Abstract
Genome stability requires tight regulation of DNA replication to ensure that the entire genome of the cell is duplicated once and only once per cell cycle. In mammalian cells, origin activation is controlled in space and time by a cell-specific and robust program called replication timing. About 100,000 potential replication origins form on the chromatin in the gap 1 (G1) phase but only 20–30% of them are active during the DNA replication of a given cell in the synthesis (S) phase. When the progress of replication forks is slowed by exogenous or endogenous impediments, the cell must activate some of the inactive or “dormant” origins to complete replication on time. Thus, the many origins that may be activated are probably key to protect the genome against replication stress. This review aims to discuss the role of these dormant origins as safeguards of the human genome during replicative stress.
1. Introduction: Eukaryotic Origins and the Replication Program
Because of their large genomes, mammalian cells need thousands of replication forks, which initiate from replication origins, to ensure the complete duplication of their DNA within a specific time frame before they can divide. In human cells, the replication process takes about 10 h and involves the activation of roughly 30,000 replication origins. In normal replication conditions, replication origins are spread over about 100 kb of DNA, and only a single origin will be active within an individual DNA unit that we call a replicon. A coordinated group of adjacent replicons, “replicon cluster”, can be visualized as DNA replication foci [1]. Several studies, which compared replication timing (RT) and genome topology, suggested the term “replication domains” for replicons clustered inside large chromatin regions (~1 Mb), close to the size of one replication foci. They are located at discrete territories of the nucleus in the gap 1 (G1) phase and replicate at the same moment during the synthesis (S) phase [2,3,4]. At any given time of the S phase, about 10% of replicons are activated and replicate simultaneously [5]. In addition, the temporal activation of origins in a specific region of the genome correlates with a distinct pattern of replication foci as cells progress from early to late S phase. The sequential activation of potential origins within replication domains is thought to play a direct role in defining the S phase program or replication program. Temporal and spatial organization of DNA replication was adopted by metazoans cells to finely control the challenging goal of replicating the entire genome in a limited time and to overcome any obstacles that replication forks may encounter.
1.1. Origin Licensing and Firing
Complete and robust DNA duplication requires loading of minichromosome maintenance DNA helicase complex (MCM2–7) onto the replication origins. This step, called origin licensing, is restricted to the G1 phase of the cell cycle. A key initial step in origin licensing is the building of pre-recognition complex (Pre-RC) which starts with loading of the origin recognition complex (ORC) onto the chromatin. This ORC complex marks all potential origins providing spatial control of origin position. In higher eukaryotes, ORC binding sites were proven to be unrelated to DNA sequence, in contrast to other organisms such as yeast and bacteria [6,7]. It is currently assumed that multiple factors can characterize an origin, such as cytosine–phosphate–guanine (CpG) islands, G-quadruplexes, epigenetic marks, chromatin accessibility, sites of active transcription, or secondary DNA structures [8,9,10,11,12,13]. This is the reason why it is so difficult to identify metazoan replication origins. In the budding yeast Saccharomyces cerevisiae, a recent structural study [14] showed that two ORC molecules are required to ensure MCM2–7 complex loading onto the chromatin. During late mitosis and the G1 phase, ORCs bind cell division cycle 6 (Cdc6), which then interacts with chromatin licensing and DNA replication factor 1 (Cdt1) to allow loading of the six MCM subunits (MCM2–MCM7) and formation of the Pre-RC. The total amount of MCM complex does not change throughout the cell cycle, but the number of MCM complexes loaded onto DNA increases from telophase to the end of the G1–S phase transition. The final step of licensing requires the loading of Cdc45 and go-ichi-ni-san (GINS) onto the MCM complex to form the pre-initiation complex (Pre-IC). This complex requires the activities of the Dbf4-dependent kinase (DDK) and cyclin-dependent kinase (CDK) for its activation at the G1–S phase transition; then, the polymerases and other replication factors are recruited to allow origin firing (Figure 1).
