HIC1 and RassF1A Methylation Attenuates Tubulin Expression and Cell Stiffness in Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
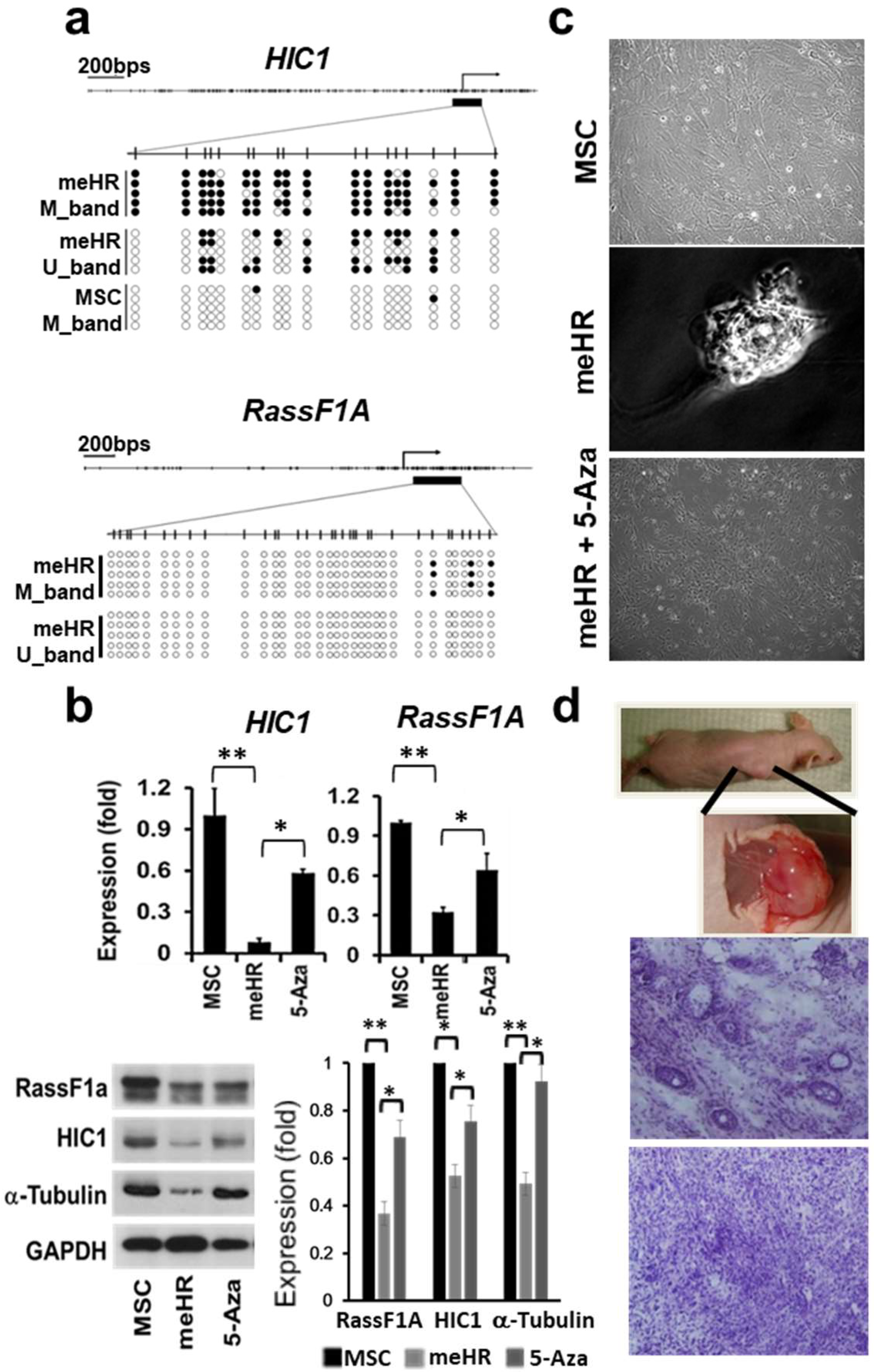
2.1. HIC1 and RassF1A Methylation Transforms MSCs
2.1.1. Repeated HIC1 and RassF1A Methylation Can Transform MSCs into CSC-Like Cells
2.1.2. Clonally Expanded me_HR-Transformed MSCs
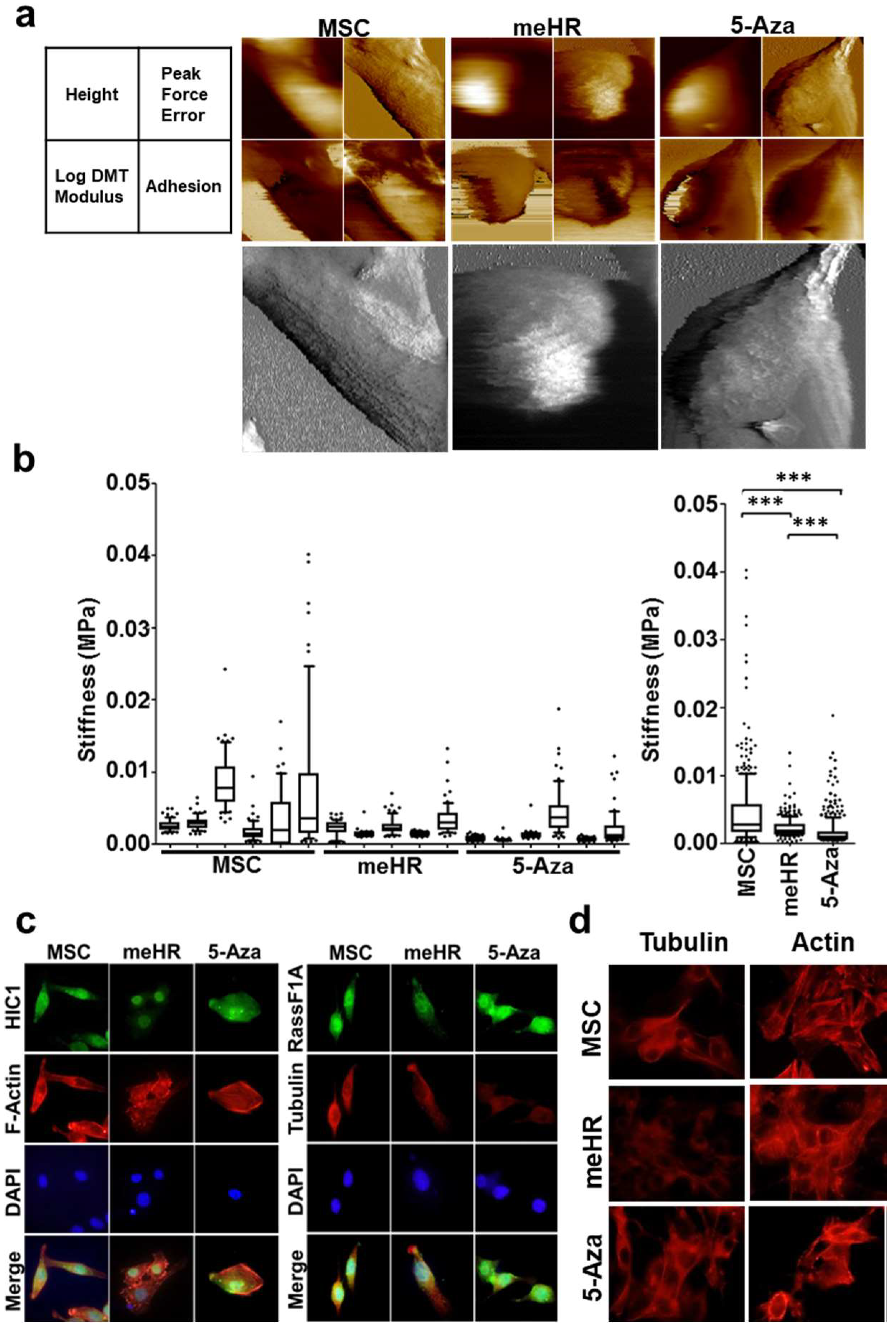
2.2. Loss of Stiffness Correlates with the Loss of Tubulin Expression in me_HR-Transformed MSCs
2.3. Reduced Tubulin Expression in me_HR Tumors
2.4. Tubulin Expression Correlates with RassF1A Expression in Liver Cancer
3. Discussion
3.1. DNA Methylation Is an Inheritable Tumor Marker
3.2. Abnormal HIC1 and RassF1A Methylation Identified in Cancers
3.3. Application of AFM in Cancer Research
3.4. Loss of Stiffness in me_HR-Transformed MSCs
3.5. Correlation between Stiffness Loss and Drug Resistance Development in Cancer
3.6. Cell Stiffness as Another Biomarker
4. Materials and Methods
4.1. Cell Culture
4.2. 5-Aza Treatment
4.3. Western Blotting
4.4. Semi-Quantitative RT-PCR, qRT-PCR
4.5. Semi-Quantitative Real-Time Methylation-Specific PCR (qMSP)
4.6. Methylation Sequencing
4.7. AFM
4.8. In Vivo Tumorigenesis and Monitoring Tumor Growth
4.9. Immunohistochemistry
4.10. Statistical Analyses
4.11. Human Subjects and Animal Care
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-Aza | 5-aza-2’-deoxycytidine |
| PCR | Polymerase chain reaction |
| AFM | Atomic Force Microscopy |
| MSP | Methylation specific PCR |
References
- Hsiao, S.H.; Huang, T.H.; Leu, Y.W. Excavating relics of DNA methylation changes during the development of neoplasia. Semin. Cancer Biol. 2009, 19, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.L.; Giannoni, E.; Comito, G.; Chiarugi, P. Microenvironment and tumor cell plasticity: An easy way out. Cancer Lett. 2013, 341, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Leu, Y.W.; Yan, P.S.; Fan, M.; Jin, V.X.; Liu, J.C.; Curran, E.M.; Welshons, W.V.; Wei, S.H.; Davuluri, R.V.; Plass, C.; et al. Loss of estrogen receptor signaling triggers epigenetic silencing of downstream targets in breast cancer. Cancer Res. 2004, 64, 8184–8192. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.J.; Zuo, T.; Lin, C.H.; Kuo, C.T.; Liyanarachchi, S.; Sun, S.; Shen, R.; Deatherage, D.E.; Potter, D.; Asamoto, L.; et al. Breast cancer-associated fibroblasts confer AKT1-mediated epigenetic silencing of Cystatin M in epithelial cells. Cancer Res. 2008, 68, 10257–10266. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum. Mol. Genet. 2007, 16, R50–R59. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, H.M.; Mohammad, H.P.; Baylin, S.B. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008, 4, e1000155. [Google Scholar] [CrossRef] [PubMed]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Teng, I.W.; Hou, P.C.; Lee, K.D.; Chu, P.Y.; Yeh, K.T.; Jin, V.X.; Tseng, M.J.; Tsai, S.J.; Chang, Y.S.; Wu, C.S.; et al. Targeted methylation of two tumor suppressor genes is sufficient to transform mesenchymal stem cells into cancer stem/initiating cells. Cancer Res. 2011, 71, 4653–4663. [Google Scholar] [CrossRef] [PubMed]
- Ikai, A.; Afrin, R.; Sekiguchi, H.; Okajima, T.; Alam, M.T.; Nishida, S. Nano-mechanical methods in biochemistry using atomic force microscopy. Curr. Protein Pept. Sci. 2003, 4, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Alonso, J.L.; Goldmann, W.H. Feeling the forces: Atomic force microscopy in cell biology. Life Sci. 2003, 72, 2553–2560. [Google Scholar] [CrossRef]
- Friedl, P.; Zallen, J.A. Dynamics of cell–cell and cell–matrix interactions in morphogenesis, regeneration and cancer. Curr. Opin. Cell Biol. 2010, 22, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, H.; Suda, T. Cancer stem cells and their niche. Cancer Sci. 2009, 100, 1166–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, S.J.; Spradling, A.C. Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Sneddon, J.B.; Werb, Z. Location, location, location: The cancer stem cell niche. Cell Stem Cell 2007, 1, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Scadden, D.T. The stem-cell niche as an entity of action. Nature 2006, 441, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Mezencev, R.; Kim, B.; Wang, L.; McDonald, J.; Sulchek, T. Cell stiffness is a biomarker of the metastatic potential of ovarian cancer cells. PLoS ONE 2012, 7, e46609. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Santiskulvong, C.; Bentolila, L.A.; Rao, J.; Dorigo, O.; Gimzewski, J.K. Correlative nanomechanical profiling with super-resolution F-actin imaging reveals novel insights into mechanisms of cisplatin resistance in ovarian cancer cells. Nanomedicine 2012, 8, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Keely, P.J. Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J. Mammary Gland Biol. Neoplasia 2011, 16, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Rolli, C.G.; Seufferlein, T.; Kemkemer, R.; Spatz, J.P. Impact of tumor cell cytoskeleton organization on invasiveness and migration: A microchannel-based approach. PLoS ONE 2010, 5, e8726. [Google Scholar] [CrossRef]
- Maller, O.; Dufort, C.C.; Weaver, V.M. YAP forces fibroblasts to feel the tension. Nat. Cell Biol. 2013, 15, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassler, S.; Donninger, H.; Birrer, M.J.; Clark, G.J. RASSF1A and the Taxol Response in Ovarian Cancer. Mol. Biol. Int. 2012, 2012, 263267. [Google Scholar] [CrossRef] [PubMed]
- Dallol, A.; Agathanggelou, A.; Fenton, S.L.; Ahmed-Choudhury, J.; Hesson, L.; Vos, M.D.; Clark, G.J.; Downward, J.; Maher, E.R.; Latif, F. RASSF1A interacts with microtubule-associated proteins and modulates microtubule dynamics. Cancer Res. 2004, 64, 4112–4116. [Google Scholar] [CrossRef] [PubMed]
- Gil, E.Y.; Jo, U.H.; Jeong, H.; Whang, Y.M.; Woo, O.H.; Cho, K.R.; Seo, J.H.; Kim, A.; Lee, E.S.; Koh, I.; et al. Promoter methylation of RASSF1A modulates the effect of the microtubule-targeting agent docetaxel in breast cancer. Int. J. Oncol. 2012, 41, 611–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, E.E.; Bachman, K.E.; Myohanen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosse-Gehling, P.; Fargeas, C.A.; Dittfeld, C.; Garbe, Y.; Alison, M.R.; Corbeil, D.; Kunz-Schughart, L.A. CD133 as a biomarker for putative cancer stem cells in solid tumours: Limitations, problems and challenges. J. Pathol. 2013, 229, 355–378. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Ohm, J.E. Epigenetic gene silencing in cancer—A mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 2006, 6, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Agathanggelou, A.; Cooper, W.N.; Latif, F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005, 65, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Fleuriel, C.; Touka, M.; Boulay, G.; Guerardel, C.; Rood, B.R.; Leprince, D. HIC1 (Hypermethylated in Cancer 1) epigenetic silencing in tumors. Int. J. Biochem. Cell Biol. 2009, 41, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leu, Y.W.; Huang, T.H.; Hsiao, S.H. Epigenetic reprogramming of mesenchymal stem cells. Adv. Exp. Med. Biol. 2013, 754, 195–211. [Google Scholar] [PubMed]
