Inflammatory Diseases and Growth: Effects on the GH–IGF Axis and on Growth Plate



Division of Paediatric Endocrinology and Diabetology, Department of Obstetrics, Gynaecology and Paediatrics, Azienda AUSL–IRCCS, Viale Risorgimento, 80, 42123 Reggio Emilia, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(9), 1878; https://doi.org/10.3390/ijms18091878
Submission received: 30 July 2017
/
Revised: 23 August 2017
/
Accepted: 29 August 2017
/
Published: 31 August 2017
(This article belongs to the Special Issue IGFs in Health and Disease)
Abstract
:This review briefly describes the most common chronic inflammatory diseases in childhood, such as cystic fibrosis (CF), inflammatory bowel diseases (IBDs), juvenile idiopathic arthritis (JIA), and intrauterine growth restriction (IUGR) that can be considered, as such, for the changes reported in the placenta and cord blood of these subjects. Changes in growth hormone (GH) secretion, GH resistance, and changes in the insulin-like growth factor (IGF) system are described mainly in relationship with the increase in nuclear factor-κB (NF-κB) and pro-inflammatory cytokines. Changes in the growth plate are also reported as well as a potential role for microRNAs (miRNAs) and thus epigenetic changes in chronic inflammation. Many mechanisms leading to growth failure are currently known; however, it is clear that further research in the field is still warranted.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Most chronic inflammatory diseases in childhood are characterised by impaired growth. The mechanism underlying the pathophysiology of this process is not clearly understood yet, although it is a complex phenomenon which comprises of chronic inflammation itself, prolonged use of glucocorticoids, and suboptimal nutrition [1,2].
Longitudinal growth in humans is under the control of multiple factors, and its regulation starts during foetal life and continues throughout childhood, with varying influence of each factor at different stages. The main determinants of growth are prenatal and perinatal health, genetic potential, adequate nutrition, and endocrine interplay. In particular, hormones involved in this process are growth hormone (GH) and insulin-like growth factor (IGF) system, thyroid hormones, insulin, and sex steroids [3].
During the last decade, attention has been focused on epigenetics and its role in normal and pathological development, and a number of epigenetic mechanisms have been recognised as regulators of growth pattern [4].
Growth retardation may, therefore, be secondary to a dysfunction in multiple systems, such as disruption of the GH–IGF axis and IGF system, changes in the growth plate, epigenetic modifications and malnutrition.
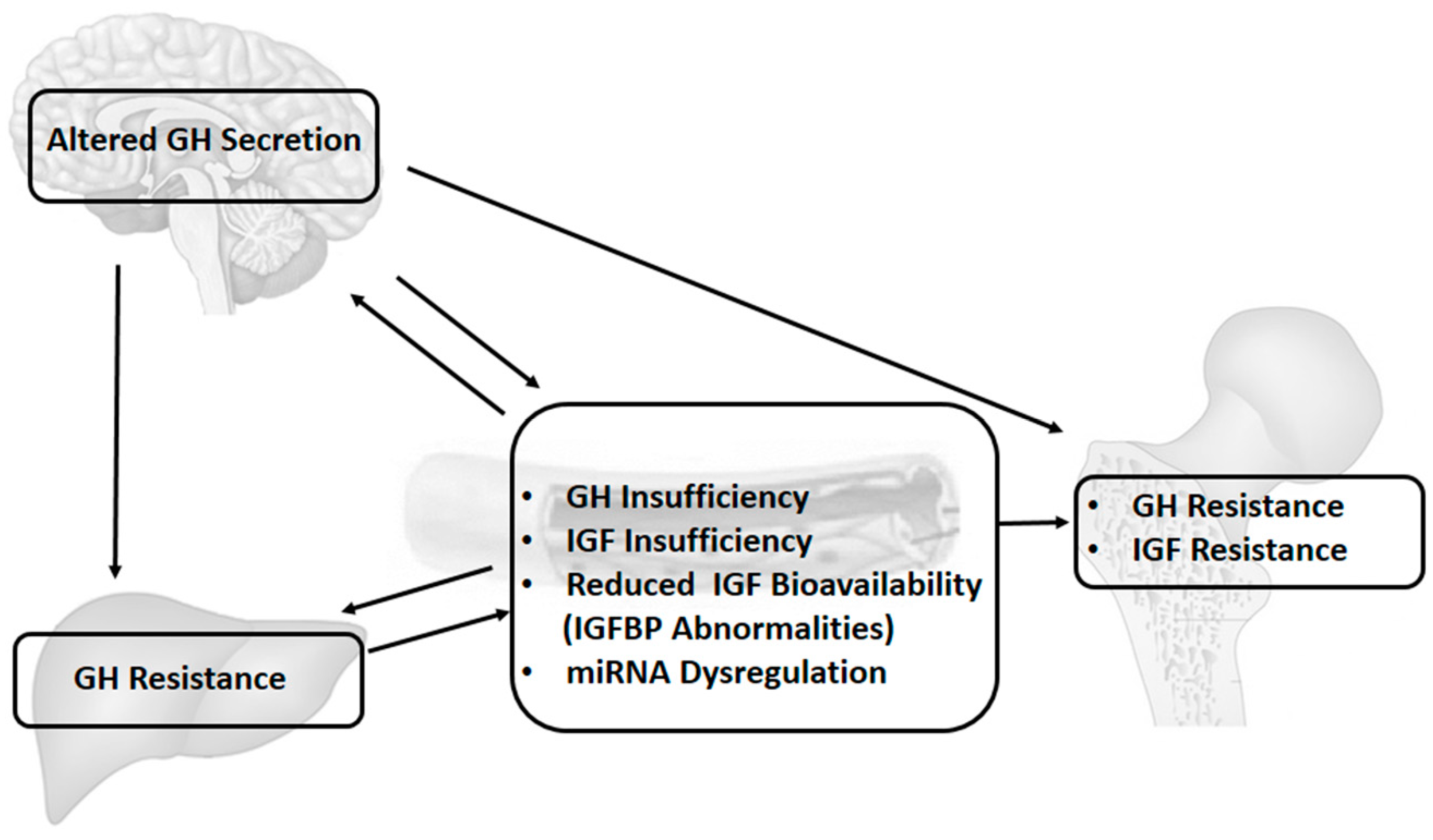
The GH–IGF axis and IGF system in children with chronic inflammation may be altered by several mechanisms, such as GH/IGF-1 insufficiency, GH/IGF-1 resistance, down-regulation of GH/IGF receptors, disruption in downstream GH/IGF signalling pathways, or dysregulation of IGF binding proteins (IGFBPs) [1]. Pro-inflammatory cytokines play a crucial role in the development of these abnormalities. Many studies over the last decade clearly demonstrate huge interactions between pro-inflammatory cytokines and the IGF system.
The aim of this review is to give an overview of the main mechanisms underlying the onset of growth impairment in chronic inflammatory disease in childhood.
2. Overview of Most Frequent Chronic Inflammatory Diseases with Impaired Growth
Growth evaluations are among the most common referrals to paediatric endocrinologists, and many are owing to growth failure in chronic inflammatory diseases.
Cystic fibrosis (CF), inflammatory bowel disease (IBD), and juvenile idiopathic arthritis (JIA) are the most common chronic inflammatory conditions in childhood associated with growth impairment. Evidence in recent years has shown that intrauterine growth restriction (IUGR) also can be considered as a chronic inflammatory condition [5,6].
2.1. Cystic Fibrosis
CF is an autosomal recessive disorder caused by mutations in a gene that encodes for the cystic fibrosis transmembrane conductance regulator (CFTR) protein, an epithelial chloride channel that is widely expressed and is involved in the homeostasis of ions and other metabolites.
CFTR is widely expressed, and therefore, CF affects different organs and systems. Lung damage is mostly responsible for high morbidity and mortality, and is characterised by bronchiectasis, small airway obstruction, and progressive respiratory impairment.
However, epithelial cell dysfunction causes important comorbidities, such as malabsorption, biliary cirrhosis, and infertility [10]. The severity of CF varies greatly from person to person, regardless of age [11]. The endocrine system is also frequently involved in CF patients, with important consequences, including poor linear growth and diabetes [12]. The prevalence of short stature in patients with CF is approximately 20% [13].
Morison et al., reporting cross-sectional data from 31 CF Centres in the UK, indicated that during the first decade of life, height and weight in patients with CF are maintained at about 0.5 SDs below those of the general population, and fall away progressively after this age [14]. Delayed puberty may also be involved in the determinism of the reduction of linear growth in these patients. Several authors have shown how adolescents with CF present lower peak height velocity, with pubertal delay and a later pubertal growth spurt [15,16].
Short stature in CF may have an impact on disease severity because it is an independent predictor of mortality. This evidence may reflect a subgroup of CF patients with poorer nutrition, or chronic inflammation and ongoing pulmonary exacerbation [17].
Fat and micronutrient malabsorption may contribute to poor growth. However, other factors involved in the determinism of growth failure in CF patients include chronic inflammation, chronic infection and treatment with inhaled and systemic glucocorticoid medications.
There is now sufficient evidence to suggest that poor growth in CF is already seen in the neonatal period, and that the CF genotype delF508 plays a contributing role [1].
2.2. Inflammatory Bowel Diseases
IBDs are conditions characterised by chronic or recurring immune response and inflammation of the gastrointestinal tract. The two most common IBDs are ulcerative colitis (UC) and Crohn’s disease (CD).
IBDs are more common in developed countries. There is north-to-south variation, and they are more common in urban communities compared with rural areas. These observations suggest that urbanisation is a potential contributing factor.
According to Centres for Disease Control and Prevention (CDC) and the USA National Health Protection Agency, although the incidence and prevalence of UC and CD are beginning to stabilise in high-incidence areas, such as northern Europe and North America, they continue to rise in low-incidence areas, such as southern Europe, Asia, and much of the developing world [18].
These conditions affect as many as 1.4 million persons in the United States, and 2.2 million persons in Europe [19].
Pathophysiology of IBDs is complex, and most authors agree on the fact that these conditions result from interactions between environmental factors, genetic predisposition, and immune response.