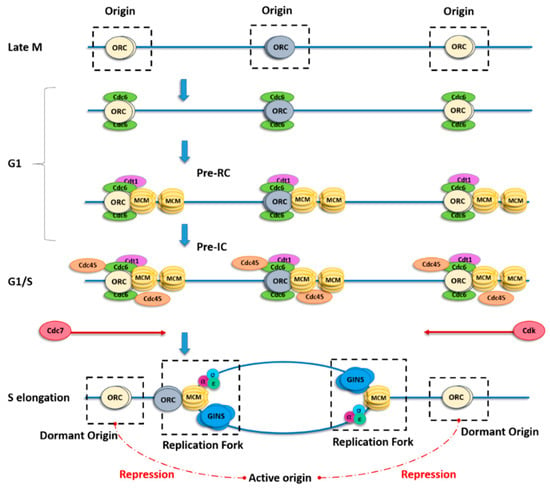
Figure 1.
Scheme describing origin licensing and firing. In late mitosis (M), the origin recognition complex (ORC) binds to origins, thus determining where replication forks might initiate, and for the subsequent recruitment of cell division cycle 6 (Cdc6) and chromatin licensing and DNA replication factor 1 (Cdt1) in the gap 1 (G1) phase. Binding of both Cdc6 and Cdt1 is necessary, in turn, for recruitment of the minichromosome maintenance DNA helicase complex (MCM) to form the pre-recognition complex (Pre-RC). Each ORC has two Cdt1-binding sites, which may explain the cooperative loading of two MCM complexes per origin. The MCM pair remains catalytically inactive until the G1–synthesis (S) phase transition, when it is phosphorylated by both cyclin-dependent kinase (CDK) and Cdc7. Once the principal origin is fired, adjacent origins from the same replicon (flexible or dormant) are repressed (red dotted lines) by a yet unclear mechanism.
During the first step of origin firing, the MCM pair slides along DNA by encircling the double helix. Recent papers proposed a switch of the MCM double hexamer from double-stranded DNA (dsDNA) to single-stranded DNA (ssDNA) mediated by N-tier ring movement, allowing the two helicases complexes to pass each other within the origin and permitting lagging-strand extrusion [15,16] (Figure 1). During the elongation step, excess MCMs that are not initiated are removed by the passage of the replication fork [17].
The cell must balance its need for sufficient origins to replicate the entire genome against the risk of re-replication of DNA in the S phase due to an excess of origins. Thus, the control of origin licensing is crucial. Repression of new origin licensing during the S phase is important to avoid re-replication, which can lead to aneuploidy, DNA double-strand breaks, gene amplification, and general genome instability [18,19,20]. DNA that is not replicated due to an insufficient number of origins or to replication fork stalling, by contrast, can also lead to genome instability and rearrangements if the DNA replication checkpoint is inactive or deficient [21,22,23].
1.2. Spatial and Temporal Organization of Replication Origins
Origin usage in eukaryotes is mainly dependent on two important factors: space and time. Replication origins fire at a defined time that remains the same among cell generations and is closely related to their spatial organization. Early replicating origins are mainly found in replication domains that are enriched in active epigenetic modifications and highly transcribed genes [24,25,26,27,28,29]. These chromosomal regions have a consequent amount of MCMs, providing potential origins that replicate early in the S phase [7,30]. Conversely, late replication occurs in origin-poor domains with low gene density, and enriched in heterochromatin hallmarks [29,31,32,33].
Replication clusters are organized in the three-dimensional (3D) nuclear space, where early-replicating domains locate mainly at the center of the nucleus while late-replicating domains are found predominantly at the nuclear periphery (Figure 2B). Chromatin conformation mapping methods such as Hi-C are very powerful for visualizing the spatial organization of early- and late-replicating domains [34,35]. Replication domains are created by topological reorganization of the chromatin in nuclear space. In metazoans, the association of particular replication domains with sub-nuclear compartments determines their replication timing. The set-up of this compartmentalization occurs at a specific time of the G1 phase and is called the timing decision point (TDP) [36,37] (Figure 2).
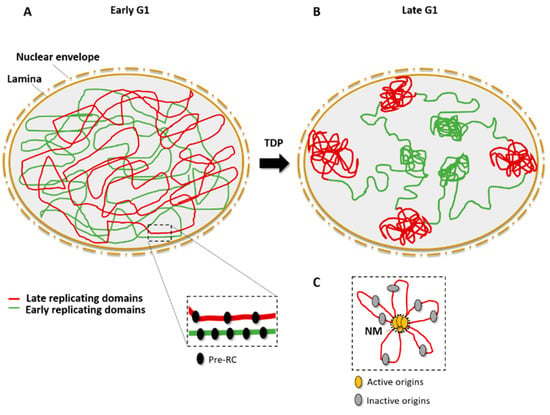
Figure 2.
