Glycans as Regulatory Elements of the Insulin/IGF System: Impact in Cancer Progression

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
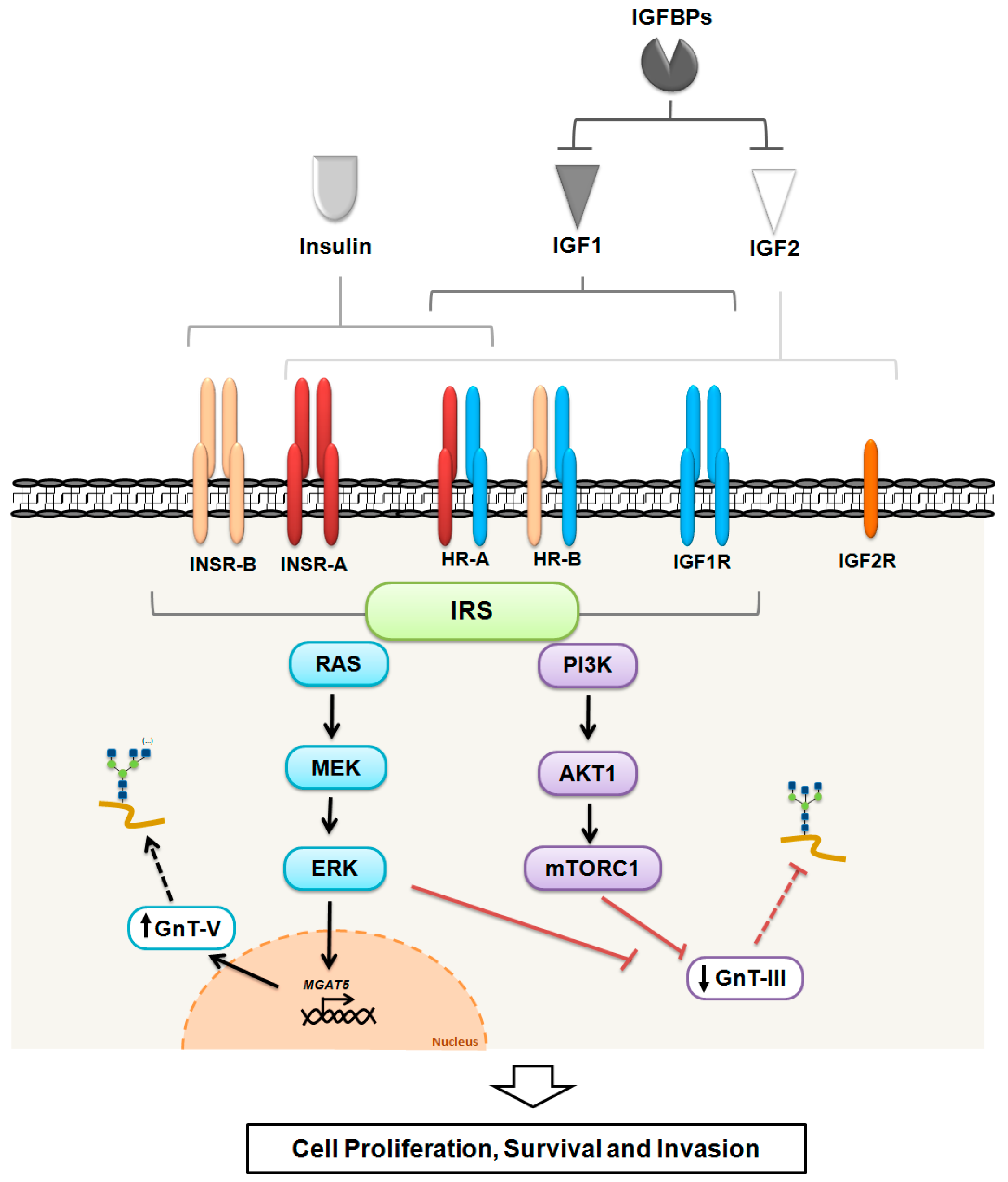
2. The Insulin/Insulin-Like Growth Factor (IGF) System
2.1. Ligands and Binding Proteins
2.2. Receptors
2.3. Downstream Proteins
3. Impact of the Insulin/IGF System in Cancer Development and Progression
3.1. Cellular Behaviour
3.2. Drug Resistance
3.3. Cell Metabolism
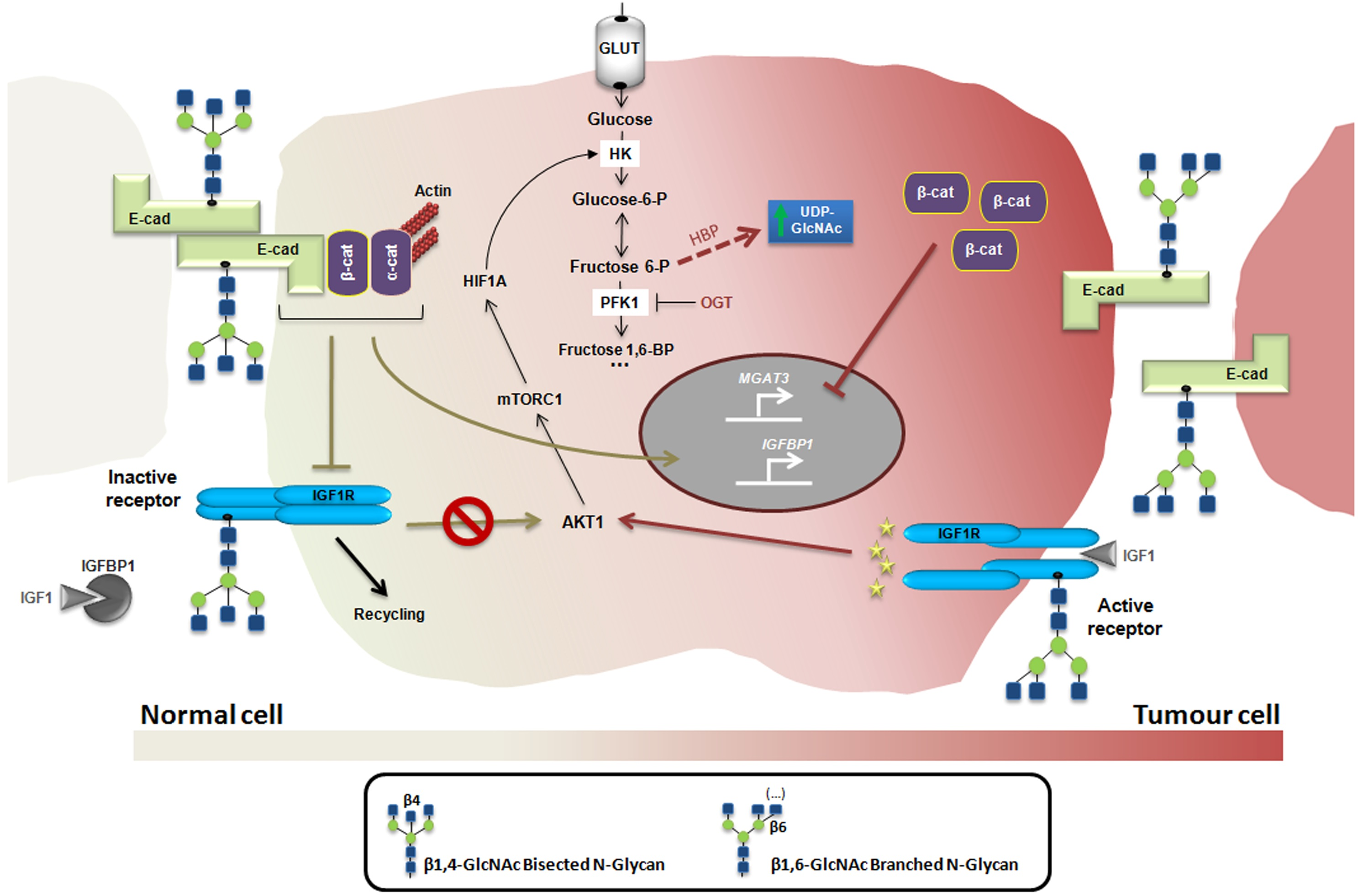
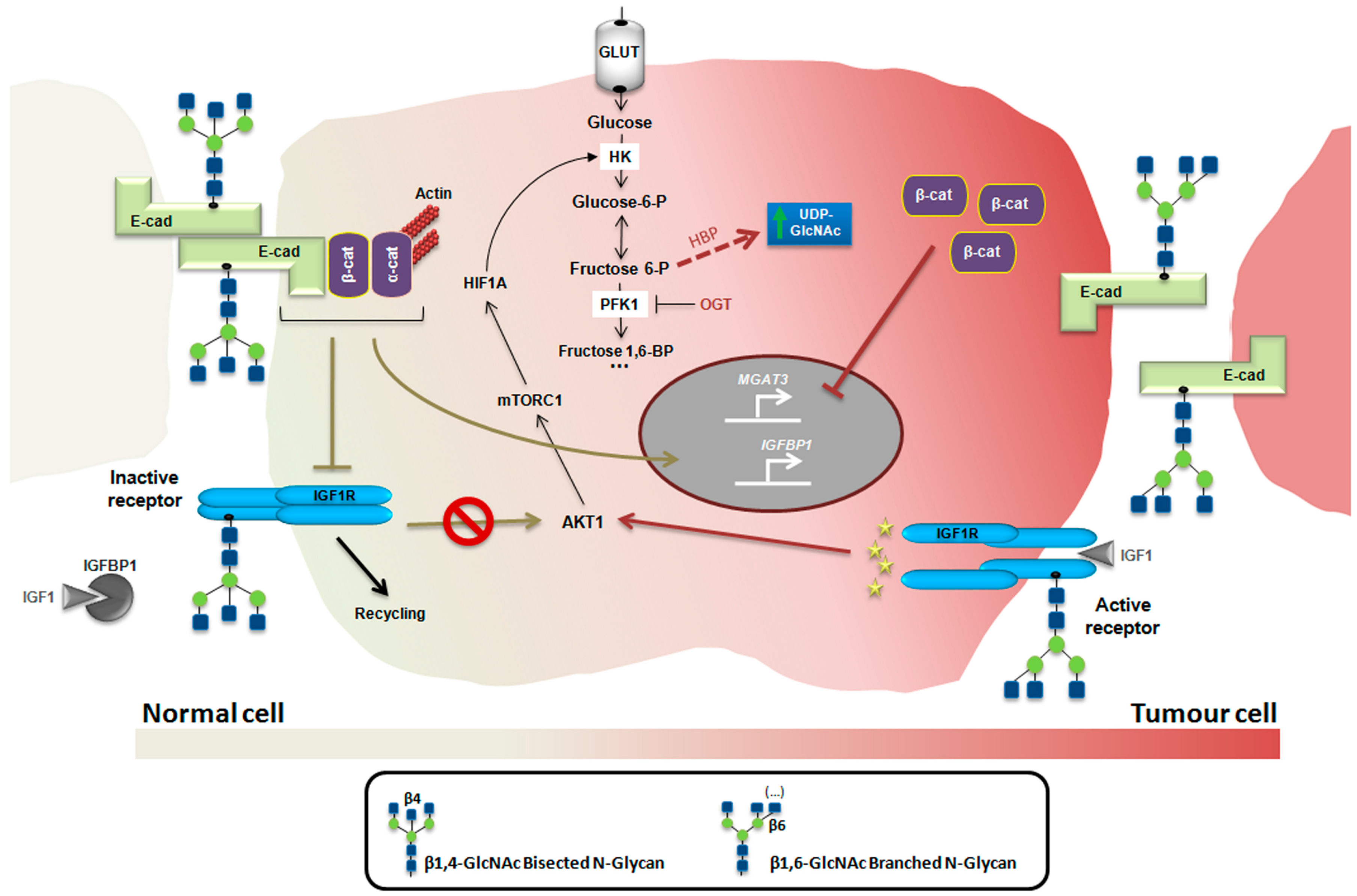
4. Glycosylation as a Regulatory Mechanism of the Insulin/IGF System in Cancer
5. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Asn | Asparagines |
| CMP | Cytidine monophosphate |
| CSC | Cancer stem cells |
| EMT | Epithelial-mesenchymal transition |
| GlcNAc | N-acetylglucosamine |
| HBP | Hexosamine biosynthetic pathway |
| HCC | Hepatocellular carcinoma |
| NADPH | Reduced nicotinamide adenine dinucleotide phosphate |
| NSCLC | Non-small cell lung cancer |
| PPP | Pentose phosphate pathway |
| PyMT | Polyomavirus middle T |
| RNA | Ribonucleic acid |
| RTK | Receptors tyrosine kinase; |
| Ser | Serine |
| Thr | Threonine |
| Tyr | Tyrosine |
| UDP | Uridine diphosphate |
| XIAP | Increasing x-linked inhibitor of apoptosis protein |
References
- Klement, R.J.; Fink, M.K. Dietary and pharmacological modification of the insulin/IGF-1 system: Exploiting the full repertoire against cancer. Oncogenesis 2016, 5, e193. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Batth, I.S.; Qu, X.; Xu, L.; Song, N.; Wang, R.; Liu, Y. IGF-IR signaling in epithelial to mesenchymal transition and targeting IGF-IR therapy: Overview and new insights. Mol. Cancer 2017, 16, 6. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhou, R.; Yuan, L.; Wang, S.; Li, X.; Ma, H.; Zhou, M.; Pan, C.; Zhang, J.; Huang, N.; et al. IGF1/IGF1R/Stat3 signaling-inducible IFITM2 promotes gastric cancer growth and metastasis. Cancer Lett. 2017, 393, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Afratis, N.A.; Bouris, P.; Skandalis, S.S.; Multhaupt, H.A.; Couchman, J.R.; Theocharis, A.D.; Karamanos, N.K. IGF-IR cooperates with erα to inhibit breast cancer cell aggressiveness by regulating the expression and localisation of ecm molecules. Sci. Rep. 2017, 7, 40138. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.L.; Agarwal, E.; Chowdhury, S.; Luo, J.; Brattain, M.G.; Black, J.D.; Wang, J. TGFΒ/SMAD3 regulates proliferation and apoptosis through IRS-1 inhibition in colon cancer cells. PLoS ONE 2017, 12, e0176096. [Google Scholar] [CrossRef] [PubMed]
- Casa, A.J.; Dearth, R.K.; Litzenburger, B.C.; Lee, A.V.; Cui, X. The type I insulin-like growth factor receptor pathway: A key player in cancer therapeutic resistance. Front. Biosci. 2008, 13, 3273–3287. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Belfiore, A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front. Endocrinol. 