription. J. Biol. Chem. 1998, 273, 11995–11998. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Cawthorne, C.J.; Williams, K.J.; Koritzinsky, M.; Wouters, B.G.; Wilson, C.; Miller, C.; Demonacos, C.; Stratford, I.J.; Dive, C. Hypoxia-mediated down-regulation of bid and bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol. Cell. Biol. 2004, 24, 2875–2889. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Q.; Zhao, C.L.; Li, W. Effect of hypoxia-inducible factor-1α on transcription of survivin in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2009, 28, 29. [Google Scholar] [CrossRef] [PubMed]
- Whelan, K.A.; Caldwell, S.A.; Shahriari, K.S.; Jackson, S.R.; Franchetti, L.D.; Johannes, G.J.; Reginato, M.J. Hypoxia suppression of Bim and Bmf blocks anoikis and luminal clearing during mammary morphogenesis. Mol. Biol. Cell 2010, 21, 3829–3837. [Google Scholar] [CrossRef] [PubMed]
- Faversani, A.; Vaira, V.; Moro, G.P.; Tosi, D.; Lopergolo, A.; Schultz, D.C.; Rivadeneira, D.; Altieri, D.C.; Bosari, S. Survivin family proteins as novel molecular determinants of doxorubicin resistance in organotypic human breast tumors. Breast Cancer Res. 2014, 16, R55. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Venkatachalam, M.A.; Wang, J.; Patel, Y.; Saikumar, P.; Semenza, G.L.; Force, T.; Nishiyama, J. Up-regulation of apoptosis inhibitory protein IAP-2 by hypoxia HIF-1-independent mechanisms. J. Biol. Chem. 2001, 276, 18702–18709. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Nishiyama, J.; Yi, X.; Venkatachalam, M.A.; Denton, M.; Gu, S.; Li, S.; Qiang, M. Gene promoter of apoptosis inhibitory protein IAP2: Identification of enhancer elements and activation by severe hypoxia. Biochem. J. 2002, 364, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S.; Duckett, C.S. Iap proteins: Blocking the road to death′s door. Nat.Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, E.C.; Baird, S.; Korneluk, R.G.; MacKenzie, A.E. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 1998, 17, 3247–3259. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.P.; Ferrara, N.; Johnson, R.S. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 2004, 6, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lin, X.; Staver, M.; Shoemaker, A.; Semizarov, D.; Fesik, S.W.; Shen, Y. Evaluating hypoxia-inducible factor-1α as a cancer therapeutic target via inducible RNA interference in vivo. Cancer Res. 2005, 65, 7249–7258. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Nagao, M.; Yamada, Y.; Narikiyo, M.; Ueno, M.; Miyagishi, M.; Taira, K.; Nakajima, Y. Small interfering RNA expression vector targeting hypoxia-inducible factor 1α inhibits tumor growth in hepatobiliary and pancreatic cancers. Cancer Gene Ther. 2006, 13, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Z.F.; Rao, J.Y.; Sato, J.D.; Brown, J.; Messadi, D.V.; Le, A.D. Treatment with siRNA and antisense oligonucleotides targeted to HIF-1α induced apoptosis in human tongue squamous cell carcinomas. Int. J. Cancer 2004, 111, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Stoeltzing, O.; McCarty, M.F.; Wey, J.S.; Fan, F.; Liu, W.; Belcheva, A.; Bucana, C.D.; Semenza, G.L.; Ellis, L.M. Role of hypoxia-inducible factor 1α in gastric cancer cell growth, angiogenesis, and vessel maturation. J. Natl. Cancer Inst. 2004, 96, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Kung, A.L.; Zabludoff, S.D.; France, D.S.; Freedman, S.J.; Tanner, E.A.; Vieira, A.; Cornell-Kennon, S.; Lee, J.; Wang, B.; Wang, J.; et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 2004, 6, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Park, E.J.; Stephen, A.G.; Calvani, M.; Cardellina, J.H.; Monks, A.; Fisher, R.J.; Shoemaker, R.H.; Melillo, G. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005, 65, 9047–9055. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Vogelzang, N.J.; Kilton, L.J.; Leibach, S.J.; Rademaker, A.W.; French, S.; Benson, A.B. A phase ii clinical trial of echinomycin in metastatic soft tissue sarcoma. Investig. New Drugs 1995, 13, 171–174. [Google Scholar] [CrossRef]
