CytoCluster: A Cytoscape Plugin for Cluster Analysis and Visualization of Biological Networks

Abstract
1. Introduction
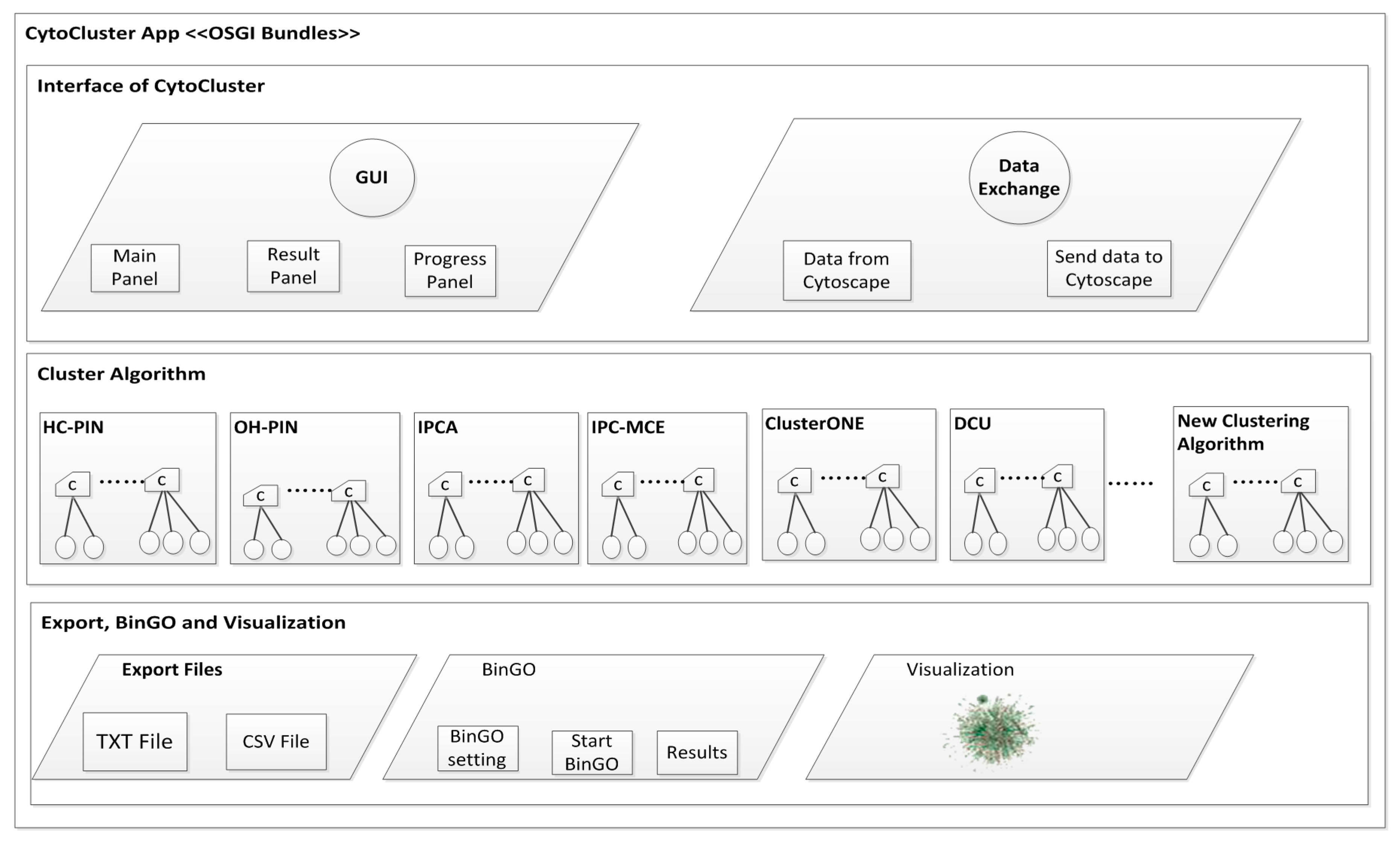
2. Architecture
3. Implementation
3.1. Calculation and Basic Analysis
3.1.1. HC-PIN (Hierarchical Clustering Algorithm in Protein Interaction Networks)
3.1.2. OH-PIN (Identifying Overlapping and Hierarchical Modules in Protein Interaction Networks)
3.1.3. IPCA (Identifying Protein Complex Algorithm)
3.1.4. IPC-MCE (Identifying Protein Complexes based on Maximal Complex Extension)
3.1.5. ClusterONE (Clustering with Overlapping Neighborhood Expansion)
3.1.6. DCU (Detecting Complexes Based on Uncertain Graph Model)
3.2. BinGO
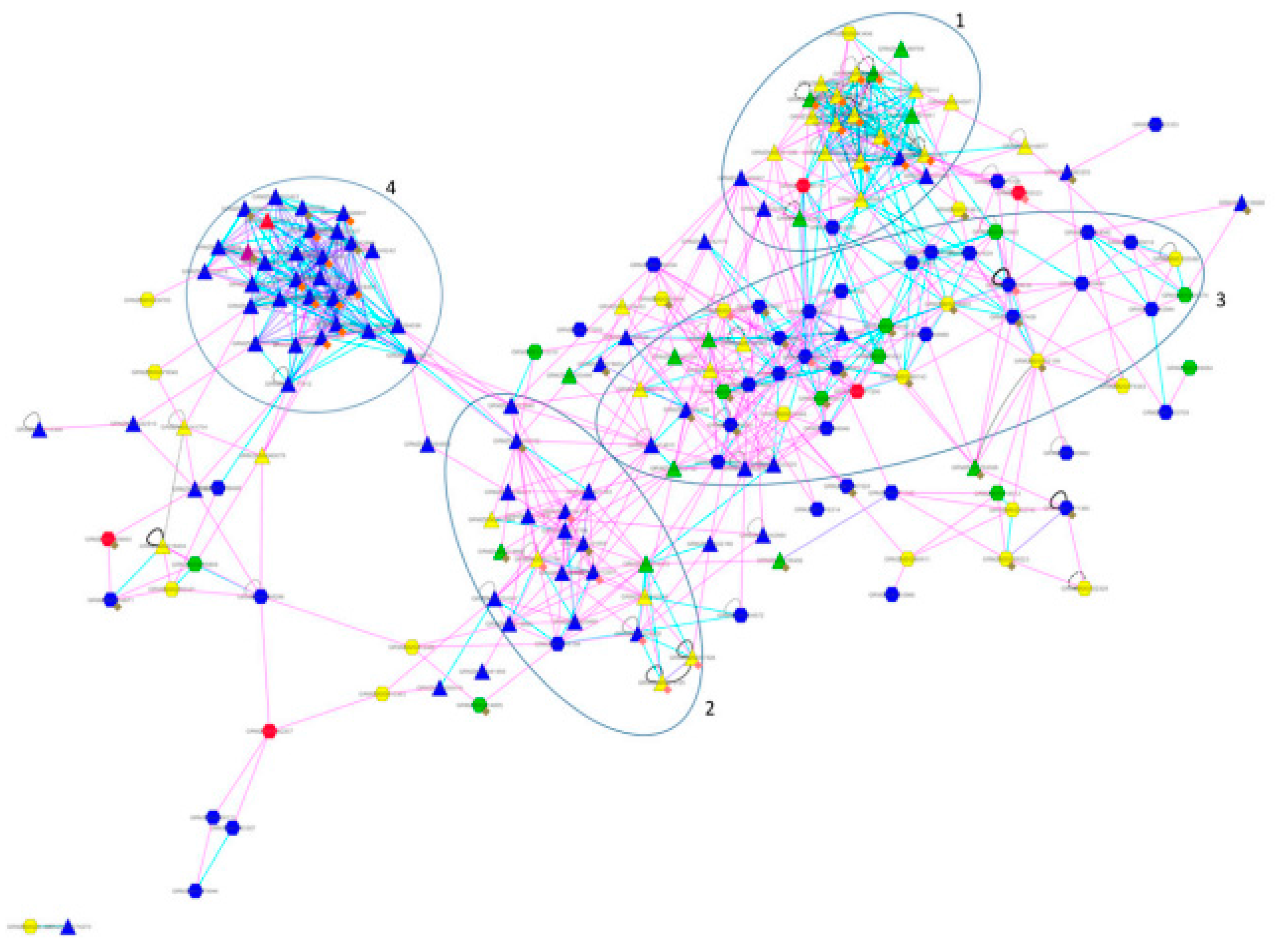
4. Cases Studies
5. Conclusions
Acknowledgment
Author Contributions
Conflicts of Interest
References
- Wang, J.; Liang, J.; Zheng, W. A graph clustering method for detecting protein complexes. J. Comput. Res. Dev. 2015, 52, 1784–1793. [Google Scholar]
- Alberts, B. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell 1998, 92, 291–294. [Google Scholar] [CrossRef]
- Lasserre, J.P.; Beyne, E.; Pyndiah, S.; Pyndiah, S.; Lapaillerie, D.; Claverol, S.; Bonneu, M. A complexomic study of Escherichia coli using two-dimensional blue native/SDS polyacrylamide gel electrophoresis. Electrophoresis 2006, 27, 3306–3321. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.J. Cell regulation: Determined to signal discrete cooperation. Trends Biochem. Sci. 2009, 3410, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Pržulj, N.; Wigle, D.A.; Jurisica, I. Functional topology in a network of protein interactions. Bioinformatics 2004, 203, 340–348. [Google Scholar] [CrossRef] [PubMed]
- King, A.D.; Pržulj, N.; Jurisica, I. Protein complex prediction via cost-based clustering. Bioinformatics 2004, 2017, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Wang, W.; Peng, X.; Wang, J. Mining protein complexes from PPI networks using the minimum vertex cut. Tsinghua Sci. Technol. 2012, 176, 674–681. [Google Scholar] [CrossRef]