2014, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Mistry, J.; Nicar, M.J.; Khosravi, M.J.; Diamandis, A.; van Doorn, J.; Juul, A. Insulin-like growth factors (IGF-I, free IGF-I and IGF-II) and insulin-like growth factor binding proteins (IGFBP-2, IGFBP-3, IGFBP-6, and ALS) in blood circulation. J. Clin. Lab. Anal. 1999, 13, 166–172. [Google Scholar] [CrossRef]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Forbes, B.E.; McCarthy, P.; Norton, R.S. Insulin-like growth factor binding proteins: A structural perspective. Front. Endocrinol. 2012, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Grimberg, A.; Cohen, P. Role of insulin-like growth factors and their binding proteins in growth control and carcinogenesis. J. Cell. Physiol. 2000, 183, 1–9. [Google Scholar] [CrossRef]
- Hu, Q.; Zhou, Y.; Ying, K.; Ruan, W. IGFBP, a novel target of lung cancer? Clin. Chim. Acta 2017, 466, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Firth, S.M.; Baxter, R.C. Cellular actions of the insulin-like growth factor binding proteins. Endocr. Rev. 2002, 23, 824–854. [Google Scholar] [CrossRef] [PubMed]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef] [PubMed]
- Lopaczynski, W.; Terry, C.; Nissley, P. Autophosphorylation of the insulin-like growth factor I receptor cytoplasmic domain. Biochem. Biophys. Res. Commun. 2000, 279, 955–960. [Google Scholar] [CrossRef] [PubMed]
- White, M.F.; Shoelson, S.E.; Keutmann, H.; Kahn, C.R. A cascade of tyrosine autophosphorylation in the β-subunit activates the phosphotransferase of the insulin receptor. J. Biol. Chem. 1988, 263, 2969–2980. [Google Scholar] [PubMed]
- Sparrow, L.G.; Lawrence, M.C.; Gorman, J.J.; Strike, P.M.; Robinson, C.P.; McKern, N.M.; Ward, C.W. N-linked glycans of the human insulin receptor and their distribution over the crystal structure. Proteins 2008, 71, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, L.G.; Gorman, J.J.; Strike, P.M.; Robinson, C.P.; McKern, N.M.; Epa, V.C.; Ward, C.W. The location and characterisation of the O-linked glycans of the human insulin receptor. Proteins 2007, 66, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Elleman, T.C.; Frenkel, M.J.; Hoyne, P.A.; McKern, N.M.; Cosgrove, L.; Hewish, D.R.; Jachno, K.M.; Bentley, J.D.; Sankovich, S.E.; Ward, C.W. Mutational analysis of the N-linked glycosylation sites of the human insulin receptor. Biochem. J. 2000, 347, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.B.; Hernandez, J.; Leduc, R.; Frost, S.C. Alternative glycosylation of the insulin receptor prevents oligomerization and acquisition of insulin-dependent tyrosine kinase activity. Biochim. Biophys. Acta 2000, 1499, 74–84. [Google Scholar] [CrossRef]
- Bastian, W.; Zhu, J.; Way, B.; Lockwood, D.; Livingston, J. Glycosylation of ASN397 or ASN418 is required for normal insulin receptor biosynthesis and processing. Diabetes 1993, 42, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Gray, A.; Tam, A.W.; Yang-Feng, T.; Tsubokawa, M.; Collins, C.; Henzel, W.; Le Bon, T.; Kathuria, S.; Chen, E. Insulin-like growth factor I receptor primary structure: Comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986, 5, 2503–2512. [Google Scholar] [PubMed]