Intestinal epithelial damage with infiltration of a large number of cells into the lamina propria, such as T and B lymphocytes, macrophages, dentritic cells, and neutrophils, is a constant occurrence along with IBDs [2,4,12]. Moreover, evidence suggests a defect of immune response regulation in these conditions, with active secretion of a large number of cytokines with both pro-inflammatory and anti-inflammatory action, including TNF, IFN-γ, IL-6, IL-12, IL-21, IL-23, IL-17, integrin, IL-10, TGFβ, and IL-35 [2,11]. The imbalance in the regulation and secretion of these cytokines plays a crucial role in initiating and sustaining intestinal inflammation and tissue injury.
Both these diseases are responsible for severe gastrointestinal symptoms, such as abdominal pain, diarrhoea, rectal bleeding, nausea/vomiting or constipation, with a potential huge reduction in quality of life.
However, the clinical presentation of IBDs in children and adolescents can be variable, and up to 22% of children may present with extra-intestinal manifestations as the only predominant initial feature. Main extra-intestinal manifestations of IBDs are growth failure, anaemia, erythema nodosum, pyoderma gangrenosum, arthritis, perianal disease, osteopenia, osteoporosis, primary sclerosing cholangitis, autoimmune hepatitis, episcleritis, uveitis, and pancreatitis [20]. Impaired linear growth is a frequent extra-intestinal complication of IBDs in children and adolescents. Up to 19–31% of children with CD and UC present with this sign as an initial feature [21,22,23]. Moreover, early growth delay has been associated with permanent growth retardation in 17% of patients [24].
2.3. Juvenile Idiopathic Arthritis
JIA is a heterogeneous group of diseases characterised by arthritis of unknown origin, with onset before age of 16 years. JIA is a common childhood rheumatic disease. The highest frequency of this condition occurs in children aged 1–3 years, and its incidence is around 20–30/100,000 below the age of 16 years [25].
JIA classification comprises different subtypes (systemic arthritis, polyarthritis, oligoarthritis, enthesitis-related arthritis, psoriatic arthritis, and undifferentiated arthritis) which are characterised by distinct clinical features and varying spectrums of disease severity [26].
Pathogenesis of JIA involves the humoral and cell-mediated immune system. T lymphocytes play a pivotal role, releasing pro-inflammatory cytokines and promoting type-1 helper T lymphocyte response. An abnormal interplay between type 1 and type 2 T helper cells has been shown [27].
Growth impairment is a well-known long-term complication in patients with JIA. Children with systemic or polyarticular JIA (about 30–40% of all JIA cases) present a higher incidence of growth disturbances among all JIA patients [28].
Children with arthritis suffer from a variety of growth disorders, ranging from general growth retardation to local acceleration of growth in the affected limb [29,30]. Leg-length discrepancy is common, as unilateral knee arthritis may result in overgrowth of the distal femur, caused by increased blood supply to the inflamed joint with consequent accelerated growth of the ossification centres.
Puberty may also be involved with retardation in the appearance of secondary sex characteristics [31]. Risk factors for impaired linear growth are represented by extended periods of active disease, and are exacerbated by long-term use of systemic steroids.
Moreover, an elevated erythrocyte sedimentation rate value seems to be a good predictor of risk for growth retardation [32].
2.4. Intrauterine Growth Restriction
IUGR is defined as the failure of a foetus to attain its expected foetal growth at any gestational age. It affects approximately 7–15% of pregnancies with an estimated prevalence of 8% in the general population [33].
The incidence of IUGR varies among countries, populations, and races, and increases with decreasing gestational age. Around 14 to 20 million infants have been affected with IUGR in developing countries annually. A large number of IUGR infants are seen in the also Asian, African and Latin American continents [34]. IUGR represents therefore one of the most important causes of perinatal mortality and morbidity worldwide.
The main determinants of foetal growth are genetic heritage of the foetus, the integrity of the materno–placento–foetal unit, adequate nutrient and oxygen supply, and the right hormonal milieu. Impaired foetal growth may be due to inadequacy of any one of these parameters.
Risk factors for the development of IUGR are, therefore, extremely variable, and may include maternal medical and social conditions, foetal abnormalities, and placental dysfunction.
IUGR puts the foetus and neonate at higher risk for perinatal mortality and morbidity [35], and it is also considered a risk factor for the future development of insulin resistance and type 2 diabetes [36].
These conditions are related by association with high serum concentrations of pro-inflammatory cytokines, such as interleukin (IL)-6 and tumour necrosis factor α (TNF-α), and low adiponectin concentrations [36,37,38,39,40,41,42].
Interestingly, IL-6 concentrations in placental lysates from IUGR pregnancies correlate with birth length, birth weight, and head circumference [36].
The increased IL-6 concentrations confirm the hypothesis that IUGR shares a common pathophysiology with other conditions characterised by chronic inflammation.
Within the first two years of life, especially in developed countries, the majority of IUGR subjects present partial or complete catch-up growth. However, approximately 13% of these subjects do not present a catch-up growth, and remain short after two years of life [43].
3. Interactions between Pro-Inflammatory Cytokines and GH–IGF Axis, IGF System, and Bone
As stated above, the mechanisms underlying the pathophysiology of growth impairment in chronic inflammatory diseases in childhood is a complex phenomenon in which chronic inflammation plays a central role [1,2].
3.1. GH–IGF Axis and IGF System
GH is a 191 amino acid protein that promotes growth by increasing cell size and cell number, and by promoting differentiation of bone and muscle cells [44]. Deficiency in either GH or the GH receptor causes severe postnatal growth retardation and subsequent dwarfism, both in humans and mice [45,46]. IGF-1 is expressed by most cells throughout development, and represents an essential factor for cell growth, intrauterine development, and postnatal growth [47,48,49,50]. IGF-1 deficiency in humans and mice causes severe intrauterine and postnatal growth retardation, and is associated with perinatal lethality, and developmental defects in the bone, muscle, and central nervous and reproductive systems. The effects of GH on postnatal body growth have been extensively described [51,52]. Briefly, GH acts on a major target organ, the liver, to stimulate the synthesis and secretion of IGF-1, which reaches its skeletal targets as a true endocrine reagent (the somatomedin hypothesis) [53], but it can stimulate longitudinal bone growth directly, also, by local production of IGF-1 (modified somatomedin hypothesis) [54,55,56,57,58,59]. Both IGF-1 and GH are necessary for postnatal growth [60]. IGF-1 regulates with a negative feedback loop mode of action on GH secretion at the pituitary level. In cartilage cells, IGF-1 has GH-independent stimulating effects, but its effects are optimised by a synergistic action with GH itself [61].
GH binds to two GH receptors (GHRs), causing a dimerisation process that activates the GHR-associated Janus kinase (JAK)2 tyrosine kinase, and tyrosine phosphorylation of both JAK2 and GHR. These events activate a series of other signalling molecules, such as mitogen-activated protein kinases (MAPKs), insulin receptor substrates, phosphatidylinositol-3-phosphate kinase, diacylglycerol, protein kinase C, intracellular calcium, and signal transducer and activator of transcription (STAT) factors. These signalling molecules lead to changes in enzymatic activity, transport function, and gene expression that determine final changes in growth and metabolism. The GH binding protein is proteolysed from the cell surface receptor, and regulates GH bioavailability, and can be used as a marker of receptor number and function [52].
The IGF system is of upmost importance for somatic growth in vertebrates. Both IGF-1 and IGF-2 signal through the IGF type 1 receptor (IGF-1R). IGF-1R regulates proliferation, differentiation, and apoptosis in many tissues and cell types. IGF-1R is a transmembrane tyrosine kinase receptor. Both ligands and the IGF-1R are similar to insulin and the insulin receptor [62,63]. Six IGF binding proteins (IGFBPs) and the mannose-6-phosphate receptor (type 2 IGF receptor) are the main regulators of IGF-1 and IGF-2 bioavailability [64,65,66].
3.2. Changes in the GH–IGF Axis and IGF System and Interactions with Pro-Inflammatory Cytokines
The GH–IGF axis may be altered by several mechanisms, and growth failure in children with chronic inflammatory conditions may be secondary to GH/IGF-1 insufficiency, GH/IGF-1 resistance, down-regulation of GH/IGF receptors, disruption in downstream GH/IGF signalling pathways, dysregulation of IGFBPs and thus of IGF bioavailability, and finally, gene regulation that is modified by changes in the microRNA system, as well as other potential epigenetic mechanisms (Figure 1).
Pro-inflammatory cytokines play a crucial role in the development of these abnormalities. Many studies over the last decade clearly demonstrate huge interactions between pro-inflammatory cytokines and the IGF system.
For example, studies in transgenic mice showed how increased IL-6 serum levels were associated with low IGF-1 serum levels and growth delay [67]. Street et al. showed also a relationship between inflammatory status and the IGF system, with a consequent effect of these interactions on longitudinal growth [36,68].
IL-6 may antagonise GH actions through disruption of JAK/STAT signalling. Recent evidence suggests a role for suppressor of cytokine signalling (SOCS) family proteins in these processes. Pro-inflammatory cytokines stimulate SOCS proteins with a consequent reduction in JAK2 and STAT activation [69,70,71]. Similarly, an abnormal expression of STAT5 and STAT3, due to the action of another crucial pro-inflammatory cytokine, IL-1β, can disrupt GH signalling [72].
The IGF-1 signalling pathway may also be altered in chronic inflammatory conditions. Several authors showed how TNF-α, IL-6, and IL-1β dysregulate IGF-1 intracellular mediators MAPK/extracellular signal-regulated kinases (ERKs), and phosphoinositide 3-kinase (PI3K), in chondrocytes [73,74,75]. Inflammation is sustained by the activation of several immune cell types which secrete soluble cytokines, as chemokines, interferons, and ILs, activating bone resorption and inhibiting bone growth/formation processes at local and systemic levels [76].
These messenger molecules influence differentiation and activity of the main skeletal cell types: osteoblasts, osteoclasts, and chondrocytes [77,78].