- Enright, A.J.; Dongen, S.V.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 41, 2. [Google Scholar]
- Palla, G.; Derényi, I.; Farkas, I.; Vicsek, T. Uncovering the overlapping community structure of complex networks in nature and society. Nature 2005, 435, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Foo, C.S.; Tan, S.H.; Ng, S.K. Interaction graph mining for protein complexes using local clique merging. Genome Inform. 2005, 16, 260–269. [Google Scholar] [PubMed]
- Altaf-Ul-Amin, M.; Shinbo, Y.; Mihara, K.; Kurokawa, K.; Kanaya, S. Development and implementation of an algorithm for detection of protein complexes in large interaction networks. BMC Bioinform. 2006, 7, 207. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, J.; Wang, J.; Hu, B.; Chen, G. Modifying the DPClus algorithm for identifying protein complexes based on new topological structures. BMC Bioinform. 2008, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wong, L.; Chua, H.N. Complex discovery from weighted PPI networks. Bioinformatics 2009, 25, 1891–1897. [Google Scholar] [CrossRef] [PubMed]
- Srihari, S.; Ning, K.; Leong, H.W. MCL-CAw: A refinement of MCL for detecting yeast complexes from weighted PPI networks by incorporating core-attachment structure. BMC Bioinform. 2010, 11, 504. [Google Scholar] [CrossRef] [PubMed]
- Nepusz, T.; Yu, H.; Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 2012, 9, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Girvan, M.; Newman, M.E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Yang, Y.; Chen, C.F.; Chang, R.; Zhou, J.; Scheuermann, R.H. Modular organization of protein interaction networks. Bioinformatics 2007, 23, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, J.; Chen, J. A fast hierarchical clustering algorithm for functional modules in protein interaction networks. In Proceedings of the IEEE 2008 International Conference on BioMedical Engineering and Informatics (BMEI), Sanya, China, 27–30 May 2008; Volume 1, pp. 3–7. [Google Scholar]