Spatial organization of origins and replication timing. (A) In the early G1 phase, Pre-RCs (black) are assembled on the chromatin and mark potential origins; early-replicating domains (green) and late-replicating domains (red) are disordered in the nuclear space. (B) After the timing decision point (TDP), in the late G1 phase, early-replicating domains are close to center of the nucleus whereas late-replication domains are associated with the lamina, close to the nuclear periphery. (C) Active origins (yellow) cluster in replication domains that are associated to the nuclear matrix (NM), leaving inactive (dormant or flexible) origins in DNA loops (gray).
Accumulating evidence indicates that DNA attachment to the nuclear matrix is important for the initiation of DNA replication [38,39,40,41,42]. The nuclear matrix permits the separation of chromosome territories and allows the formation of replication clusters [39]. The organization of replicon clusters might, thus, reflect chromatin looping to bring the origins from different replicons into a single domain and to exclude the flexible and/or dormant origins from this replication factory (Figure 2C). The cohesin complex may be a key player in chromatin looping because it was found to interact physically with the MCM complex and to be enriched at origin sites [43].
1.3. Techniques to Detect and Identify Origins
The first quantitative method for determining origin density in the genomes of bacteria and mammalians was DNA fiber autoradiography [44,45]. This time-consuming technique is now replaced by other assays, such as DNA combing or spreading, which label newly replicated DNA with nucleosides analogs, including bromo-, chloro-, and iododeoxyuridine, and visualize the newly replicated DNA by immunofluorescence microscopy using antibodies specific for the analog [46].
The use of next-generation DNA sequencing led to the discovery of tens of thousands of potential replication origins in the human genome. Several independent approaches were used that exploit the direct identification of DNA replication initiation intermediates. The first approach is based on the purification and quantification of short nascent strands (SNS) of DNA [26]. In this method, 1.5–2.5-kb nascent strands specific to replication origins are purified thanks to their resistance to λ-exonuclease digestion due to the incorporation, by the primase, of small RNA primers at their 5′ ends [47]. The exonuclease digests the large excess of broken genomic DNA that would generate a background signal if not correctly removed. These genome-wide SNS analyses showed that active origins often co-localize with transcription start sites (TSS) and are located in GC-rich regions, close to CpG islands or G-quadruplexes, confirming previous microarray hybridization results [24,25,48]. A second approach [29] is based on the sequencing of an early intermediate called the DNA replication bubble, which forms when two replication forks diverge from a single origin. The technique consists of fragmenting the replicating DNA via a restriction endonuclease, and then trapping the circular replication bubbles in agarose gel [29]. This so-called “bubble-seq” method led to the mapping of more than 100,000 origins in the human genome. A third genome-wide approach relies on sequencing purified Okazaki fragments (“OK-seq”) to determine replication fork polarity, which allows the identification of initiation and termination sites [49]. With this approach, between 5000 and 10,000 broad initiation zones of up to 150 kb were detected. These sites are mainly non-transcribed but often surrounded by active genes, and they contain a single randomly located initiation event. Finally, a fourth method for identifying metazoan replication origins is called initiation-site sequencing (“ini-seq”) [50]. In this method, initiation events are synchronized biochemically in a cell-free system in which newly replicated DNA, synthesized a few minutes after initiation, is directly labeled and subsequently immuno-precipitated. This original approach has the important advantage of allowing functional genome-wide studies of origin activation. As these approaches become more and more accurate and complementary to each other, they provide an increasingly large, novel dataset on the characteristics of replication origins.
1.4. Origin Flexibility, Dormancy, and Efficiency
The replication initiation program of metazoan cells is remarkably flexible, with many origins firing at disparate frequencies depending on the cell lineage. MCM complexes and all the components of the Pre-RC are loaded in excess onto the chromatin in the G1 phase to provide this flexibility. In addition to differences between cell lineages, origin flexibility is also observed within a cell population [42,51].
Very few origins are activated almost all the time; they are called “constitutive” origins [52]. The majority of origins do not initiate replication in all cell cycles; these are called “flexible” origins. Origins that are activated only when replication from adjacent origins is compromised are called “dormant” origins. Unlike constitutive and flexible origins, dormant origins are not detectable in whole-genome analyses. Inter-origin distances measured by whole-genome sequencing are shorter than those measured by single-fiber analyses. This discrepancy may be explained by the flexibility of origin choice within replicons [53], which might also help coordinate DNA replication with transcription [54,55] and other nuclear processes, such as DNA repair, in order to facilitate recovery when replication is compromised. Given that there is no DNA consensus sequence for metazoan origins and that there exists such a flexibility in establishing which potential origins are activated, one might wonder how initiation ever occurs accurately and at consistent origins [56].