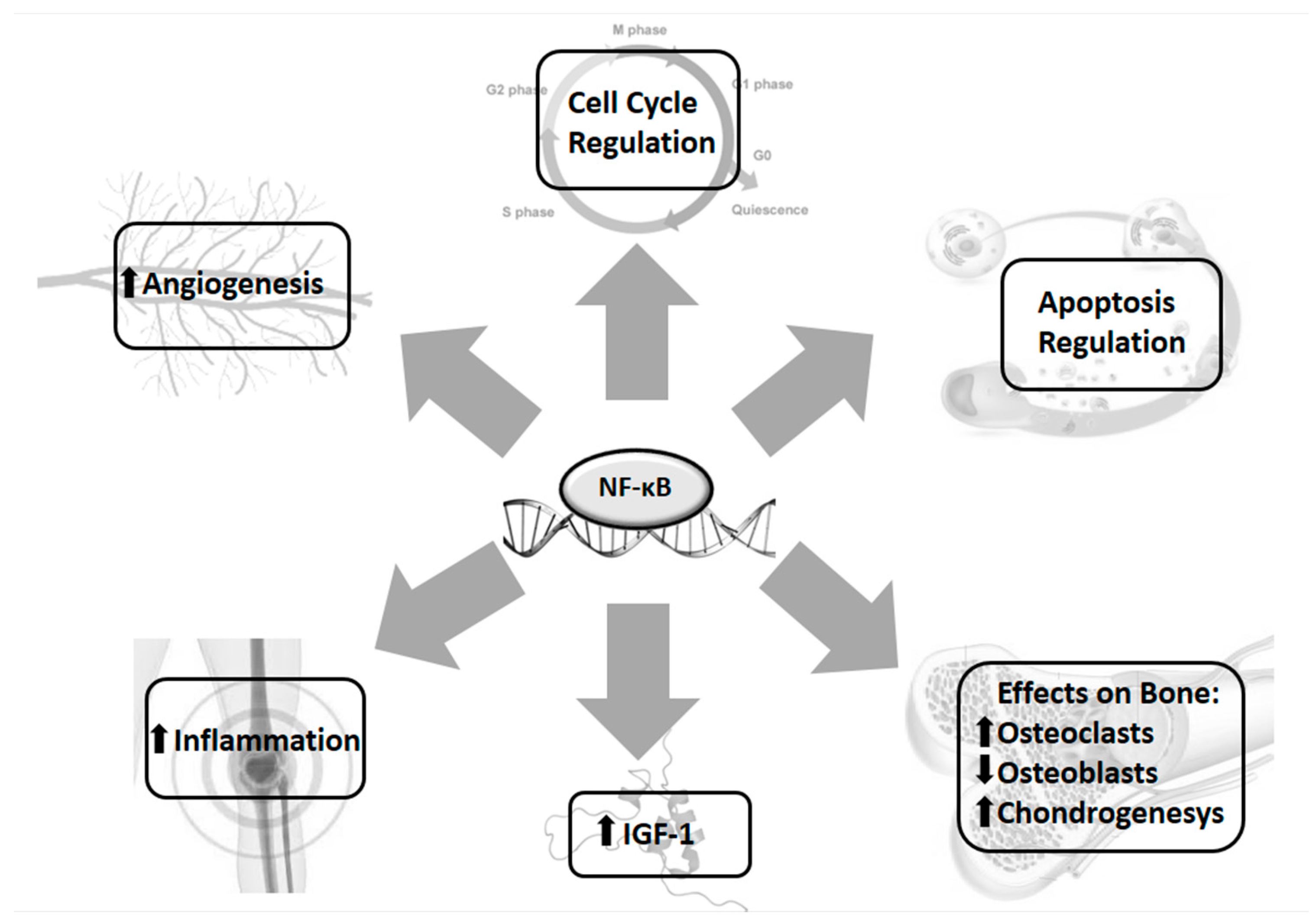
Experimental and clinical evidence suggests that the nuclear factor-κB (NF-κB) pathway plays a role in IGF1-GH signalling [79,80], and exerts a regulatory role in bone growth and development [81,82,83]. NF-κB is a family of transcription factors which can form homo- and heterodimers, including the five members, p50 (NF-κB1), p52 (NF-κB2), p65 (RelA), RelB, and c-Rel. All of these proteins are structurally homologous and share functional domains: N-terminal Rel homology domain (RHD), responsible for dimerisation as well as DNA-binding, and a transactivation domain (TA) relevant for the transcriptional activity. Moreover, they present a nuclear localisation signal (NLS) that promotes translocation into the nucleus from the cytoplasm after stimulation [84].
In resting cells, NF-κB dimers are retained in the cytoplasm, where they are covalently bound to the inhibitor of NF-κB (IκB) proteins, which mask their NLSs.
The main and most well studied NF-κB activation pathways are canonical, and alternative pathways differ mainly in composition of downstream dimeric effectors.
NF-κB pathways are activated by many extracellular signals, e.g., pro-inflammatory cytokines, hormones, growth factors, viral proteins, lipopolysaccharide (LPS), and RANKL.
In the activated classic pathway, the IκB kinase (IKK) induces the ubiquitination and subsequent degradation of IκB. Subsequently, NF-κB, predominantly p50/p65, translocates into the nucleus, where it regulates genes involved in cell proliferation, differentiation, and death [85].
3.3. Inflammation and miRNAs
Epigenetics, defined as “the inheritance of variation (-genetics) above and beyond (epi-) changes in the DNA sequence” [88], refers to inheritable changes of gene function, which do not imply a change in the DNA sequence [89].
MicroRNAs (miRNAs) are a recent chapter in the study of epigenetic regulation. They are endogenous small non-coding RNAs, approximately 22 nucleotides long that act as post transcriptional regulators [90]. Mature miRNAs hybridise to partially complementary binding sites that are typically localised in the 3′ untranslated regions (3′UTR) of target mRNAs [91].
Upon binding, miRNA can cause the degradation of the mRNA target if the complementarity between them is perfect; when their complementarity is only partial, the target’s translational repression takes place [92,93]. In these ways, each single miRNA regulates several hundreds of transcripts, and each mRNA can be regulated by many miRNAs [94,95].
In turn, miRNAs themselves are targets of transcription factors and molecular signals, thus explaining the complexity of the regulatory network existing on and controlled by miRNAs [96,97].
The complexity of miRNA action, and their strict regulation, provides the evidence that miRNAs are crucial in various physiological and pathological processes; in fact, they are involved in inflammation, apoptosis, differentiation, and proliferation [98].
Moreover, miRNAs have a role in the antigen-presenting capacity and costimulation activity of macrophages and dendritic cells [99,100], and they are also essential in the development and actions of B and T cells [84,101,102].
In particular, under inflammatory stimuli, miRNAs act as post-transcriptional regulators of genes involved in the adaptive and innate immune response; for example, they control the expression of mediators involved in the Toll like receptor (TLR) signalling pathways, which all culminate in the activation of NF-κB [103,104,105].
During many biological processes, like inflammation or innate and adaptive immunity, NF-κB regulates the transcription of miRNAs through the induction of genes codifying for regulatory proteins [106].
NF-κB targets miRNA sequences, including miR-9, miR-21, miR-143, miR-146, and miR-224 [103,107,108,109,110,111], which are involved in feedback mechanisms regulating the transcription of NF-κB itself.
miRNAs have the potential to influence the expression of the proteins involved in the regulation of GH–IGF axis. Their deregulation has been well documented in association with chronic paediatric diseases in relationship to the inflammatory state, as in CF [112,113,114,115], IBD [116,117,118,119,120,121,122], JIA [123,124], and IUGR [125,126].
3.4. Bone Growth and Inflammation
Bone has a lot of functions, such as structural support, calcium reserve, and many others. During life, bone undergoes modelling and remodelling, which is due to the action of two major cell types: osteoblasts and osteoclasts. Modelling is an adaptive process by which bones answer to external influences to adjust the skeleton to events occurring during life [127].
Bone renewal occurs physiologically by bone remodelling. This process is necessary for maintaining bone strength and mineral homeostasis [127].
Longitudinal growth occurs only during childhood. It is only in this period that bone formation can occur independently of bone resorption [128]. The process of longitudinal growth occurs in growth plates. An alteration between bone formation and bone resorption occurs in chronic inflammatory conditions. Inflammation is a primary cause of bone loss. This bone loss is both local and systemic, and is associated with an enhancement of bone resorption or an inhibition of bone formation.
Most of chronic paediatric inflammatory diseases are associated with a catabolic state that reduces bone formation [128].
The epiphyseal growth plate is the final target organ of the above described growth-regulating mechanisms. The epiphyseal growth plates are located in the proximal and distal parts of the long bones and have a definite cellular organisation according to stage of maturation, with germinative, proliferative, hypertrophic, and degenerative cell layers. The germinative cell layer consists of stem cells or progenitor cells, which rarely divide. During the process of longitudinal bone growth, stem cells enter the proliferative cell layer and begin to divide frequently, forming continuous cell columns parallel to the longitudinal axis of the bone. Subsequently, these cells stop dividing, mature, and become part of the hypertrophic cell layer [129]. Finally, calcification occurs as cartilaginous matrix is transformed into bone matrix. Longitudinal bone growth is due to the recruitment of new progenitor cells from the stem cell layer that undergo divisions in the proliferative layer, and then increase in size in the hypertrophic layer. The growth plate is a constantly renewing tissue that pushes the epiphysis further and further away from the centre of the long bone [130].
Linear growth occurs during development and the childhood years until epiphyseal fusion occurs. This is due to endochondral ossification, and is regulated by systemic hormones and paracrine or autocrine factors. Childhood growth requires GH, IGF-1, glucocorticoids, and thyroid hormone to be present and normally active. Sex steroids are then additionally necessary for the pubertal growth spurt and epiphyseal fusion [131]. Furthermore, during linear growth, GH, IGF-1, glucocorticoids, and thyroid hormone interact at the level of the hypothalamus and pituitary. However, recent evidence suggests that these hormones also act directly on peripheral target tissues, such as liver and growth plate [132].
GH action has both direct and indirect effects on the growth plate. GH acts indirectly, stimulating the production of IGF-1 that promotes chondrocyte hypertrophy, which in turn exerts its effects on the growth plate. The direct effect of GH on the growth plate stimulates chondrocyte proliferation [133].
The balance between bone degradation and bone building is critical for the physiological bone homeostasis. NF-κB and cytokines controlled by this factor may perturb this equilibrium in paediatric chronic inflammatory diseases [134,135,136].
NF-κB activation is a relevant component for osteoclast development, differentiation, and survival, cooperating with other pro-inflammatory cytokines. Loss of NF-κB signalling prevents osteoclastogenesis [82]. NF-κB knockout mice showed severe osteopetrosis [137].
Furthermore, NF-κB inhibits osteoblast differentiation by blocking transcription factors induced by several extracellular signals, including bone morphogenic proteins (BMPs), fibroblast growth factor (FGF), transforming growth factor β (TGF-β), and transducers of GH–IGF-1 axis. The inhibition of bone formation by NF-κB is well documented by in vivo and in vitro experiments [138,139].