- Shen, H.; Cheng, X.; Cai, K.; Hu, M.B. Detect overlapping and hierarchical community structure in networks. Phys. A Stat. Mech. Appl. 2009, 388, 1706–1712. [Google Scholar] [CrossRef]
- Wang, J.; Li, M.; Chen, J.; Pan, Y. A fast hierarchical clustering algorithm for functional modules discovery in protein interaction networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ren, J.; Li, M.; Wu, F.X. Identification of hierarchical and overlapping functional modules in PPI networks. IEEE Trans. Nanobiosci. 2012, 11, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Fu, Y.; Shang, M. A fast and efficient heuristic algorithm for detecting community structures in complex networks. Phys. A Stat. Mech. Appl. 2009, 388, 2741–2749. [Google Scholar] [CrossRef]
- Inoue, K.; Li, W.; Kurata, H. Diffusion model based spectral clustering for protein-protein interaction networks. PLoS ONE 2010, 5, e12623. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, Y. Semi-supervised consensus clustering for gene expression data analysis. BioData Min. 2014, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wu, X.; Wang, J.; Pan, Y. Progress on graph-based clustering methods for the analysis of protein-protein interaction networks. Comput. Eng. Sci. 2012, 34, 124–136. [Google Scholar]
- Ji, J.; Liu, Z.; Liu, H.; Liu, C. An overview of research on functional module detection for protein-protein interaction networks. Acta Autom. Sin. 2014, 40, 577–593. [Google Scholar]
- Protein-Protein Interaction Networks Co-Clustering. Available online: http://wwwinfo.deis.unical.it/rombo/co-clustering/ (accessed on 21 April 2017).