There are currently two theories to explain how origins are selected. One relies on the idea of an origin decision point (ODP)—which occurs in the G1 phase, after the timing decision point—that determines which origins are activated during replication [57]. The second theory postulates increasing origin efficiency based on the random use of replication origins [58], with the idea that the efficiency of origin firing increases throughout the S phase as the replicative DNA polymerases recycle to new origins. Moreover, replication origin efficiency also depends on their location in the nucleus, epigenetic marks, and mainly on the amount of loaded MCM complexes [7,59,60] or nucleosome occupancy [61]. Chromosome architecture also plays an important role in the regulation of DNA replication origin localization and activation [62], although chromosomal loops and loop anchors are still poorly defined biochemically. Further studies using single-cell technologies will be required in the future to better understand the mechanism of origin choice.
2. Dormant Origin Activation in Response to Replicative Stress
2.1. The Notion of DNA Replication Stress
During DNA replication, the appearance of endogenous or exogenous sources of stress leads to replication forks slowing or stalling. Exogenous sources of stress comprise mainly genotoxic chemicals, and ultraviolet and ionizing radiation. Endogenous sources of stress that are considered to be barriers to replication include repetitive sequences, G-quadruplexes, telomeres, DNA–RNA hybrids, errors in the incorporation of ribonucleotides, collisions between replication and transcription machineries, compaction of chromatin, deregulation of origin activity, and reduction of the deoxyribonucleotide triphosphate (dNTP) pool. Some regions of the genome, such as early-replicating fragile sites (ERFSs) and common fragile sites (CFSs), are more prone than others to replicative stress. Moreover, evidence is emerging that constitutive activation or overexpression of oncogenes, such as Harvey rat sarcoma (HRas) and myelocytomatosis (c-Myc), are a potential source of replication stress [63]. These oncogenes promote replication initiation or origin firing, leading to an elevated risk of nucleotide pool depletion and/or increased collisions with transcription complexes [64,65]. This may explain why supplementing cancer cells with exogenous nucleosides helps decrease chromosomal instability [66].
The first consequence of replication stress is fork collapse, creating DNA single-strand breaks and/or double-strand breaks. These lesions must be resolved before cell division by repair mechanisms such as homologous recombination (HR), non-homologous end-joining (NHEJ). or micro-homology mediated end-joining (MMEJ). In normal cells, the ataxia telangiectasia mutated (ATM) and ataxia telangiectasia Rad3-related (ATR) checkpoint signaling pathways prevent cell division when the genome is damaged. When some proteins of the checkpoint pathway, for example p53, are mutated, the cell can divide despite the presence of DNA lesions (including breaks and unreplicated DNA), which may lead to chromosome fragmentation, rearrangements, and genomic instability [67,68,69,70].
2.2. The Discovery of Dormant Origins and Their Link to Replicative Stress
In 1977, J. Herbert Taylor [71] first described the firing of new origins in response to replication fork stalling during DNA replication in Chinese hamster ovary (CHO) cells, a finding that later suggested the existence of dormant origins. Moreover, several studies in a range of eukaryotes, including S. cerevisiae, Xenopus laevis, and human cells, demonstrated that MCM complexes are loaded onto DNA in a large excess when compared to the number of DNA-bound ORCs and the number of active replication origins [72,73,74,75,76,77]. It was later shown in X. laevis [78] and in human cells that this excess of MCM provides a reservoir of dormant origins, which are activated when replication forks are arrested by agents such as aphidicolin (APH) or hydroxyurea (HU) [79,80]. These studies also showed that depletion of MCM by small interfering RNAs leads to hypersensitivity to replication inhibitors due to the lack of dormant origins [79,80]. Moreover, checkpoint kinase 1 (Chk1) activation is required for firing of dormant origins within active replication clusters, as well as for repression of other replicons that are not yet active [81], suggesting a link between the DNA damage response and dormant origin activation. Indeed, in vertebrates, inactivation or depletion of various proteins involved in genome maintenance, such as ATR [82,83], Chk1 [84,85,86,87], Wee1 [88,89], bloom syndrome protein (BLM) [90], Claspin [91,92], breast cancer type 2 susceptibility protein (BRCA2), and Rad51 [93], slows replication forks and also increases the number of initiation events, at least in studies where initiation events were examined. This finding indicates a link between fork speed and the number of active origins, as we examine further below.