NF-κB is also involved in the regulation of growth plate chondrogenesis by IGF-1, which promotes bone longitudinal growth during childhood and foetal development by stimulating chondrocytes proliferation and preventing apoptosis [140,141]. Various stimuli, such as TNF-α, LPS, and hypoxia, increase the expression of IGF-1, vascular endothelial growth factor (VEGF), and FGF-2 by an NF-κB dependent mechanism [142] (Figure 2).
Another system involved in growth failure in the chronic inflammation state is represented by the adrenal axis. Circulating levels of endogenous glucocorticoids (GC) increase during inflammation, leading to growth suppression. Steroids act both at systemic and local level, as well as the GH system. Localised GC directly inhibits chondrocyte proliferation and bone mineralisation, as well as increases apoptosis. By systemic action, GC inhibits GH secretion, with a consequent reduction of IGF-1 production and activity [143].
Glucocorticoids are frequently used for the treatment for many inflammatory diseases. Initially, steroids could have beneficial effects on bone growth, suppressing inflammation, but long-term therapy frequently has adverse effects on bone [144]. High levels of circulating glucocorticoids disrupt bone remodelling, disrupting the balance between formation and resorption. Glucocorticoids act directly on osteoblasts, decreasing their proliferation and the consequent production of specific proteins as osteocalcin, a bone specific alkaline phosphatase [145,146]. Enhanced adverse effects of glucocorticoids are partly dependent on the underlying illness [147,148].
4. Specific Changes in the GH–IGF Axis, IGF System, and Growth Plate in Individual Inflammatory Conditions
In CF patients, studies of GH secretion are limited. Arginine and clonidine GH stimulation tests, performed in a group of adolescents with CF, revealed how approximately 50% of these patients have peak GH levels <6 µg/L, and IGF-1 levels of −0.5 SDs, suggesting the potential co-existence of GH insufficiency and GH resistance [149].
A significant positive correlation was indeed found between insulin secretion and height growth velocity and serum IGFBP-3 levels [149]. According to data published by our group, CF patients have higher serum concentrations of IL-1β, IL-6, TNF-α, and IGFBP-2. Conversely, serum concentrations of IGF-1 and IGF-2 are significantly lower. IGFBP-3 serum concentrations are similar, with comparable IGF-1/IGFBP-3, and decreased IGF-1/IGFBP-2 and IGF-2/IGFBP-2 molar ratios.
Statistical analyses revealed a significant positive correlation between IGFBP-2 and IL-6 and a negative correlation between IGFBP-2 and IGFBP-3, suggesting that inflammation is an important modulator of the IGF-IGFBP system with an overall reduction in IGF bioactivity in CF [68].
Moreover, circulating levels of TNF-α, IL-6, insulin, and the IGF system were found to be related to linear growth in children with CF [36].
IGF-1 concentrations in patients with CF are significantly lower than those in a healthy control population. IGF-1 reduction in these patients may reflect their catabolic state and play a part in their abnormal growth pattern [150,151].
Animal models have shown that chondrocytes express functional CFTR [152], and cartilage abnormalities in tracheal ring structure have been reported in CF, both in humans and in animal models [153,154]. Both CFTR loss of function and local and systemic inflammation are hypothesised to be responsible for these changes, however, it is yet unclear whether cartilage abnormalities involve growth plate chondrocytes also, as these data are missing to date in the literature.
The precise mechanisms underlying growth failure in IBDs are not well known. The relative roles of impaired nutrition and active inflammation in disturbing GH–IGF axis remain controversial. Nutritional status regulates the IGF system, with both caloric and protein restriction resulting in low serum IGF-1 and IGFBP-3 levels. Serum IGFBP-2 regulation is more dependent on protein intake [155]. However, it was demonstrated that linear growth impairment occurs, independent of undernutrition, as a direct result of the inflammatory process. According to Ballinger and his group, in a rat experimental model of colitis, approximately 30–40% of linear growth impairment was directly related to inflammation [156]. These same authors suggested a normal stimulated and spontaneous GH production in children with CD, and growth failure with a low IGF-1 plasma concentration, conditions compatible with GH resistance [157]. Moreover, we previously described low IGF-1 and high IGFBP-2 levels related to disease activity and anatomical distribution, consistent with active inflammation modifying the IGF–IGFBP system.
Intestinal inflammation is well known to have a negative impact on bone health. Scientific evidence has confirmed a reduction in bone mineral density in patients with IBD, and changes in the growth plate cartilage, in addition. In detail, induction of moderate intestinal inflammation in young male mice has been reported to reduce growth plate thickness and induce a hypertrophic response in chondrocyte matrix [158]. Specific data in humans are still missing in the literature.
Impairment of GH–IGF axis in JIA may be due to several mechanisms, ranging from GH secretion abnormalities to GH resistance or increased IGF-1 clearance [159]. Templ et al. showed that GH response to GHRH was reduced in patients with newly diagnosed rheumatoid arthritis, compared to healthy controls [160], and others showed how an inflammatory cytokine milieu caused impairment of target cell sensitivity to GH. GH resistance in JIA seems to be a consequence of reduced GHR expression, or changes in intracellular signalling (deactivation of GHR/JAK2 complex, inhibition of JAK/STAT signalling by SOCS) [161,162].
GHR mRNA expression has been reported to be significantly reduced in mononuclear cells of JIA patients at the onset of the disease, with a restoration of GHR expression after two years of treatment [163]. However, these data remain controversial, as other authors did not report changes in GHR gene expression in a rat experimental arthritis model [164]. Granado et al. reported a downregulation in liver IGF-1 gene expression in another experimental arthritis model [165], while others showed that circulating IGFBPs are increased in arthritis, resulting in reduction in IGF-1 bioavailability [166]. Furthermore, acid labile subunit (ALS) seems to be decreased in children with JIA [167].
JIA is characterised by significant changes in the articular microenvironment. Immune cell proliferation causes localised hypoxia and a reduction of pH. As a consequence, osteoblast function is impaired and bone mineralisation reduced [168].
In addition, negative direct effects on growth plate mediated by inflammatory cytokines have been described, in particular, TNF-α and IL-1β inhibit chondrocyte proliferation and function [169]. Moreover, an additive negative effect of IL-1β and TNF-α on bone growth has been described [74].
The IGF system is central to foetal growth, and relationships between cytokines and the IGF system have been shown in the placenta and cord serum of IUGR foetuses. Changes are reported also in children born with IUGR in the following years.
During foetal life, an adequate nutrient supply mainly determines foetal growth, while the intervening growth factors have rather a paracrine or autocrine role to play at the local level, to assure the availability of a nutrient supply, and to promote functional differentiation of the different tissues and organs. These growth factors are mainly insulin, IGF-1, and IGF-2. The IGF axis plays a critical role also in placental development and function. IGF signalling (specifically IGF-2, IGFBP-1, and IGF-1R) plays a critical role in trophoblast invasion and increased utero–placental blood flow during implantation, while imbalances or abnormalities in this signalling lead to adverse pregnancy outcomes, and have been associated with IUGR [170]. Defects in IGF signalling, may lead to impaired foetal growth [171].
Cord serum IGF-2 was described to be lower in IUGR compared with adequate for gestational age newborns, whereas IGFBP-2 was higher [172]. IUGR neonates present higher placental concentrations of IGF-2, IGFBP-1, IGFBP-2, and IL-6 [6].
During childhood, GH status in IUGR is not completely clear. GH responses to provocative stimulation tests and serum levels of IGF-1 and IGFBP-3 are reported to be normal in the majority of patients with normal IGF bioavailability [173,174]. However, in some studies, high pulse frequency and attenuated pulse amplitude related to GH secretion, have been reported [173,174]. These authors described reduced IGF-1, IGF-2, and IGFBP-3 serum concentrations, and reduced spontaneous GH secretion also [175,176].
5. Conclusions
Inflammation is a clear cause of growth impairment. Mechanisms related to GH secretion and resistance, changes in the IGF system, and some changes in the growth plate, have been quite extensively studied, but are not fully elucidated yet. Furthermore, new mechanisms are arising, such as changes reported in the miRNA system that need to be addressed in the near future, in order to improve treatment of inflammation and growth.
Acknowledgments
The authors thank Cecilia Catellani for technical assistance and graphic support to the manuscript.