- Batagelj, V.; Mrvar, A. Pajek-program for large network analysis. Connections 1998, 21, 47–57. [Google Scholar]
- Adamcsek, B.; Palla, G.; Farkas, I.J.; Derényi, I.; Vicsek, T. CFinder: Locating cliques and overlapping modules in biological networks. Bioinformatics 2006, 22, 1021–1023. [Google Scholar] [CrossRef] [PubMed]
- Moschopoulos, C.N.; Pavlopoulos, G.A.; Schneider, R.; Likothanassis, S.D.; Kossida, S. GIBA: A clustering tool for detecting protein complexes. BMC Bioinform. 2009, 10, S11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Xu, Y.; Zhang, X.; Liu, Z.P.; Wang, Z.; Chen, L.; Zhu, X.G. CMIP: A software package capable of reconstructing genome-wide regulatory networks using gene expression data. BMC Bioinform. 2016, 17, 137. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Tang, Y.; Wu, X.; Wang, J.; Wu, F.X.; Pan, Y. C-DEVA: Detection, evaluation, visualization and annotation of clusters from biological networks. Biosystems 2016, 150, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhong, J.; Chen, G.; Li, M.; Wu, F.X.; Pan, Y. ClusterViz: A Cytoscape APP for cluster analysis of biological network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.H.; Apeltsin, L.; Newman, A.M.; Baumbach, J.; Wittkop, T.; Su, G.; Bader, G.D.; Ferrin, T.E. clusterMaker: A multi-algorithm clustering plugin for Cytoscape. BMC Bioinform. 2011, 12, 436. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, J.; Li, M.; Wu, F.X.; Pan, Y. Detecting protein complexes based on uncertain graph model. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, J.X.; Liu, B.B.; Chen, J.E. An algorithm for identifying protein complexes based on maximal clique extension. J. Cent. South Univ. 2010, 41, 560–565. [Google Scholar]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Nishizawa, T.; Hayakumo, M.; Hikosaka, S.; Saito, K.; Goto, E.; Kusano, M. Exploring tomato gene functions based on coexpression modules using graph clustering and differential coexpression approaches. Plant Physiol. 2012, 158, 1487–1502. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, R.J.; Michno, J.M.; Myers, C.L. Unraveling gene function in agricultural species using gene co-expression networks. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2017, 1860, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, J.; Li, L.; Xu, X.; Zhang, Y.; Teng, Z.; Wu, F. Identification of molecular targets for Predicting Colon Adenocarcinoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 460–468. [Google Scholar] [CrossRef]
- Wang, M.; Chen, G.; Lu, C.; Xiao, C.; Li, L.; Niu, X.; He, X.; Jiang, M.; Lu, A. Rheumatoid arthritis with deficiency pattern in traditional Chinese medicine shows correlation with cold and hot patterns in gene expression profiles. Evid.-Based Complement. Altern. Med. 2013, 2013, 248650. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Niu, X.; Xiao, C.; Chen, G.; Zha, Q.; Guo, H.; Jiang, M.; Lu, A. Network-based gene expression biomarkers for cold and heat patterns of rheumatoid arthritis in traditional Chinese medicine. Evid.-Based Complement. Altern. Med. 2012, 2012, 203043. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xiao, C.; Chen, G.; Jiang, M.; Zha, Q.; Yan, X.; Kong, W.; Lu, A. Cold and heat pattern of rheumatoid arthritis in traditional Chinese medicine: Distinct molecular signatures indentified by microarray expression profiles in CD4-positive T cell. Rheumatol. Int. 2012, 32, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Lu, C.; Zha, Q.; Xiao, C.; Xu, S.; Ju, D.; Zhou, Y.; Jia, W.; Lu, A. A network-based analysis of traditional Chinese medicine cold and hot patterns in rheumatoid arthritis. Complement. Ther. Med. 2012, 20, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, B.; Jiang, M.; Tan, Y.; Lu, A.P. Functional networks for Salvia miltiorrhiza and Panax notoginseng in combination explored with text mining and bioinformatical approach. J. Med. Plants Res. 2011, 5, 4030–4040. [Google Scholar]