- Dehennaut, V.; Leprince, D. Implication of HIC1 (Hypermethylated in Cancer 1) in the DNA damage response. Bull. Cancer 2009, 96, E66–E72. [Google Scholar] [PubMed]
- Rood, B.R.; Leprince, D. Deciphering HIC1 control pathways to reveal new avenues in cancer therapeutics. Expert Opin. Ther. Targets 2013, 17, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Vos, M.D.; Clark, G.J. The RASSF1A tumor suppressor. J. Cell Sci. 2007, 120 Pt 1, 3163–3172. [Google Scholar] [CrossRef]
- Van der Weyden, L.; Adams, D.J. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim. Biophys. Acta 2007, 1776, 58–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuba, S.H.; Bennink, M.L.; Zlatanova, J. Single-molecule analysis of chromatin. Methods Enzym. 2004, 376, 73–105. [Google Scholar]
- Morkvenaite-Vilkonciene, I.; Ramanaviciene, A.; Ramanavicius, A. Atomic force microscopy as a tool for the investigation of living cells. Medicina 2013, 49, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Yallapu, M.M.; Katti, K.S.; Katti, D.R.; Mishra, S.R.; Khan, S.; Jaggi, M.; Chauhan, S.C. The roles of cellular nanomechanics in cancer. Med. Res. Rev. 2015, 35, 198–223. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.J.; Dryden, D.T.; Berge, T.; Edwardson, J.M.; Henderson, R.M. Direct observation of DNA translocation and cleavage by the EcoKI endonuclease using atomic force microscopy. Nat. Struct. Biol. 1999, 6, 15–17. [Google Scholar] [PubMed]
- Marszalek, P.E. DNA sequencing: Detecting methylation with force. Nat. Nanotechnol. 2010, 5, 765–766. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Howorka, S.; Proll, J.; Kienberger, F.; Preiner, J.; Hesse, J.; Ebner, A.; Pastushenko, V.P.; Gruber, H.J.; Hinterdorfer, P. Nanomechanical recognition measurements of individual DNA molecules reveal epigenetic methylation patterns. Nat. Nanotechnol. 2010, 5, 788–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plodinec, M.; Loparic, M.; Monnier, C.A.; Obermann, E.C.; Zanetti-Dallenbach, R.; Oertle, P.; Hyotyla, J.T.; Aebi, U.; Bentires-Alj, M.; Lim, R.Y.; et al. The nanomechanical signature of breast cancer. Nat. Nanotechnol. 2012, 7, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Hesson, L.B.; Cooper, W.N.; Latif, F. The role of RASSF1A methylation in cancer. Dis. Mark. 2007, 23, 73–87. [Google Scholar] [CrossRef]