2.3. The Density of Active Origins Depends on Replication Fork Speed
Under normal conditions, dormant origins do not fire and are passively replicated by the fork coming from adjacent activated origins. Thus, it makes sense to assume that replication fork speed can be a regulator of active origin density. In two complementary studies on CHO cells [62,94], it was demonstrated that replication fork speed has a direct impact on the number of active origins. When the fork is slowed down by HU treatment, the density of active origins increases. In contrast, in conditions that accelerate fork speed (addition of adenine and uridine to the culture medium), fewer origins are active. These studies further showed that the cell starts compensating for the decrease in fork speed within half an hour of treatment by activating dormant origins, which are then able to change their status within the S phase. Regulation of the number of initiation events occurs at the level of individual clusters, consistent with the functional organization of origins into replicon clusters [95]. Another study demonstrated that, in the absence of Cdc7 or ORC1, replication forks progress more rapidly than in control cells and fewer origins fire [96], again suggesting that the number of active origins and the fork rate are interdependent. Similarly, using chemical inhibitors of origin activity (a Cdc7 kinase inhibitor) and of DNA synthesis (APH), a more recent study found that the primary effects of replicative stress on fork rate can be distinguished from those on origin firing [97]. Collectively, these results support the conclusion that the density of origin firing depends on fork speed and, thus, is affected by endogenous or exogenous replicative stress.
2.4. CFS Fragility Due to the Lack of Dormant Origins
CFSs play a major role in cancer initiation because of their instability in conditions of replication stress. CFSs were first described as gaps and constrictions in the metaphase chromosomes of human lymphocytes grown under mild replication stress conditions (i.e., a low dose of APH) [98]. These observations were since seen in other organisms and are very likely to be the consequence of under-replication and/or DNA breaks caused by replication stress [99,100].
Although CFSs have been known for over two decades, the cause of their fragility is still controversial [55,101]. CFS fragility was first linked to non-B DNA sequences, such as AT-rich sequences, which are able to adopt secondary structures, constituting barriers to replication forks [102,103,104,105]. Deletion of these sequences from some cancer cell lines does not prevent breaks at these loci [106,107,108], suggesting that DNA sequence is not the sole reason for the instability of CFSs. Genome-wide analysis of replication and DNA combing experiments found a paucity of replication origins within the core of CFSs [109,110] and an incapacity to activate additional origins in response to replicative stress [111]. This suggests that, in order to replicate these regions, the fork must pass through long stretches of DNA containing multiple non-B DNA conformation sequences, and that their fragility correlates with the absence of additional replication origin firing when replication is slowed down. Most CFSs correspond to long genes (>300 kb), which might increase the risk of collision between the transcription and replication machineries [112]. Although one study showed that the transcription of large genes does not systematically dictate CFS fragility [113], other studies found that replication stress induces locus- and cell-type-specific genomic instability at active, large transcription units corresponding to CFSs [114,115]. Moreover, it is thought that fragility of these sites result from entry into mitosis before their complete replication [116,117]. Taken together, these observations suggest that replication defects at fragile sites may be due to a low density of licensed origins or may reflect inefficient or delayed activation of replication forks under replication stress.
3. Regulation of Dormant Origins: A Passive or Active Mechanism?
3.1. Activation of Dormant Origins by a “Passive” Mechanism
It is currently not clear what drives the firing of dormant origins when forks are slowed down or inhibited. One first hypothesis could be that it does not involve an active mechanism, but occurs as a consequence of the stochastic nature of origin firing [18,79]. Dormant origins have a precise lap of time to fire before being passively replicated then inactivated by forks from adjacent origins. When fork progression is impeded, the replication at dormant origins is delayed and, therefore, they have an increased probability to fire. By means of computational modeling, a study showed that the same levels of dormant origin activation seen in vivo can be reproduced by a passive mechanism [118]. In this model, the mechanism relies simply on the stochastic nature of origin firing, without any need for additional regulatory pathways.
This simple theory can be sufficient to explain the activation of dormant origins in response to replicative stress. Nonetheless, it cannot be ruled out that dormant origins may also be regulated by active mechanisms, involving DNA damage response and other replication-related pathways.