- Jiang, M.; Lu, C.; Chen, G.; Xiao, C.; Zha, Q.; Niu, X.; Chen, S.; Lu, A. Understanding the molecular mechanism of interventions in treating rheumatoid arthritis patients with corresponding traditional Chinese medicine patterns based on bioinformatics approach. Evid.-Based Complement. Altern. Med. 2012, 2012, 129452. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, B.; Jiang, M.; Aiping, L. System Analysis of the Synergistic Mechanisms between Salvia Miltiorrhiza and Panax Notoginseng in Combination. World Sci. Technol. 2010, 12, 566–570. [Google Scholar]
- Kalenitchenko, D.; Fagervold, S.K.; Pruski, A.M.; Vétion, G.; Yücel, M.; Le Bris, N.; Galand, P.E. Temporal and spatial constraints on community assembly during microbial colonization of wood in seawater. ISME J. 2015, 9, 2657–2670. [Google Scholar] [CrossRef] [PubMed]
- Meistertzheim, A.L.; Lartaud, F.; Arnaud-Haond, S.; Kalenitchenko, D.; Bessalam, M.; Le Bris, N.; Galand, P.E. Patterns of bacteria-host associations suggest different ecological strategies between two reef building cold-water coral species. Deep Sea Res. Part I Oceanogr. Res. Pap. 2016, 114, 12–22. [Google Scholar] [CrossRef]
- Guo, H.; Chen, J.; Meng, F. Identification of novel diagnosis biomarkers for lung adenocarcinoma from the cancer genome atlas. Orig. Artic. 2016, 9, 7908–7918. [Google Scholar]
- Atan, N.A.D.; Yekta, R.F.; Nejad, M.R.; Nikzamir, A. Pathway and network analysis in primary open angle glaucoma. J. Paramed. Sci. 2014, 5. [Google Scholar] [CrossRef]
- Wang, H.; Wei, Z.; Mei, L.; Gu, J.; Yin, S.; Faust, K.; Raes, J.; Deng, Y.; Wang, Y.; Shen, Q.; Yin, S. Combined use of network inference tools identifies ecologically meaningful bacterial associations in a paddy soil. Soil Biol. Biochem. 2017, 105, 227–235. [Google Scholar] [CrossRef]
- Havugimana, P.C.; Hart, G.T.; Nepusz, T.; Yang, H.; Turinsky, A.L.; Li, Z.; Wang, P.I.; Boutz, D.R.; Fong, V.; Phanse, S.; et al. A census of human soluble protein complexes. Cell 2012, 150, 1068. [Google Scholar] [CrossRef] [PubMed]
- Van Landeghem, S.; de Bodt, S.; Drebert, Z.J.; Inzé, D.; van de Peer, Y. The potential of text mining in data integration and network biology for plant research: A case study on Arabidopsis. Plant Cell 2013, 25, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Gudivada, R.C.; Aronow, B.J.; Jegga, A.G. Computational drug repositioning through heterogeneous network clustering. BMC Syst. Biol. 2013, 7, S6. [Google Scholar] [CrossRef] [PubMed]
- Baute, J.; Herman, D.; Coppens, F.; de Block, J.; Slabbinck, B.; dell’Aqcua, M.; Pè, M.E.; Maere, S.; Nelissen, H.; Inzé, D. Combined large-scale phenotyping and transcriptomics in maize reveals a robust growth regulatory network. Plant Physiol. 2016, 170, 1848–1867. [Google Scholar] [CrossRef] [PubMed]
- Czerwinska, U.; Calzone, L.; Barillot, E.; Zinovyev, A. DeDaL: Cytoscape 3 app for producing and morphing data-driven and structure-driven network layouts. BMC Syst. Biol. 2015, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2010, 39, D691–D697. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, J.; Chen, J.; Cai, Z.; Chen, G. Identifying the overlapping complexes in protein interaction networks. Int. J. Data Min. Bioinform. 2010, 4, 91–108. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J.; Zhao, B.; Wu, F.X.; Pan, Y. Identification of protein complexes from multi-relationship protein interaction networks. Hum. Genom. 2016, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Ding, Y.; Wu, F.X. Detecting protein complexes from DPINs by density based clustering with Pigeon-Inspired Optimization Algorithm. Sci. China Inf. Sci. 2016, 59, 070103. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, J.; Li, M.; Li, X.; Li, Y.; Wu, F.X.; Pan, Y. A new method for predicting protein functions from dynamic weighted interactome networks. IEEE Trans. Nanobiosci. 2016, 15, 131–139. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Algorithms | Application | Network | Description | Reference |
|---|---|---|---|---|
| IPCA | Exploring tomato gene functions | The tomato co-expression network was chosen and 465 complexes were found | IPCA was used to identity a densely connected network | [40] |