- Minet, E.; Mottet, D.; Michel, G.; Roland, I.; Raes, M.; Remacle, J.; Michiels, C. Hypoxia-induced activation of HIF-1: Role of HIF-1α-Hsp90 interaction. FEBS Lett. 1999, 460, 251–256. [Google Scholar] [CrossRef]
- Mabjeesh, N.J.; Post, D.E.; Willard, M.T.; Kaur, B.; Van Meir, E.G.; Simons, J.W.; Zhong, H. Geldanamycin induces degradation of hypoxia-inducible factor 1α protein via the proteosome pathway in prostate cancer cells. Cancer Res. 2002, 62, 2478–2482. [Google Scholar] [PubMed]
- Vaishampayan, U.N.; Burger, A.M.; Sausville, E.A.; Heilbrun, L.K.; Li, J.; Horiba, M.N.; Egorin, M.J.; Ivy, P.; Pacey, S.; Lorusso, P.M. Safety, efficacy, pharmacokinetics, and pharmacodynamics of the combination of sorafenib and tanespimycin. Clin. Cancer Res. 2010, 16, 3795–3804. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D′Andrea, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. Hsp90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Chun, Y.S.; Lee, D.S.; Huang, L.E.; Park, J.W. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood 2008, 111, 3131–3136. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.; Williams, R.; Kirkpatrick, L.; Paine-Murrieta, G.; Powis, G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1α. Mol. Cancer Ther. 2004, 3, 233–244. [Google Scholar] [PubMed]
- Chau, N.M.; Rogers, P.; Aherne, W.; Carroll, V.; Collins, I.; McDonald, E.; Workman, P.; Ashcroft, M. Identification of novel small molecule inhibitors of hypoxia-inducible factor-1 that differentially block hypoxia-inducible factor-1 activity and hypoxia-inducible factor-1α induction in response to hypoxic stress and growth factors. Cancer Res. 2005, 65, 4918–4928. [Google Scholar] [CrossRef] [PubMed]
- Carroll, V.A.; Ashcroft, M. Role of hypoxia-inducible factor (HIF)-1α versus HIF-2α in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-i, or loss of von hippel-lindau function: Implications for targeting the hif pathway. Cancer Res. 2006, 66, 6264–6270. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Cerniglia, G.J.; Lindsten, T.; Koumenis, C.; Maity, A. Dual PI3K/mtor inhibitor NVP-BEZ235 suppresses hypoxia-inducible factor (HIF)-1α expression by blocking protein translation and increases cell death under hypoxia. Cancer Biol. Ther. 2012, 13, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Fang, J.; Xia, C.; Shi, X.; Jiang, B.H. Trans-3,4,5′-trihydroxystibene inhibits hypoxia-inducible factor 1α and vascular endothelial growth factor expression in human ovarian cancer cells. Clin. Cancer Res. 2004, 10, 5253–5263. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Kloog, Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1α, causing glycolysis shutdown and cell death. Cancer Res. 2005, 65, 999–1006. [Google Scholar] [PubMed]
- Fang, J.; Cao, Z.; Chen, Y.C.; Reed, E.; Jiang, B.H. 9-β-d-Arabinofuranosyl-2-fluoroadenine inhibits expression of vascular endothelial growth factor through hypoxia-inducible factor-1 in human ovarian cancer cells. Mol. Pharmacol. 2004, 66, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Zage, P.E.; Zeng, L.; Xu, L.; Cascone, T.; Wu, H.K.; Saigal, B.; Zweidler-McKay, P.A.; Heymach, J.V. Multiple receptor tyrosine kinases regulate HIF-1α and HIF-2α in normoxia and hypoxia in neuroblastoma: Implications for antiangiogenic mechanisms of multikinase inhibitors. Oncogene 2010, 29, 2938–2949. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Puri, N.; Khramtsov, A.; Ahmed, S.; Nallasura, V.; Hetzel, J.T.; Jagadeeswaran, R.; Karczmar, G.; Salgia, R. A selective small molecule inhibitor of c-MET, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. 2007, 67, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, Y.; Mukohara, T.; Tomioka, H.; Ekyalongo, R.C.; Kataoka, Y.; Inui, Y.; Kawamori, Y.; Toyoda, M.; Kiyota, N.; Fujiwara, Y.; et al. Excessive MET signaling causes acquired resistance and addiction to MET inhibitors in the MKN45 gastric cancer cell line. Investig. New Drugs 2013, 31, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. Erbb receptors and cancer: The complexity of targeted inhibitors. Nature Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Franovic, A.; Gunaratnam, L.; Smith, K.; Robert, I.; Patten, D.; Lee, S. Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 13092–13097. [Google Scholar] [CrossRef] [PubMed]