Conflicts of Interest
The authors declare that there is no conflict of interests. This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
References
- Wong, S.C.; Dobie, R.; Altowati, M.A.; Werther, G.A.; Farquharson, C.; Ahmed, S.F. Growth and the growth hormone-insulin like growth factor 1 axis in children with chronic inflammation: Current evidence, gaps in knowledge, and future directions. Endocr. Rev. 2015, 37, 62–110. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, I.R. Growth problems in children with IBD. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, B. Growth. Pediatr. Rev. 2011, 32, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Hussain, N. Epigenetic influences that modulate infant growth, development, and disease. Antioxid. Redox Signal. 2012, 17, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Mullins, E.; Prior, T.; Roberts, I.; Kumar, S. Changes in the fetal and neonatal cytokine profile in pregnancies complicated by fetal growth restriction. Am. J. Reprod. Immunol. 2013, 69, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Street, M.E.; Seghini, P.; Fieni, S.; Ziveri, M.A.; Volta, C.; Martorana, D.; Viani, I.; Gramellini, D.; Bernasconi, S. Changes in interleukin-6 and IGF system and their relationships in placenta and cord blood in newborns with fetal growth restriction compared with controls. Eur. J. Endocrinol. 2006, 155, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Montanini, L.; Smerieri, A.; Gullì, M.; Cirillo, F.; Pisi, G.; Sartori, C.; Amarri, S.; Bernasconi, S.; Marmiroli, N.; Street, M.E. miR-146a, miR-155, miR-370, and miR-708 Are CFTR-Dependent, Predicted FOXO1 Regulators and Change at Onset of CFRDs. J. Clin. Endocrinol. Metab. 2016, 101, 4955–4963. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, F.; Kubik, Ł.; Oblak, A.; Lorè, N.I.; Cigana, C.; Lanzetta, R.; Parrilli, M.; Hamad, M.A.; De Soyza, A.; Silipo, A.; et al. Activation of human toll-like receptor 4 (TLR4) myeloid differentiation factor 2 (MD-2) by hypoacylated lipopolysaccharide from a clinical isolate of Burkholderia cenocepacia. J. Biol. Chem. 2015, 290, 21305–21319. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, D.; O’Connell, J. Cystic fibrosis related diabetes. Curr. Diabetes Rep. 2014, 4, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Schluchter, M.D.; Konstan, M.W.; Drumm, M.L.; Yankaskas, J.R.; Knowles, M.R. Classifying severity of cystic fibrosis lung disease using longitudinal pulmonary function data. Am. J. Respir. Crit. Care Med. 2006, 174, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Blackman, S.M.; Tangpricha, V. Endocrine disorders in cystic fibrosis. Pediatr. Clin. N. Am. 2016, 63, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.C.; Kosorok, M.R.; Sondel, S.A.; Chen, S.T.; FitzSimmons, S.C.; Green, C.G.; Shen, G.; Walker, S.; Farrell, P.M. Growth status in children with cystic fibrosis based on the National Cystic Fibrosis Patient Registry data: Evaluation of various criteria used to identify malnutrition. J. Pediatr. 1998, 132, 478–485. [Google Scholar] [CrossRef]
- Morison, S.; Dodge, J.A.; Cole, T.J.; Lewis, P.A.; Coles, E.C.; Geddes, D.; Russell, G.; Littlewood, J.M.; Scott, M.T. Height and weight in cystic fibrosis: A cross sectional study. Arch. Dis. Child. 1997, 77, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Bournez, M.; Bellis, G.; Huet, F. Growth during puberty in cystic fibrosis: A retrospective evaluation of a French cohort. Arch. Dis. Child. 2012, 97, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Aswani, N.; Taylor, C.J.; McGaw, J.; Pickering, M.; Rigby, A.S. Pubertal growth and development in cystic fibrosis: A retrospective review. Acta Paediatr. 2003, 92, 1029–1032. [Google Scholar] [CrossRef] [PubMed]
- Vieni, G.; Faraci, S.; Collura, M.; Lombardo, M.; Traverso, G.; Cristadoro, S.; Termini, L.; Lucanto, M.C.; Furnari, M.L.; Trimarchi, G.; et al. Stunting is an independent predictor of mortality in patients with cystic fibrosis. Clin. Nutr. 2013, 32, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Hanauer, S.B. Inflammatory bowel disease: Epidemiology, pathogenesis, and therapeutic opportunities. Inflamm. Bowel Dis. 2006, 12, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Loftus, E.V., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.J.; Dhawan, A.; Saeed, S.A. Inflammatory bowel disease in children and adolescents. JAMA Pediatr. 2015, 169, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.M.; Nguyen, P.; Smith, C.; MacMillan, J.H.; Sherman, P.M. Growth and clinical course of children with Crohn’s disease. Gut 1993, 34, 939–943. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, D.P.; Dawson, A.M. Crohn’s disease in childhood. Arch. Dis. Child. 1977, 52, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Gribetz, D.; Korelitz, B.I. Growth retardation in children with ulcerative colitis: The effect of medical and surgical therapy. Paediatrics 1975, 55, 459–467. [Google Scholar]
- Kirschner, B.S. Growth and development in chronic inflammatory bowel disease. Acta Paediatr. 1990, 79, 98–104. [Google Scholar] [CrossRef]
- Sullivan, D.B.; Cassidy, J.T.; Petty, R.E. Pathogenic implications of age of onset in juvenile rheumatoid arthritis. Arthritis Rheumatol. 1975, 18, 251–255. [Google Scholar] [CrossRef]
- Petty, R.E.; Southwood, T.R.; Baum, J.; Bhettay, E.; Glass, D.N.; Manners, P.; Maldonado-Cocco, J.; Suarez-Almazor, M.; Orozco-Alcala, J.; Prieur, A.M. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J. Rheumatol. 1998, 25, 1991–1994. [Google Scholar] [PubMed]
- Bruck, N.; Schnabel, A.; Hedrich, C.M. Current understanding of the pathophysiology of systemic juvenile idiopathic arthritis (sJIA) and target-directed therapeutic approaches. Clin. Immunol. 2015, 159, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Bechtold, S.; Simon, D. Growth abnormalities in children and adolescents with juvenile idiopathic arthritis. Rheumatol. Int. 2014, 34, 1483–1488. [Google Scholar] [CrossRef] [PubMed]
- MacRae, V.E.; Ahmed, S.F.; Mushtaq, T.; Farquharson, C. IGF-I signalling in bone growth: Inhibitory actions of dexameth- asone and IL-1β. Growth Horm. IGF Res. 2007, 17, 435–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packham, J.C.; Hall, M.A. Long-term follow-up of 246 adults with juvenile idiopathic arthritis: Social function, relationships and sexual activity. Rheumatology 2002, 41, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.A.; Hoch, S.; Erlandson, D.; Partridge, R.; Jackson, J.M. The timing of menarche in juvenile rheumatoid arthritis. J. Adolesc. Health Care 1988, 9, 483–487. [Google Scholar] [CrossRef]
- Padeh, S.; Pinhas-Hamiel, O.; Zimmermann-Sloutskis, D.; Berkun, Y. Children with oligoarticular juvenile idiopathic arthritis are at considerable risk for growth retardation. J. Pediatr. 2011, 159, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Panera, N.; Agostoni, C.; Nobili, V. Intrauterine growth retardation and nonalcoholic Fatty liver disease in children. Int. J. Endocrinol. 2011, 2011, 269853. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Shastri, S.; Sharma, P. Intrauterine growth restriction: Antenatal and postnatal aspects. Clin. Med. Insights Pediatr. 2016, 10, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, I.M.; Horbar, J.D.; Badger, G.J.; Ohlsson, A.; Golan, A. Morbidity and mortality among very-low-birth-weight neonates with intrauterine growth restriction. Am. J. Obstet. Gynecol. 2000, 182, 198–206. [Google Scholar] [CrossRef]
- Street, M.E.; Volta, C.; Ziveri, M.A.; Viani, I.; Bernasconi, S. Markers of insulin sensitivity in placentas and cord serum of intrauterine growth-restricted newborns. Clin. Endocrinol. 2009, 71, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, S.; Martinez, C.; Maiorana, A.; Scirè, G.; Spadoni, G.L.; Boemi, S. Adiponectin levels are reduced in children born small for gestational age and are inversely related to postnatal catch-up growth. J. Clin. Endocrinol. Metab. 2004, 89, 1346–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, R.; Dziura, J.; Burgert, T.S.; Tamborlane, W.V.; Taksali, S.E.; Yeckel, C.W.; Allen, K.; Lopes, M.; Savoye, M.; Morrison, J.; et al. Obesity and the metabolic syndrome in children and adolescents. N. Engl. J. Med. 2004, 350, 2362–2374. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, S.; Maiorana, A.; Geremia, C.; Scirè, G.; Spadoni, G.L.; Germani, D. Blood glucose concentrations are reduced in children born small for gestational age (SGA), and thyroid-stimulating hormone levels are increased in SGA with blunted postnatal catch-up growth. J. Clin. Endocrinol. Metab. 2003, 88, 2699–2705. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.; Kroke, A.; Möhlig, M.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Adiponectin and protection against type 2 diabetes mellitus. Lancet 2003, 361, 226–228. [Google Scholar] [CrossRef]
- Veening, M.A.; Van Weissenbruch, M.M.; Delemarre-Van De Waal, H.A. Glucose tolerance, insulin sensitivity, and insulin secretion in children born small for gestational age. J. Clin. Endocrinol. Metab. 2002, 87, 4657–4661. [Google Scholar] [CrossRef] [PubMed]
- Hofman, P.L.; Cutfield, W.S.; Robinson, E.M.; Bergman, R.N.; Menon, R.K.; Sperling, M.A.; Gluckman, P.D. Insulin resistance in short children with intrauterine growth retardation. J. Clin. Endocrinol. Metab. 1997, 82, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Karlberg, J.; Albertsson-Wikland, K. Growth in full-term small-for-gestational-age infants: From birth to final height. Pediatr. Res. 1995, 38, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, O.G.; Eden, S.; Jansson, J.O. Mode of action of pituitary growth hormone on target cells. Annu. Rev. Physiol. 1985, 47, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, B.C.; Maheshwari, H.G.; He, L.; Reed, M.; Lozykowski, M.; Okada, S.; Cataldo, L.; Coschigamo, K.; Wagner, T.E.; et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc. Natl. Acad. Sci. USA 1997, 94, 13215–13220. [Google Scholar] [CrossRef] [PubMed]
- Donahue, L.R.; Beamer, W.G. Growth hormone deficiency in ‘little’ mice results in aberrant body composition, reduced insulin-like growth factor-I and insulin-like growth factor-binding protein-3 (IGFBP-3), but does not affect IGFBP-2, -1 or -4. J. Endocrinol. 1993, 136, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.A.; Camacho-Hubner, C.; Savage, M.O.; Clark, A.J. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 1996, 335, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Liu, J.P.; Robertson, E.J.; Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993, 75, 73–82. [Google Scholar] [CrossRef]