| Unravelling gene function | The tomato co-expression network was chosen and 465 complexes were found | IPCA was choosen to identify thick connected nodes | [41] | |
| Predicting colon adenocarcinoma | The networks from IntAct and reactome were merged | IPCA was used to identify highly connected subnetworks | [42] | |
| The correlation between cold and heat patterns | The network from RA 18 was diagnosed with defciency pattern and 15 others were diagnosed with nondefciency pattern | IPCA was used to analyze the characteristics of networks | [43] | |
| Evidence-based complementary and alternative medicine | PPI network from genes was chosen so that the ratio of cold patterns to heat patterns in patients with RA was more or less than 1:1.4 | IPCA was used to detect highly connected subnetworks | [44] | |
| Cold and heat patterns of rheumatoid arthritis | PPI network from these genes was chose that the ratio of cold patterns to heat patterns in patients with RA was more or less than 1:2 | Highly connected regions associated with typical TCM cold patterns and heat patterns were identified | [45] | |
| Cold and heat pattern of rheumatoid arthritis | Network for differentially expressed genes between RA patients with TCM cold and heat patterns | IPCA was used to infer significant complexes or pathways in the PPI network | [46] | |
| Functional networks | Network contained some gene expressions or regulated proteins | Then eight highly connected regions were found by IPCA to infer complexes or pathways | [47] | |
| The molecular mechanism of interventions | PPI networks of biomedical combination was chosen and 11 complexes were found | IPCA was used to analyze the characteristics of the network | [48] | |
| The synergistic sechanisms | Network associated with Salvia miltiorrhiza and Panax notoginseng | Significant complexes or pathways were inferred | [49] | |
| HC-PIN | Constraints on community | Associations between bacteria OTUs and four subnetworks were found | Subnetworks of OTUs were detected | [50] |
| Strategies between two reef building cold-water coral species | Association network of the cold-water scleractinian corals bacterial communities | HC-PIN was used to identify OTUs | [51] | |
| Biomarkers | The network was extracted from the TCGA database | miRNA-gene clusters were identified | [52] | |
| Finding the candidate biomarkers for POAG disease | Network was extracted from previous studies with 474 proteins and nine subnetworks were found | HC-PIN was choosen to perform the clustering with a complex size threshold of 3 | [53] | |
| OH-PIN | Bacterial associations | Bulk soil DNA was extracted | The subnetworks were partitioned into modulars | [54] |
| ClusterONE | A census of human soluble protein complexes | Network was extracted from human HeLa S3 and HEK293 cells grown | ClusterONE was used to detect protein complexes | [55] |
| An arabidopsis | A network with 8900 nodes and 6382 edges was chosen and 701 clusters were found | ClusterONE was used to obtain subnetworks | [56] | |
| Fndinge disease-drug modules | Disease-gene and drug-target associations were found from drug-target data | Overlapping subnetworks were identified | [57] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Li, D.; Tang, Y.; Wu, F.; Wang, J. CytoCluster: A Cytoscape Plugin for Cluster Analysis and Visualization of Biological Networks. Int. J. Mol. Sci. 2017, 18, 1880. https://doi.org/10.3390/ijms18091880
Li M, Li D, Tang Y, Wu F, Wang J. CytoCluster: A Cytoscape Plugin for Cluster Analysis and Visualization of Biological Networks. International Journal of Molecular Sciences. 2017; 18(9):1880. https://doi.org/10.3390/ijms18091880
Chicago/Turabian StyleLi, Min, Dongyan Li, Yu Tang, Fangxiang Wu, and Jianxin Wang. 2017. "CytoCluster: A Cytoscape Plugin for Cluster Analysis and Visualization of Biological Networks" International Journal of Molecular Sciences 18, no. 9: 1880. https://doi.org/10.3390/ijms18091880
APA StyleLi, M., Li, D., Tang, Y., Wu, F., & Wang, J. (2017). CytoCluster: A Cytoscape Plugin for Cluster Analysis and Visualization of Biological Networks. International Journal of Molecular Sciences, 18(9), 1880. https://doi.org/10.3390/ijms18091880