- Kiess, W.; Greenstein, L.A.; Lee, L.; Thomas, C.; Nissley, S.P. Biosynthesis of the insulin-like growth factor-II (IGF-II)/mannose-6-phosphate receptor in rat c6 glial cells: The role of N-linked glycosylation in binding of IGF-II to the receptor. Mol. Endocrinol. 1991, 5, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Robajac, D.; Masnikosa, R.; Miković, Ž.; Nedić, O. Gestation-associated changes in the glycosylation of placental insulin and insulin-like growth factor receptors. Placenta 2016, 39, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Ikink, G.J.; Boer, M.; Bakker, E.R.; Hilkens, J. IRS4 induces mammary tumorigenesis and confers resistance to HER2-targeted therapy through constitutive PI3K/AKT-pathway hyperactivation. Nat. Commun. 2016, 7, 13567. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.J.; Ahmad, F.; Ding, W.; Li, P.M.; Zhang, W.R. Regulation of the insulin signalling pathway by cellular protein-tyrosine phosphatases. Mol. Cell. Biochem. 1998, 182, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Rui, L.; Aguirre, V.; Kim, J.K.; Shulman, G.I.; Lee, A.; Corbould, A.; Dunaif, A.; White, M.F. Insulin/IGF-1 and TNF-α stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J. Clin. Investig. 2001, 107, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Cai, W.; Zheng, Y.; Evers, B.M.; She, Q.B. ERK and AKT signaling cooperate to translationally regulate survivin expression for metastatic progression of colorectal cancer. Oncogene 2014, 33, 1828–1839. [Google Scholar] [CrossRef] [PubMed]
- Tamemoto, H.; Kadowaki, T.; Tobe, K.; Yagi, T.; Sakura, H.; Hayakawa, T.; Terauchi, Y.; Ueki, K.; Kaburagi, Y.; Satoh, S. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 1994, 372, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Lipes, M.A.; Patti, M.E.; Brüning, J.C.; Haag, B.; Johnson, R.S.; Kahn, C.R. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 1994, 372, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Björnholm, M.; He, A.R.; Attersand, A.; Lake, S.; Liu, S.C.; Lienhard, G.E.; Taylor, S.; Arner, P.; Zierath, J.R. Absence of functional insulin receptor substrate-3 (IRS-3) gene in humans. Diabetologia 2002, 45, 1697–1702. [Google Scholar] [PubMed]
- Denduluri, S.K.; Idowu, O.; Wang, Z.; Liao, Z.; Yan, Z.; Mohammed, M.K.; Ye, J.; Wei, Q.; Wang, J.; Zhao, L.; et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015, 2, 13–25. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining emt during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.R.; Zhau, H.E.; Odero-Marah, V.A.; Osunkoya, A.O.; Kimbro, K.S.; Tighiouart, M.; Liu, T.; Simons, J.W.; O’Regan, R.M. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008, 68, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, J.; Zeng, Y.; Zhang, X.; Hu, Q.; Zheng, J.; Chen, B.; Xie, B.; Zhang, W.M. Implication of epithelial-mesenchymal transition in IGF1R-induced resistance to EGFR-TKIs in advanced non-small cell lung cancer. Oncotarget 2015, 6, 44332–44345. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.W.; Lin, R.J.; Yu, J.; Chang, W.Y.; Fu, C.H.; Lai, A.; Yu, J.C.; Yu, A.L. The expression and significance of insulin-like growth factor-1 receptor and its pathway on breast cancer stem/progenitors. Breast Cancer Res. 2013, 15, R39. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Frasca, F.; Garozzo, A.; Gianì, F.; Pandini, G.; Vella, V.; Vigneri, R.; Belfiore, A. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J. Clin. Endocrinol. Metab. 2011, 96, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Shimamura, T.; Kimura, N.; Murayama, T.; Matsubara, D.; Kanauchi, H.; Niida, A.; Shimizu, S.; Nishioka, K.; Tsuji, E.I.; et al. Addiction to the IGF2-ID1-IGF2 circuit for maintenance of the breast cancer stem-like cells. Oncogene 2017, 36, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Shen, J.; Liu, L.; Xia, F.; Xu, C.; Duan, G.; Xu, Y.; Ma, Q.; Yang, Z.; Zhang, Q.; et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology 2012, 56, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Ruan, B.; Wu, J.; Wang, J.; Shang, R.; Sun, W.; Li, X.; Dou, K.; Wang, D.; Li, Y. Insulin-like growth factor binding protein-1 inhibits cancer cell invasion and is associated with poor prognosis in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 5645–5654. [Google Scholar] [PubMed]
- Li, H.; Xu, L.; Li, C.; Zhao, L.; Ma, Y.; Zheng, H.; Li, Z.; Zhang, Y.; Wang, R.; Liu, Y.; et al. Ubiquitin ligase Cbl-b represses IGF-I-induced epithelial mesenchymal transition via ZEB2 and microRNA-200c regulation in gastric cancer cells. Mol. Cancer 2014, 13, 136. [Google Scholar] [CrossRef] [PubMed]
- Taliaferro-Smith, L.; Oberlick, E.; Liu, T.; McGlothen, T.; Alcaide, T.; Tobin, R.; Donnelly, S.; Commander, R.; Kline, E.; Nagaraju, G.P.; et al. Fak activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget 2015, 6, 4757–4772. [Google Scholar] [CrossRef] [PubMed]
- Davison, Z.; de Blacquière, G.E.; Westley, B.R.; May, F.E. Insulin-like growth factor-dependent proliferation and survival of triple-negative breast cancer cells: Implications for therapy. Neoplasia 2011, 13, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Girnita, L.; Wang, M.; Xie, Y.; Nilsson, G.; Dricu, A.; Wejde, J.; Larsson, O. Inhibition of N-linked glycosylation down-regulates insulin-like growth factor-1 receptor at the cell surface and kills ewing’s sarcoma cells: Therapeutic implications. Anti-Cancer Drug Des. 2000, 15, 67–72. [Google Scholar]
- Li, Z.H.; Xiong, Q.Y.; Xu, L.; Duan, P.; Yang, Q.O.; Zhou, P.; Tu, J.H. miR-29a regulated ER-positive breast cancer cell growth and invasion and is involved in the insulin signaling pathway. Oncotarget 2017, 8, 32568–32575. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, H.; Guo, X.; Wang, Z.; Liang, S.; Dang, C. IGF-I induces epithelial-to-mesenchymal transition via the IGF-IR-Src-microRNA-30a-E-cadherin pathway in nasopharyngeal carcinoma cells. Oncol. Res. 2016, 24, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.H.; Chen, P.H.; Hsi, E.; Shih, C.M.; Chang, W.C.; Cheng, C.H.; Lin, C.W.; Chen, K.C. Identification of IGF-1-enhanced cytokine expressions targeted by miR-181d in glioblastomas via an integrative miRNA/mRNA regulatory network analysis. Sci. Rep. 2017, 7, 732. [Google Scholar] [CrossRef] [PubMed]