- Liu, J.P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (IGF-1) and type 1 IGF receptor (IGF1R). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef]
- Powell-Braxton, L.; Hollingshead, P.; Warburton, C.; Dowd, M.; Pitts-Meek, S.; Dalton, D.; Gillett, N.; Stewart, T.A. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993, 7, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Daughaday, W.H. From sulphation factor to IGF-I, 40 years of research on the regulation of cartilage growth. In Proceedings of the 4th International Symposium on Insulin-like Growth Factors, Tokyo, Japan, 21–24 October 1997; Takano, K., Hizuka, N., Takahashi, S.-I., Eds.; Elsevier: Tokyo, Japan, 1997. [Google Scholar]
- Carter-Su, C.; Schwartz, J.; Smit, L.S. Molecular mechanism of growth hormone action. Annu. Rev. Physiol. 1996, 58, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Daughaday, W.H.; Hall, K.; Raben, M.S.; Salmon, W.D., Jr.; van Den Brande, J.L.; Van Wyk, J.J. Somatomedin: Proposed designation for sulphation factor. Nature 1972, 235, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Bartold, P.M.; Zhang, C.Z.; Clarkson, R.W.; Young, W.G.; Waters, M.J. Growth hormone and insulin-like growth factor I induce bone morphogenetic proteins 2 and 4: A mediator role in bone and tooth formation? Endocrinology 1998, 139, 3855–3862. [Google Scholar] [CrossRef] [PubMed]
- Billestrup, N.; Nielsen, J.H. The stimulatory effect of growth hormone, prolactin, and placental lactogen on b-cell proliferation is not mediated by insulin-like growth factor-I. Endocrinology 1991, 129, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, O.G.; Lindahl, A.; Nilsson, A.; Isgaard, J. Mechanism of the stimulatory effect of growth hormone on longitudinal bone growth. Endocr. Rev. 1987, 8, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, A.; Nilsson, A.; Isaksson, O.G. Effects of growth hormone and insulin-like growth factor-I on colony formation of rabbit epiphyseal chondrocytes at different stages of maturation. J. Endocrinol. 1987, 115, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Schlechter, N.L.; Russell, S.M.; Spencer, E.M.; Nicoll, C.S. Evidence suggesting that the direct growth-promoting effect of growth hormone on cartilage in vivo is mediated by local production of somatomedin. Proc. Natl. Acad. Sci. USA 1986, 83, 7932–7934. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, O.G.; Jansson, J.O.; Gause, I.A. Growth hormone stimulates longitudinal bone growth directly. Science 1982, 216, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; LeRoith, D. Insulin-like growth factor I is essential for postnatal growth in response to growth hormone. Endocrinology 1999, 140, 5178–5184. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z. Insulin-like growth factor 1 (IGF-1): A growth hormone. Mol. Pathol. 2001, 54, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Blakesley, V.A.; Butler, A.A.; Koval, A.P.; Okubo, Y.; LeRoith, D. IGF-I receptor function: Transducing the IGF-1 signal into intracellular events. In The IGF System; Rosenfeld, R., Roberts, C., Jr., Eds.; Humana Press: Totowa, NJ, USA, 1999; pp. 143–164. [Google Scholar]
- White, M.F.; Kahn, C.R. The insulin signalling system. J. Biol. Chem. 1994, 269, 1–4. [Google Scholar] [PubMed]
- Martin, J.L.; Baxter, R.C. IGF binding proteins and extracellular matrix. In The IGF System; Rosenfeld, R., Roberts, C., Jr., Eds.; Humana Press: Totowa, NJ, USA, 1999; pp. 227–256. [Google Scholar]
- Jones, J.I.; Clemmons, D.R. Insulin-like growth factors and their binding proteins: Biological actions. Endocr. Rev. 1995, 16, 3–34. [Google Scholar] [PubMed]
- Holzenberger, M.; Leneuve, P.; Hamard, G.; Ducos, B.; Perin, L.; Binoux, M.; Le Bouc, Y. A targeted partial invalidation of the insulin-like growth factor I receptor gene in mice causes a postnatal growth deficit. Endocrinology 2000, 141, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Alonzi, T.; Moretta, A.; Lazzaro, D.; Costa, P.; Poli, V.; Martini, A.; Ciliberto, G.; Fattori, E. Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin-like growth factor-I-A model for stunted growth in children with chronic inflammation. J. Clin. Investig. 1997, 99, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Street, M.E.; Ziveri, M.A.; Spaggiari, C.; Viani, I.; Volta, C.; Grzincich, G.L.; Virdis, R.; Bernasconi, S. Inflammation is a modulator of the insulin-like growth factor (IGF)/IGF-binding protein system inducing reduced bioactivity of IGFs in cystic fibrosis. Eur. J. Endocrinol. 2006, 154, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Pass, C.; MacRae, V.E.; Huesa, C.; Faisal Ahmed, S.; Farquharson, C. SOCS2 is the critical regulator of GH action in murine growth plate chondrogenesis. J. Bone Miner Res. 2012, 27, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Farquharson, C. The effect of GH and IGF1 on linear growth and skeletal development and their modu- lation by SOCS proteins. J. Endocrinol. 2010, 206, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Denson, L.A.; Held, M.A.; Menon, R.K.; Frank, S.J.; Parlow, A.F.; Arnold, D.L. Interleukin-6 inhibits hepatic growth hormone signaling via upregulation of Cis and Socs-3. Am. J. Physiol. 2003, 284, G646–G654. [Google Scholar] [CrossRef] [PubMed]
- Boisclair, Y.R.; Wang, J.; Shi, J.; Hurst, K.R.; Ooi, G. Role of the suppressor of cytokine signaling-3 in mediating the inhib- itory effects of interleukin-1β on the growth hormone-de- pendent transcription of the acid-labile subunit gene in liver cells. J. Biol. Chem. 2000, 275, 3841–3847. [Google Scholar] [CrossRef] [PubMed]
- Choukair, D.; Hügel, U.; Sander, A.; Uhlmann, L.; Tönshoff, B. Inhibition of IGF1-related intracellular signaling path- ways by proinflammatory cytokines in growth plate chondrocytes. Pediatr. Res. 2014, 76, 245–251. [Google Scholar] [CrossRef] [PubMed]
- MacRae, V.E.; Farquharson, C.; Ahmed, S.F. The restricted potential for recovery of growth plate chondrogenesis and longitudinal bone growth following exposure to pro-inflammatory cytokines. J. Endocrinol. 2006, 189, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Broussard, S.R.; McCusker, R.H.; Novakofski, J.E.; Strle, K.; Shen, W.H.; Johnson, R.W.; Dantzer, R.; Kelley, K.W. IL-1β impairs insulin-like growth factor i-induced differentiation and downstream activation signals of the insulin-like growth factor I receptor in myoblasts. J. Immunol. 2004, 172, 7713–7720. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I.M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Baum, R.; Gravallese, E.M. Impact of Inflammation on the Osteoblast in Rheumatic Diseases. Curr. Osteoporos. Rep. 2014, 12, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.P.; Lerner, U.H. The role of cytokines in inflammatory bone loss. Immunol. Investig. 2013, 42, 555–622. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Levenson, A.; Kharitonenkov, A.; De Luca, F. Fibroblast growth factor 21 (FGF21) inhibits chondrocyte function and growth hormone action directly at the growth plate. J. Biol. Chem. 2012, 287, 26060–26067. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Guo, N.J.; Tian, H.; Marohn, M.; Gearhart, S.; Bayless, T.M.; Brant, S.R.; Kwon, J.H. Peripheral blood microRNAs distinguish active ulcerative colitis and Crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.; van Wengen, A.; Hoeve, M.A.; ten Dam, M.; van der Burg, M.; van Dongen, J.; van de Vosse, E.; van Tol, M.; Bredius, R.; Ottenhoff, T.H.; et al. The same IκBα mutation in two related individuals leads to completely different clinical syndromes. J. Exp. Med. 2004, 200, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Franzoso, G.; Carlson, L.; Xing, L.; Poljak, L.; Shores, E.W.; Brown, K.D.; Leonardi, A.; Tran, T.; Boyce, B.F.; Siebenlist, U. Requirement for NF-κB in osteoclast and B-cell development. Nature 1998, 392, 611–614. [Google Scholar]
- Kanegae, Y.; Tavares, A.T.; Izpisua Belmonte, J.C.; Verma, I.M. Role of Rel/NF-κB transcription factors during the outgrowth of the vertebrate limb. Genes Dev. 1997, 11, 3482–3496. [Google Scholar]
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Beinke, S.; Ley, S.C. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem. J. 2004, 382, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Karin, M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular signals of epigenetic states. Science 2010, 330, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Stoffel, M. MicroRNAs: A new class of regulatory genes affecting metabolism. Cell Metab. 2006, 4, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Iborra, M.; Bernuzzi, F.; Invernizzi, P.; Danese, S. MicroRNAs in autoimmunity and inflammatory bowel disease: Crucial regulators in immune response. Autoimmun. Rev. 2012, 11, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Antonetti, F.R.; Cardone, J.; Bonmassar, E. miR-155 gene: A typical multifunctional microRNA. Biochim. Biophys. Acta 2009, 1792, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Sharp, P.A. Specificity of microRNA target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of β-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A.; Good, L. Does everything now make (anti)sense? Biochem. Soc. Trans. 2006, 34, 1148–1150. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Ceppi, M.; Pereira, P.M.; Dunand-Sauthier, I.; Barras, E.; Reith, W.; Santos, M.A.; Pierre, P. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocytederived dendritic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2735–2740. [Google Scholar] [CrossRef] [PubMed]
- O’ Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.F.; Liston, A. MicroRNA in the immune system, microR NA as an immune system. Immunology 2009, 127, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wang, S.; Mayr, C.; Bartel, D.P.; Lodish, H.F. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc. Natl. Acad. Sci. USA 2007, 104, 7080–7085. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-κB signalling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Dunne, A.; O’Neill, L.A. Adaptor usage and Toll-like receptor signaling specificity. FEBS Lett. 2005, 579, 3330–3335. [Google Scholar] [CrossRef] [PubMed]