- Karakashev, S.V.; Reginato, M.J. Hypoxia/HIF1A induces lapatinib resistance in ERBB2-positive breast cancer cells via regulation of DUSP2. Oncotarget 2015, 6, 1967–1980. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.I.; Fehrenbacher, L.; Hainsworth, J.D.; Heim, W.; Berlin, J.; Holmgren, E.; Hambleton, J.; Novotny, W.F.; Kabbinavar, F. Bevacizumab in combination with fluorouracil and leucovorin: An active regimen for first-line metastatic colorectal cancer. J. Clin. Oncol. 2005, 23, 3502–3508. [Google Scholar] [CrossRef] [PubMed]
- Shih, T.; Lindley, C. Bevacizumab: An angiogenesis inhibitor for the treatment of solid malignancies. Clin. Ther. 2006, 28, 1779–1802. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, G.; Rangaswami, H.; Jain, S.; Kundu, G.C. Hypoxia regulates cross-talk between Syk and Lck leading to breast cancer progression and angiogenesis. J. Biol. Chem. 2006, 281, 11322–11331. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Subarsky, P.; Hill, R.P. The hypoxic tumour microenvironment and metastatic progression. Clin. Exp. Metastasis 2003, 20, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Udelnow, A.; Kreyes, A.; Ellinger, S.; Landfester, K.; Walther, P.; Klapperstueck, T.; Wohlrab, J.; Henne-Bruns, D.; Knippschild, U.; Wurl, P. Omeprazole inhibits proliferation and modulates autophagy in pancreatic cancer cells. PLoS ONE 2011, 6, e20143. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Joshua, A.M.; Saggar, J.K.; Yu, M.; Wang, M.; Kanga, N.; Zhang, J.Y.; Chen, X.; Wouters, B.G.; Tannock, I.F. Effect of pantoprazole to enhance activity of docetaxel against human tumour xenografts by inhibiting autophagy. Br. J. Cancer 2015, 112, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Saggar, J.K.; Yu, M.; Wang, M.; Tannock, I.F. Mechanisms of drug resistance related to the microenvironment of solid tumors and possible strategies to inhibit them. Cancer J. 2015, 21, 254–262. [Google Scholar] [CrossRef] [PubMed]
- De Milito, A.; Iessi, E.; Logozzi, M.; Lozupone, F.; Spada, M.; Marino, M.L.; Federici, C.; Perdicchio, M.; Matarrese, P.; Lugini, L.; et al. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res. 2007, 67, 5408–5417. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.; Kim, D.K.; Kim, Y.B.; Oh, T.Y.; Lee, J.E.; Cho, S.W.; Kim, H.C.; Hahm, K.B. Selective induction of apoptosis with proton pump inhibitor in gastric cancer cells. Clin. Cancer Res. 2004, 10, 8687–8696. [Google Scholar] [CrossRef] [PubMed]
- De Milito, A.; Canese, R.; Marino, M.L.; Borghi, M.; Iero, M.; Villa, A.; Venturi, G.; Lozupone, F.; Iessi, E.; Logozzi, M.; et al. Ph-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int. J. Cancer 2010, 127, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Mittal, D.; Stagg, J.; Smyth, M.J. Targeting cancer-derived adenosine: New therapeutic approaches. Cancer Discov. 2014, 4, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Page, D.B.; Wolchok, J.D. Immune modulation for cancer therapy. Br. J. Cancer 2014, 111, 2214–2219. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Belikoff, B.; Schreiber, T.H.; Sethumadhavan, S.; Abbott, R.; Philbrook, P.; Thayer, M.; Shujia, D.; et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J. Mol. Med. 2014, 92, 1283–1292. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-Y.; Lee, J.-Y. Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome. Int. J. Mol. Sci. 2017, 18, 1854. https://doi.org/10.3390/ijms18091854
Kim J-Y, Lee J-Y. Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome. International Journal of Molecular Sciences. 2017; 18(9):1854. https://doi.org/10.3390/ijms18091854
Chicago/Turabian StyleKim, Jae-Young, and Joo-Yong Lee. 2017. "Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome" International Journal of Molecular Sciences 18, no. 9: 1854. https://doi.org/10.3390/ijms18091854
APA StyleKim, J.-Y., & Lee, J.-Y. (2017). Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome. International Journal of Molecular Sciences, 18(9), 1854. https://doi.org/10.3390/ijms18091854