- Dallas, N.A.; Xia, L.; Fan, F.; Gray, M.J.; Gaur, P.; van Buren, G.; Samuel, S.; Kim, M.P.; Lim, S.J.; Ellis, L.M. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-i receptor inhibition. Cancer Res. 2009, 69, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Gaikwad, S.M.; Jinager, A.; Chaudhury, S.; Maheshwari, A.; Ray, P. IGF-1R inhibition potentiates cytotoxic effects of chemotherapeutic agents in early stages of chemoresistant ovarian cancer cells. Cancer Lett. 2014, 354, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Bang, Y.J.; Qin, S.K.; Chung, H.C.; Xu, J.M.; Park, J.O.; Jeziorski, K.; Shparyk, Y.; Hoff, P.M.; Sobrero, A.; et al. Lapatinib in combination with capecitabine plus oxaliplatin in human epidermal growth factor receptor 2-positive advanced or metastatic gastric, esophageal, or gastroesophageal adenocarcinoma: TRIO-013/LOGIC—A randomized phase III trial. J. Clin. Oncol. 2016, 34, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, J.; Ji, D.; Wang, C.; Liu, R.; Wu, Z.; Liu, L.; Zhu, D.; Chang, J.; Geng, R.; et al. Functional genetic approach identifies MET, HER3, IGF1R, INSR pathways as determinants of lapatinib unresponsiveness in HER2-positive gastric cancer. Clin. Cancer Res. 2014, 20, 4559–4573. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Kang, M.J.; Yoon, Y.K.; Kim, H.P.; Park, J.; Song, S.H.; Han, S.W.; Park, J.W.; Kang, G.H.; Kang, K.W.; et al. Heterodimerization of glycosylated insulin-like growth factor-1 receptors and insulin receptors in cancer cells sensitive to anti-IGF1R antibody. PLoS ONE 2012, 7, e33322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, C.; Wu, D. Activation of insulin-like growth factor 1 receptor regulates the radiation-induced lung cancer cell apoptosis. Immunobiology 2015, 220, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Osuka, S.; Sampetrean, O.; Shimizu, T.; Saga, I.; Onishi, N.; Sugihara, E.; Okubo, J.; Fujita, S.; Takano, S.; Matsumura, A.; et al. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells 2013, 31, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lee, H.J.; Seo, W.D.; Park, K.H.; Lee, Y.S. Sialylation of integrin β1 is involved in radiation-induced adhesion and migration in human colon cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- De-Freitas-Junior, J.C.; Bastos, L.G.; Freire-Neto, C.A.; Rocher, B.D.; Abdelhay, E.S.; Morgado-Díaz, J.A. N-glycan biosynthesis inhibitors induce in vitro anticancer activity in colorectal cancer cells. J. Cell. Biochem. 2012, 113, 2957–2966. [Google Scholar] [CrossRef] [PubMed]
- Contessa, J.N.; Bhojani, M.S.; Freeze, H.H.; Ross, B.D.; Rehemtulla, A.; Lawrence, T.S. Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin. Cancer Res. 2010, 16, 3205–3214. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Simopoulos, C.; Polychronidis, A.; Sivridis, E. Lactate dehydrogenase 5 (LDH5) relates to up-regulated hypoxia inducible factor pathway and metastasis in colorectal cancer. Clin. Exp. Metastasis 2005, 22, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”?—Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Treins, C.; Giorgetti-Peraldi, S.; Murdaca, J.; Monthouël-Kartmann, M.N.; Van Obberghen, E. Regulation of hypoxia-inducible factor (HIF)-1 activity and expression of hif hydroxylases in response to insulin-like growth factor I. Mol. Endocrinol. 