- Kracht, M.; Saklatvala, J. Transcriptional and post-transcriptional control of gene expression in inflammation. Cytokine 2002, 20, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Shi, Y.; Tan, G.; Yang, C.H.; Fan, M.; Pfeffer, L.M.; Wu, Z.-H. DNA damage induces NF-κB-dependent microRNA-21 up-regulation and promotes breast cancer cell invasion. J. Biol. Chem. 2012, 287, 21783–21795. [Google Scholar] [CrossRef] [PubMed]
- Scisciani, C.; Vossio, S.; Guerrieri, F.; Schinzari, V.; De Laco, R.; D’Onorio De Meo, P.; Cervello, M.; Montalto, G.; Pollicino, T.; Raimondo, G.; et al. Transcriptional regulation of miR-224 upregulated in human HCCs by NFκB inflammatory pathways. J. Hepatol. 2012, 56, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, F.; Rossato, M.; Fabbri, M.; Gaudiosi, D.; Mirolo, M.; Mori, L.; Tamassia, N.; Mantovani, A.; Cassatella, M.A.; Locati, M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. USA 2009, 106, 5282–5287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, S.; Hu, T.; Liu, S.; He, Y.; Sun, S. Up-regulated microRNA-143 transcribed by nuclear factor κ B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology 2009, 50, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Vencken, S.F.; Agrawal, R.; Gaughan, K.; Molloy, K.; Higgins, G.; McNally, P.; McElvaney, N.G.; Mall, M.A.; Greene, C.M. miR-17 overexpression in cystic fibrosis airway epithelial cells decreases interleukin-8 production. Eur. Respir. J. 2015, 46, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; Cialfi, S.; Cimino, G.; De Biase, R.V.; Dominici, C.; Quattrucci, S.; Pizzuti, A. Elevated levels of miR-145 correlate with SMAD3 down-regulation in cystic fibrosis patients. J. Cyst. Fibros. 2013, 12, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Balakathiresan, N.S.; Dalgard, C.; Gutti, U.; Armistead, D.; Jozwik, C.; Srivastava, M.; Pollard, H.B.; Biswas, R. Elevated miR-155 promotes inflammation in cystic fibrosis by driving hyperexpression of interleukin-8. J. Biol. Chem. 2011, 286, 11604–11615. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Bray, I.M.; Chotirmall, S.H.; Stallings, R.L.; O’Neill, S.J.; McElvaney, N.G.; Greene, C.M. MiR-126 is downregulated in cystic fibrosis airway epithelial cells and regulates TOM1 expression. J. Immunol. 2010, 184, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, S.; Yu, Q.; Yang, G.; Guo, J.; Li, M.; Zeng, Z.; He, Y.; Chen, B.; Chen, M. Circulating MicroRNA223 is a New Biomarker for Inflammatory Bowel Disease. Medicine 2016, 95, e2703. [Google Scholar] [CrossRef] [PubMed]
- Polytarchou, C.; Oikonomopoulos, A.; Mahurkar, S.; Touroutoglou, A.; Koukos, G.; Hommes, D.W.; Iliopoulos, D. Assessment of circulating microRNAs for the diagnosis and disease activity evaluation in patients with ulcerative colitis by using the nanostring technology. Inflamm. Bowel Dis. 2015, 21, 2533–2539. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, J.S.; Attumi, T.; Opekun, A.R.; Abraham, B.; Hou, J.; Shelby, H.; Graham, D.Y.; Streckfus, C.; Klein, J.R. MicroRNA signatures differentiate Crohn’s disease from ulcerative colitis. BMC Immunol. 2015, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Nakamichi, I.; Esaki, M.; Asano, K.; Matsumoto, T.; Kitazono, T. Serum microRNA levels in patients with Crohn’s disease during induction therapy by infliximab. J. Gastroenterol. Hepatol. 2014, 29, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Plank, M.; Maltby, S.; Mattes, J.; Foster, P.S. Targeting translational control as a novel way to treat inflammatory disease: The emerging role of microRNAs. Clin. Exp. Allergy 2013, 43, 981–999. [Google Scholar] [CrossRef] [PubMed]
- Duttagupta, R.; DiRienzo, S.; Jiang, R.; Bowers, J.; Gollub, J.; Kao, J.; Kearney, K.; Rudolph, D.; Dawany, N.B.; Showe, M.K.; et al. Genome-wide maps of circulating miRNA biomarkers for ulcerative colitis. PLoS ONE 2012, 7, e31241. [Google Scholar] [CrossRef] [PubMed]
- Paraskevi, A.; Theodoropoulos, G.; Papaconstantinou, I.; Mantzaris, G.; Nikiteas, N.; Gazouli, M. Circulating MicroRNA in inflammatory bowel disease. J. Crohn’s Colitis 2012, 6, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wu, F.; Xin, L.; Su, G.; He, F.; Yang, Y.; Sun, J.; Liu, Z. Differential plasma microRNAs expression in juvenile idiopathic arthritis. Mod. Rheumatol. 2016, 26, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, Y.; Kawada, J.; Kawano, Y.; Torii, Y.; Kawabe, S.; Iwata, N.; Ito, Y. Serum microRNAs as potential biomarkers of juvenile idiopathic arthritis. Clin. Rheumatol. 2015, 34, 1705–1712. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Tsai, S.Q.; Hardison, N.E.; James, A.H.; Motsinger-Reif, A.A.; Thames, B.; Stone, E.A.; Deng, C.; Piedrahita, J.A. Differentially expressed microRNAs and affected biological pathways revealed by modulated modularity clustering (MMC) analysis of human preeclamptic and IUGR placentas. Placenta 2013, 34, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Shen, Z.; Xu, Q.; Huang, X.; Chen, Q.; Li, D. Increased levels of microRNA-424 are associated with the pathogenesis of fetal growth restriction. Placenta 2013, 34, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B. Normal bone anatomy and physiology. Clin. J. Am. Soc. Nephrol. 2008, 3, S131–S139. [Google Scholar] [CrossRef] [PubMed]
- Redlich, K.; Smolen, J.S. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 2012, 11, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, C.; Isgaard, J.; Tornell, J.; Nilsson, A.; Isaksson, O.G.P.; Lindahl, A. Endocrine regulation of longitudinal bone growth. Acta Paediatr. 1993, 391, 33–40. [Google Scholar] [CrossRef]
- Wit, J.M.; Boersma, B. Catch-up growth: Definition, mechanisms, and models. J. Pediatr. Endocrinol. Metab. 2002, 15, 1229–1241. [Google Scholar] [PubMed]
- Wit, J.M.; Camacho-Hübner, C. Endocrine regulation of longitudinal bone growth. Endocr. Dev. 2011, 21, 30–41. [Google Scholar] [PubMed]
- Sederquist, B.; Fernandez-Vojvodich, P.; Zaman, F.; Sävendahl, L. Recent research on the growth plate: Impact of inflammatory cytokines on longitudinal bone growth. J. Mol. Endocrinol. 2014, 53, T35–T44. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, J.; Cheng, C.M.; Kopchick, J.J.; Bondy, C.A. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J. Endocrinol. 2004, 180, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Shead, E.F.; Haworth, C.S.; Barker, H.; Bilton, D.; Compston, J.E. Osteoclast function, bone turnover and inflammatory cytokines during infective exacerbations of cystic fibrosis. J. Cyst. Fibros. 2010, 9, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Paganelli, M.; Albanese, C.; Borrelli, O.; Civitelli, F.; Canitano, N.; Viola, F.; Passariello, R.; Cucchiara, S. Inflammation is the main determinant of low bone mineral density in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Romas, E.; Gillespie, M.T. Inflammation-induced bone loss: Can it be prevented? Rheum. Dis. Clin. N. Am. 2006, 32, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Iotsova, V.; Caamaño, J.; Loy, J.; Yang, Y.; Lewin, A.; Bravo, R. Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat. Med. 1997, 3, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Jimi, E.; Hirata, S.; Shin, M.; Yamazaki, M.; Fukushima, H. Molecular mechanisms of BMP-induced bone formation: Cross-talk between BMP and NF-κB signaling pathways in osteoblastogenesis. Jpn. Dent. Sci. Rev. 2010, 46, 33–42. [Google Scholar] [CrossRef]
- Gilbert, L.C.; Rubin, J.; Nanes, M.S. The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis. Am. J. Physiol. Endocrinol. Metab. 2005, 13, 720–730. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F. Role of Nuclear Factor κ B (NF-κB) in Growth Plate Chondrogenesis. Pediatr. Endocrinol. Rev. 2016, 13, 720–730. [Google Scholar] [PubMed]
- Wu, S.; Flint, J.K.; Rezvani, G.; De Luca, F. Nuclear factor-κB p65 facilitates longitudinal bone growth by inducing growth plate chondrocyte proliferation and differentiation and by preventing apoptosis. J. Biol. Chem. 2007, 282, 33698–33706. [Google Scholar] [CrossRef] [PubMed]
- Crisostomo, P.R.; Wang, Y.; Markel, T.A.; Wang, M.; Lahm, T.; Meldrum, D.R. Human mesenchymal stem cells stimulated by TNF-α, LPS, or hypoxia produce growth factors by an NF-κB- but not JNK-dependent mechanism. Am. J. Physiol. Cell Physiol. 2008, 294, C675–C682. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.S.; Ko, J.Y.; Yeh, D.W.; Ke, H.C.; Wu, H.L. Modulation of dickkopf-1 attenuates glucocorticoid induction of osteoblast apoptosis, adipocytic differentiation, and bone mass loss. Endocrinology 2008, 149, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Marcovecchio, M.L.; Mohn, A.; Chiarelli, F. Inflammatory cytokines and growth in childhood. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Eijken, M.; Koedam, M.; van Driel, M.; Buurman, C.J.; Pols, H.A.; van Leeuwen, J.P. The essential role of glucocorticoids for proper human osteoblast differentiation and matrix mineralization. Mol. Cell. Endocrinol. 2006, 248, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, R.S.; Jilka, R.L.; Parfitt, A.M.; Manolagas, S.C. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J. Clin. Investig. 1998, 102, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Burnham, J.M.; Shults, J.; Petit, M.A.; Semeao, E.; Beck, T.J.; Zemel, B.S.; Leonard, M.B. Alterations in proximal femur geometry in children treated withglucocorticoids for Crohn disease or nephrotic syndrome: Impact of the underlying disease. J. Bone Miner. Res. 2007, 22, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Leonard, M.B.; Feldman, H.I.; Shults, J.; Zemel, B.S.; Foster, B.J.; Stallings, V.A. Long-term, high-dose glucocorticoids and bone mineral content in childhood glucocorticoid-sensitive nephrotic syndrome. N. Engl. J. Med. 2004, 351, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Ciro, D.; Padoan, R.; Blau, H.; Marostica, A.; Fuoti, M.; Volpi, S.; Pilotta, A.; Meyerovitch, J.; Sher, D.; Assael, B.M. Growth retardation and reduced growth hormone secretion in cystic fibrosis. Clinical observations from three CF centers. J. Cyst. Fibros. 2013, 12, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Ripa, P.; Robertson, I.; Cowley, D.; Harris, M.; Masters, I.B.; Cotterill, A.M. The relationship between insulin secretion, the insulin-like growth factor axis and growth in children with cystic fibrosis. Clin. Endocrinol. 2002, 56, 383–389. [Google Scholar] [CrossRef]