- Leu, Y.W.; Rahmatpanah, F.; Shi, H.; Wei, S.H.; Liu, J.C.; Yan, P.S.; Huang, T.H. Double RNA interference of DNMT3b and DNMT1 enhances DNA demethylation and gene reactivation. Cancer Res. 2003, 63, 6110–6115. [Google Scholar] [PubMed]
- Yan, P.S.; Venkataramu, C.; Ibrahim, A.; Liu, J.C.; Shen, R.Z.; Diaz, N.M.; Centeno, B.; Weber, F.; Leu, Y.W.; Shapiro, C.L.; et al. Mapping geographic zones of cancer risk with epigenetic biomarkers in normal breast tissue. Clin. Cancer Res. 2006, 12, 6626–6636. [Google Scholar] [CrossRef] [PubMed]
- Young, T.J.; Monclus, M.A.; Burnett, T.L.; Broughton, W.R.; Ogin, S.L.; Smith, P.A. The use of the PeakForceTM quantitative nanomechanical mapping AFM-based method for high-resolution Young’s modulus measurement of polymers. Meas. Sci. Technol. 2011, 22, 125703. [Google Scholar] [CrossRef]
- Goodall, A.R.; Wilkinson, M.C.; Hearn, J. Formation of anomalous particles during the emulsifier-free polymerization of styrene. J. Colloid Interface Sci. 1975, 53, 327–331. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-C.; He, B.-C.; Chen, Y.-L.; Lee, K.-D.; Tung, C.-H.; Hsu, C.-C.; Lin, P.-Y.; Chu, P.-Y.; Leu, Y.-W.; Fu, W.-E.; et al. HIC1 and RassF1A Methylation Attenuates Tubulin Expression and Cell Stiffness in Cancer. Int. J. Mol. Sci. 2018, 19, 2884. https://doi.org/10.3390/ijms19102884
Chen C-C, He B-C, Chen Y-L, Lee K-D, Tung C-H, Hsu C-C, Lin P-Y, Chu P-Y, Leu Y-W, Fu W-E, et al. HIC1 and RassF1A Methylation Attenuates Tubulin Expression and Cell Stiffness in Cancer. International Journal of Molecular Sciences. 2018; 19(10):2884. https://doi.org/10.3390/ijms19102884
Chicago/Turabian StyleChen, Chih-Cheng, Bo-Ching He, Yao-Li Chen, Kuan-Der Lee, Chun-Hsin Tung, Chia-Chen Hsu, Ping-Yi Lin, Pei-Yi Chu, Yu-Wei Leu, Wei-En Fu, and et al. 2018. "HIC1 and RassF1A Methylation Attenuates Tubulin Expression and Cell Stiffness in Cancer" International Journal of Molecular Sciences 19, no. 10: 2884. https://doi.org/10.3390/ijms19102884