2005, 19, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, J.; Zhang, S.; Xu, X.; Zheng, M.; Jiang, G.; Li, F. IGF-1 induces hypoxia-inducible factor 1α-mediated GLUT3 expression through PI3K/Akt/mTOR dependent pathways in PC12 cells. Brain Res. 2012, 1430, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Nurwidya, F.; Takahashi, F.; Kobayashi, I.; Murakami, A.; Kato, M.; Minakata, K.; Nara, T.; Hashimoto, M.; Yagishita, S.; Baskoro, H.; et al. Treatment with insulin-like growth factor 1 receptor inhibitor reverses hypoxia-induced epithelial-mesenchymal transition in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2014, 455, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.; Kim, K.W. Diabetes and cancer: Is diabetes causally related to cancer? Diabetes Metab. J. 2011, 35, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Carstensen, B.; Witte, D.; Bowker, S.L.; Lipscombe, L.; Renehan, A.G.; Consortium, D.a.C.R. Diabetes and cancer (1): Evaluating the temporal relationship between type 2 diabetes and cancer incidence. Diabetologia 2012, 55, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Alisson-Silva, F.; Freire-de-Lima, L.; Donadio, J.L.; Lucena, M.C.; Penha, L.; Sá-Diniz, J.N.; Dias, W.B.; Todeschini, A.R. Increase of O-glycosylated oncofetal fibronectin in high glucose-induced epithelial-mesenchymal transition of cultured human epithelial cells. PLoS ONE 2013, 8, e60471. [Google Scholar] [CrossRef] [PubMed]
- Masur, K.; Vetter, C.; Hinz, A.; Tomas, N.; Henrich, H.; Niggemann, B.; Zänker, K.S. Diabetogenic glucose and insulin concentrations modulate transcriptome and protein levels involved in tumour cell migration, adhesion and proliferation. Br. J. Cancer 2011, 104, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos-Dos-Santos, A.; Loponte, H.F.; Mantuano, N.R.; Oliveira, I.A.; de Paula, I.F.; Teixeira, L.K.; de-Freitas-Junior, J.C.; Gondim, K.C.; Heise, N.; Mohana-Borges, R.; et al. Hyperglycemia exacerbates colon cancer malignancy through hexosamine biosynthetic pathway. Oncogenesis 2017, 6, e306. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, S.Y.; Hu, Z.F.; Winslet, M.C.; Wang, W.; Seifalian, A.M. Growth factors enhance endothelial progenitor cell proliferation under high-glucose conditions. Med. Sci. Monit. 2009, 15, BR357–BR363. [Google Scholar] [PubMed]
- Kalaany, N.Y.; Sabatini, D.M. Tumours with PI3K activation are resistant to dietary restriction. Nature 2009, 458, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991, 266, 4706–4712. [Google Scholar] [PubMed]
- Carvalho, S.; Oliveira, T.; Bartels, M.F.; Miyoshi, E.; Pierce, M.; Taniguchi, N.; Carneiro, F.; Seruca, R.; Reis, C.A.; Strahl, S.; et al. O-mannosylation and N-glycosylation: Two coordinated mechanisms regulating the tumour suppressor functions of E-cadherin in cancer. Oncotarget 2016, 7, 65231–65246. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Catarino, T.A.; Dias, A.M.; Kato, M.; Almeida, A.; Hessling, B.; Figueiredo, J.; Gärtner, F.; Sanches, J.M.; Ruppert, T.; et al. Preventing E-cadherin aberrant N-glycosylation at Asn-554 improves its critical function in gastric cancer. Oncogene 2016, 35, 1619–1631. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Figueiredo, J.; Cabral, J.; Carvalho, S.; Dourado, J.; Magalhães, A.; Gärtner, F.; Mendonfa, A.M.; Isaji, T.; Gu, J.; et al. E-cadherin and adherens-junctions stability in gastric carcinoma: Functional implications of glycosyltransferases involving N-glycan branching biosynthesis, N-acetylglucosaminyltransferases III and V. Biochim. Biophys. Acta 2013, 1830, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- De-Freitas-Junior, J.C.; Morgado-Díaz, J.A. The role of N-glycans in colorectal cancer progression: Potential biomarkers and therapeutic applications. Oncotarget 2016, 7, 19395–19413. [Google Scholar] [CrossRef] [PubMed]
- De Freitas Junior, J.C.; Silva, B.u.R.; de Souza, W.F.; de Araújo, W.M.; Abdelhay, E.S.; Morgado-Díaz, J.A. Inhibition of N-linked glycosylation by tunicamycin induces E-cadherin-mediated cell-cell adhesion and inhibits cell proliferation in undifferentiated human colon cancer cells. Cancer Chemother. Pharmacol. 2011, 68, 227–238. [Google Scholar] [CrossRef] [PubMed]
- De-Freitas-Junior, J.C.; Carvalho, S.; Dias, A.M.; Oliveira, P.; Cabral, J.; Seruca, R.; Oliveira, C.; Morgado-Díaz, J.A.; Reis, C.A.; Pinho, S.S. Insulin/IGF-I signaling pathways enhances tumor cell invasion through bisecting glcnac N-glycans modulation. An interplay with E-cadherin. PLoS ONE 2013, 8, e81579. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chaney, W. Induction of N-acetylglucosaminyltransferase V by elevated expression of activated or proto-Ha-ras oncogenes. Mol. Cell. Biochem. 1993, 122, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Reis, C.A.; Paredes, J.; Magalhães, A.M.; Ferreira, A.C.; Figueiredo, J.; Xiaogang, W.; Carneiro, F.; Gärtner, F.; Seruca, R. The role of N-acetylglucosaminyltransferase III and V in the post-transcriptional modifications of E-cadherin. Hum. Mol. Genet. 2009, 18, 2599–2608. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Carvalho, S.; Marcos-Pinto, R.; Magalhães, A.; Oliveira, C.; Gu, J.; Dinis-Ribeiro, M.; Carneiro, F.; Seruca, R.; Reis, C.A. Gastric cancer: Adding glycosylation to the equation. Trends Mol. Med. 2013, 19, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Korekane, H. Branched N-glycans and their implications for cell adhesion, signaling and clinical applications for cancer biomarkers and in therapeutics. BMB Rep. 2011, 44, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Granovsky, M.; Fata, J.; Pawling, J.; Muller, W.J.; Khokha, R.; Dennis, J.W. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat. Med. 2000, 6, 306–312. [Google Scholar] [PubMed]
- Webster, M.A.; Hutchinson, J.N.; Rauh, M.J.; Muthuswamy, S.K.; Anton, M.; Tortorice, C.G.; Cardiff, R.D.; Graham, F.L.; Hassell, J.A.; Muller, W.J. Requirement for both Shc and phosphatidylinositol 3′ kinase signaling pathways in polyomavirus middle T-mediated mammary tumorigenesis. Mol. Cell. Biol. 1998, 18, 2344–2359. [Google Scholar] [CrossRef] [PubMed]