- Laursen, T.; Jørgensen, J.O.; Jakobsen, G.; Hansen, B.L.; Christiansen, J.S. Continuous infusion versus daily injections of growth hormone (GH) for 4 weeks in GH-deficient patients. J. Clin. Endocrinol. Metab. 1995, 80, 2410–2418. [Google Scholar] [PubMed]
- Liang, H.; Yang, L.; Ma, T.; Zhao, Y. Functional expression of cystic fibrosis transmembrane conductance regulator in mouse chondrocytes. Clin. Exp. Pharmacol. Physiol. 2010, 37, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Diwakar, A.; Adam, R.J.; Michalski, A.S.; Tamegnon, M.M.; Fischer, A.J.; Launspach, J.L.; Horan, R.A.; Kao, S.C.; Chaloner, K.; Meyerholz, D.K.; et al. Sonographic evidence of abnormal tracheal cartilage ring structure in cystic fibrosis. Laryngoscope 2015, 125, 2398–2404. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, E.; Le Rouzic, P.; Bernaudin, J.F.; Cottart, C.H.; Vandebrouck, C.; Crié, A.; Leal, T.; Clement, A.; Bonora, M. Congenital tracheal malformation in cystic fibrosis transmembrane conductance regulatordeficient mice. J. Physiol. 2008, 586, 3231–3243. [Google Scholar] [CrossRef] [PubMed]
- Ketelslegers, J.M.; Maiter, D.; Maes, M.; Underwood, L.E.; Thissen, J.P. Nutritional regulation of the growth hormone and insulin-like growth factor-binding proteins. Horm. Res. 1996, 45, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, A.B.; Azooz, O.; El-Haj, T.; Poole, S.; Farthing, M.J. Growth failure occurs through a decrease in insulin-like growth factor 1 which is independent of undernutrition in a rat model of colitis. Gut 2000, 46, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, A. Fundamental mechanisms of growth failure in inflammatory bowel disease. Horm. Res. 2002, 58, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Irwin, R.; Raehtz, S.; Parameswaran, N.; McCabe, L.R. Intestinal inflammation without weight loss decreases bone density and growth. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R1149–R1157. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Meazza, C.; Oliveri, M.; Pignatti, P.; Vivarelli, M.; Alonzi, T.; Fattori, E.; Garrone, S.; Barreca, A.; Martini, A. Effect of IL-6 on IGF binding protein-3: A study in IL-6 trans- genic mice and in patients with systemic juvenile idiopathic arthritis. Endocrinology 2001, 142, 4818–4826. [Google Scholar] [CrossRef] [PubMed]
- Templ, E.; Koeller, M.; Riedl, M.; Wagner, O.; Graninger, W.; Luger, A. Anterior pituitary function in patients with newly diagnosed rheumatoid arthritis. Br. J. Rheumatol. 1996, 35, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Bergad, P.L.; Schwarzenberg, S.J.; Humbert, J.T.; Morrison, M.; Amar- asinghe, S.; Towle, H.C.; Berry, S.A. Inhibition of growth hor- mone action in models of inflammation. Am. J. Physiol. Cell Physiol. 2000, 279, C1906–C1917. [Google Scholar] [PubMed]
- Von Laue, S.; Ross, R.J. Inflammatory cytokines and acquired growth hormone resistance. Growth Horm. IGF Res. 2000, 10, S9–S14. [Google Scholar] [CrossRef]
- Bozzola, E.; Pagani, S.; Meazza, C.; Cortis, E.; Lisini, D.; Laarej, K.; Bozzola, M. Changes in growth hormone receptor gene expression during therapy in children with juvenile idiopathic arthritis. Horm. Res. Paediatr. 2012, 77, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Calderon, A.; Ibáñez de Caceres, I.; Soto, L.; Priego, T.; Martin, A.I.; Villanua, M.A. The decrease in hepatic IGF-I gene expression in arthritic rats is not associated with modifications in hepatic GH receptor mRNA. Eur. J. Endocrinol. 2001, 144, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Granado, M.; Martín, A.I.; Villanúa, M.A.; López-Calderón, A. Experimental arthritis inhibits the insulin-like growth factor-I axis and induces muscle wasting through cyclooxygenase-2 activation. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1656–E1665. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Calderón, A.; Soto, L.; Martín, A.I. Chronic inflammation inhibits GH secretion and alters the serum insulin-like growth factor system in rats. Life Sci. 1999, 65, 2049–2060. [Google Scholar] [CrossRef]
- Wong, S.C.; MacRae, V.E.; Gracie, J.A.; McInnes, I.B.; Galea, P.; Gardner-Medwin, J.; Ahmed, S.F. Inflammatory cytokines in juvenile idiopathic arthritis: Effects on physical growth and the insulin-like-growth factor axis. Growth Horm. IGF Res. 2008, 18, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Wehmeyer, C.; Pap, T.; Buckley, C.D.; Naylor, A.J. The role of stromal cells in inflammatory bone loss. Clin. Exp. Immunol. 2017, 189, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, S.; Marcovecchio, M.L.; Breda, L.; Chiarelli, F. Growth in juvenile idiopathic arthritis: The role of inflammation. Clin. Exp. Immunol. 2011, 29, 104–110. [Google Scholar]
- Bowman, C.J.; Streck, R.D.; Chapin, R.E. Maternal-placental insulin-like growth factor (IGF) signaling and its importance to normal embryo-fetal development. Birth Defects Res. B 2010, 89, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Kanaka-Gantenbein, C.; Mastorakos, G.; Chrousos, G.P. Endocrine-related causes and consequences of intrauterine growth retardation. Ann. N. Y. Acad. Sci. 2003, 997, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Smerieri, A.; Petraroli, M.; Ziveri, M.A.; Volta, C.; Bernasconi, S.; Street, M.E. Effects of cord serum insulin, IGF-II, IGFBP-2, IL-6 and cortisol concentrations on human birth weight and length: Pilot study. PLoS ONE 2011, 6, e29562. [Google Scholar] [CrossRef] [PubMed]
- Toure, D.M.; Baccaglini, L.; Opoku, S.T.; Barnes-Josiah, D.; Cox, R.; Hartman, T.; Klinkebiel, D. Epigenetic dysregulation of Insulin-like growth factor (IGF)-related genes and adverse pregnancy outcomes: A systematic review. J. Matern. Fetal. Neonatal. Med. 2016, 29, 3542–3552. [Google Scholar] [CrossRef] [PubMed]
- Völkl, T.M.; Schwöbel, K.; Simm, D.; Beier, C.; Rohrer, T.R.; Dörr, H.G. Spontaneous growth hormone secretion and IGF1:IGFBP3 molar ratios in children born small for gestational age (SGA). Growth Horm. IGF Res. 2004, 14, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Boguszewski, M.; Rosberg, S.; Albertsson-Wikland, K. Spontaneous 24-hour growth hormone profiles in prepubertal small for gestational age children. J. Clin. Endocrinol. Metab. 1995, 80, 2599–2606. [Google Scholar] [PubMed]
- De Waal, W.J.; Hokken-Koelega, A.C.; Stijnen, T.; de Muinck Keizer-Schrama, S.M.; Drop, S.L. Endogenous and stimulated GH secretion, urinary GH excretion, and plasma IGF-I and IGF-II levels in prepubertal children with short stature after intrauterine growth retardation. Clin. Endocrinol. 1994, 41, 621–630. [Google Scholar] [CrossRef]
- Philip, A.G. Fetal growth retardation: Femurs, fontanels, and follow-up. Pediatrics 1978, 62, 446–453. [Google Scholar] [PubMed]
- Wilson, M.G.; Meyers, H.I.; Peters, A.H. Postnatal bone growth of infants with fetal growth retardation. Pediatrics 1967, 40, 213–223. [Google Scholar] [PubMed]
Figure 1.
Overview of actions of pro-inflammatory cytokines on the growth hormone (GH)–insulin-like growth factor (IGF) axis and IGF system. Pro-inflammatory cytokines determine a dysregulation in GH–IGF axis and IGF system, both at a central and peripheral level. In the brain, inflammation determines a dysregulation of GH secretion from the pituitary gland. In the liver, a pro-inflammatory cytokine milieu results in a down-regulation of GH receptors and in impaired downstream signalling, with subsequent GH resistance and IGF-1 insufficiency. GH and IGF-1 resistance are present in the growth plate. Abnormalities in IGF binding proteins (IGFBPs), with reduction in IGF bioavailability, are a constant feature. MicroRNAs (miRNAs) targeting genes within the GH–IGF axis and IGF system are dysregulated, and potential key mediators of these processes.
Figure 1.
Overview of actions of pro-inflammatory cytokines on the growth hormone (GH)–insulin-like growth factor (IGF) axis and IGF system. Pro-inflammatory cytokines determine a dysregulation in GH–IGF axis and IGF system, both at a central and peripheral level. In the brain, inflammation determines a dysregulation of GH secretion from the pituitary gland. In the liver, a pro-inflammatory cytokine milieu results in a down-regulation of GH receptors and in impaired downstream signalling, with subsequent GH resistance and IGF-1 insufficiency. GH and IGF-1 resistance are present in the growth plate. Abnormalities in IGF binding proteins (IGFBPs), with reduction in IGF bioavailability, are a constant feature. MicroRNAs (miRNAs) targeting genes within the GH–IGF axis and IGF system are dysregulated, and potential key mediators of these processes.

Figure 2.
NF-κB as a central transcriptional factor in the inflammatory process. Nuclear factor-κB (NF-κB) is a transcriptional factor with a crucial role in the control of mechanisms mediated by inflammation. It orchestrates several processes, such as angiogenesis, apoptosis, and cell cycle regulation, and is important as a regulator of bone remodelling, enhancing osteoclast and reducing osteoblast activity. Moreover, it influences IGF-1 secretion, and it represents one of the factors linking inflammation with growth.
Figure 2.
NF-κB as a central transcriptional factor in the inflammatory process. Nuclear factor-κB (NF-κB) is a transcriptional factor with a crucial role in the control of mechanisms mediated by inflammation. It orchestrates several processes, such as angiogenesis, apoptosis, and cell cycle regulation, and is important as a regulator of bone remodelling, enhancing osteoclast and reducing osteoblast activity. Moreover, it influences IGF-1 secretion, and it represents one of the factors linking inflammation with growth.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cirillo, F.; Lazzeroni, P.; Sartori, C.; Street, M.E. Inflammatory Diseases and Growth: Effects on the GH–IGF Axis and on Growth Plate. Int. J. Mol. Sci. 2017, 18, 1878. https://doi.org/10.3390/ijms18091878
AMA Style
Cirillo F, Lazzeroni P, Sartori C, Street ME. Inflammatory Diseases and Growth: Effects on the GH–IGF Axis and on Growth Plate. International Journal of Molecular Sciences. 2017; 18(9):1878. https://doi.org/10.3390/ijms18091878
Chicago/Turabian StyleCirillo, Francesca, Pietro Lazzeroni, Chiara Sartori, and Maria Elisabeth Street. 2017. "Inflammatory Diseases and Growth: Effects on the GH–IGF Axis and on Growth Plate" International Journal of Molecular Sciences 18, no. 9: 1878. https://doi.org/10.3390/ijms18091878
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.