- Partridge, E.A.; Le Roy, C.; Di Guglielmo, G.M.; Pawling, J.; Cheung, P.; Granovsky, M.; Nabi, I.R.; Wrana, J.L.; Dennis, J.W. Regulation of cytokine receptors by golgi N-glycan processing and endocytosis. Science 2004, 306, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Toscano, M.A.; Jackson, S.S.; Vasta, G.R. Functions of cell surface galectin-glycoprotein lattices. Curr. Opin. Struct. Biol. 2007, 17, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, J.; Hashidate, T.; Arata, Y.; Nishi, N.; Nakamura, T.; Hirashima, M.; Urashima, T.; Oka, T.; Futai, M.; Muller, W.E.; et al. Oligosaccharide specificity of galectins: A search by frontal affinity chromatography. Biochim. Biophys. Acta 2002, 1572, 232–254. [Google Scholar] [CrossRef]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A.; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Slawson, C.; Copeland, R.J.; Hart, G.W. O-glcnac signaling: A metabolic link between diabetes and cancer? Trends Biochem. Sci. 2010, 35, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-glcnacylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Vosseller, K. Cancer metabolism and elevated O-glcnac in oncogenic signaling. J. Biol. Chem. 2014, 289, 34457–34465. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.L.; Berkaw, M.N.; Buse, M.G.; Ball, L.E. O-linked N-acetylglucosamine modification of insulin receptor substrate-1 occurs in close proximity to multiple SH2 domain binding motifs. Mol. Cell. Proteomics 2009, 8, 2733–2745. [Google Scholar] [CrossRef] [PubMed]
- Gandy, J.C.; Rountree, A.E.; Bijur, G.N. Akt1 is dynamically modified with O-glcnac following treatments with pugnac and insulin-like growth factor-1. FEBS Lett. 2006, 580, 3051–3058. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Reis, C.A.; Pinho, S.S. Cadherins glycans in cancer: Sweet players in a bitter process. Trends Cancer 2016, 2, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, M.; Harris, T.; Sundaram, S.; Johnson, L.; Guttenplan, J.; Rogler, C.; Stanley, P. Progression of hepatic neoplasms is severely retarded in mice lacking the bisecting n-acetylglucosamine on N-glycans: Evidence for a glycoprotein factor that facilitates hepatic tumor progression. Cancer Res. 1998, 58, 2881–2887. [Google Scholar] [PubMed]
- Ishimura, H.; Takahashi, T.; Nakagawa, H.; Nishimura, S.; Arai, Y.; Horikawa, Y.; Habuchi, T.; Miyoshi, E.; Kyan, A.; Hagisawa, S.; et al. N-acetylglucosaminyltransferase V and β1-6 branching N-linked oligosaccharides are associated with good prognosis of patients with bladder cancer. Clin. Cancer Res. 2006, 12, 2506–2511. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De-Freitas-Junior, J.C.M.; Andrade-da-Costa, J.; Silva, M.C.; Pinho, S.S. Glycans as Regulatory Elements of the Insulin/IGF System: Impact in Cancer Progression. Int. J. Mol. Sci. 2017, 18, 1921. https://doi.org/10.3390/ijms18091921
De-Freitas-Junior JCM, Andrade-da-Costa J, Silva MC, Pinho SS. Glycans as Regulatory Elements of the Insulin/IGF System: Impact in Cancer Progression. International Journal of Molecular Sciences. 2017; 18(9):1921. https://doi.org/10.3390/ijms18091921
Chicago/Turabian StyleDe-Freitas-Junior, Julio Cesar M., Jéssica Andrade-da-Costa, Mariana Costa Silva, and Salomé S. Pinho. 2017. "Glycans as Regulatory Elements of the Insulin/IGF System: Impact in Cancer Progression" International Journal of Molecular Sciences 18, no. 9: 1921. https://doi.org/10.3390/ijms18091921