3.2. Regulation of Dormant Origins by “Active” Mechanisms
3.2.1. ATR/Chk1 Kinases as Modulators of Origin Activation
The inhibition of replication forks activates the DNA damage checkpoint kinases ATR–Chk1 and ATM–Chk2, which have many different functions, including stabilizing replication forks, delaying or blocking the progress of the cell cycle, and promoting DNA lesion repair [119,120,121]. It may seem surprising that, in response to replication stress, the cell can both activate dormant origins and suppress overall origin initiation; however, when replication forks stall, it makes sense that dormant origins should be activated in their vicinity and not elsewhere in the genome.
In the normal S phase, Chk1 affects replication fork speed by inhibiting excess origin firing [23,85,86]. In response to low levels of replication stress induced by APH or HU, ATR and Chk1 impede the activation of new replicon clusters while allowing dormant origins to fire within those already activated and affected by the drug [79,81], thereby avoiding the deleterious impact of replication fork stalling (Figure 3). The mechanism responsible for this phenomenon is not yet elucidated, but one possibility is that ATR and Chk1 mildly reduce CDK levels, resulting in activation of fewer replication clusters [122]. Alternatively, Chk1 might directly inhibit the initiation process through an interaction with Treslin, which is required to stabilize Cdc45, GINS, and the MCM complex together with topoisomerase 2-binding protein 1 (TOPBP1) [123,124,125,126,127]. Moreover, a recent study found that an ATR inhibitor not only induced unscheduled origin firing, but also revealed another mechanism of origin regulation through a Cdc7-dependent phosphorylation of GINS [128]. Finally, a very recent study found that the ATR-activation domain of TOPBP1 is required to suppress origin firing during the S phase [129], further supporting an important role for the ATR–Chk1 pathway in regulating the activation of origins.
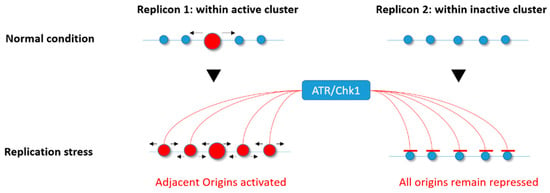
Figure 3.
Ataxia telangiectasia Rad3-related (ATR)/checkpoint kinase 1 (Chk1) involvement in the differential regulation of origin firing under replicative stress. In response to replication stress, the ATR/Chk1 kinases allow the activation of dormant origins within active replicon clusters (active origin(s) in red) while repressing any firing within those that are not yet activated (inactive origins in blue).
3.2.2. Mannose Receptor C-Type 1 (Mrc1)/Claspin Is a Central Regulator of Origin Firing under Normal and Stressed Replication
S. cerevisiae Mrc1 and its metazoan ortholog Claspin are not only involved in the S phase checkpoint signaling pathway, but are also important components of replication forks. They interact with many factors known to function in or to regulate DNA replication, including MCM4, MCM10, ATR, Chk1, Cdc7, Cdc45, DNA polymerases α, δ, and ε, and proliferating cell nuclear antigen (PCNA) [130,131,132,133]. The presence of Mrc1/Claspin is necessary for normal DNA replication [91,92,134,135], probably by making a connection between the helicase components and replicative polymerases at the replication fork. Also, Claspin plays another role in the initiation of DNA replication in human cells during the normal S phase by recruiting Cdc7 to facilitate phosphorylation of MCM proteins [136]. It was recently discovered in yeast that Mrc1 has two crucial functions in regulating the firing of origins: a checkpoint independent-role to activate early-firing origins during normal replication, and a checkpoint dependent-function to inhibit late/dormant origins in the presence of HU [137].
3.2.3. Fanconi Anemia Proteins in the Regulation of Dormant Origins
The role of the Fanconi anemia (FA) pathway in the DNA repair of interstrand cross-links (ICLs) was studied for many years. A clear model emerged describing that FA proteins orchestrate the interplay between multiple DNA repair pathways, including homologous recombination (HR) and translesion synthesis (TLS) [138,139,140]. However, treatment of cells with a low dose APH robustly activates the FA pathway, indicating a role of the FA proteins during DNA replication [141].
FA complementation group 1 (FANCI) was shown to be involved in dormant origin firing upon low replication stress through a FA pathway-independent mechanism [142]. FANCI associates with MCM3 and MCM5, localizes with replication origins, and acts as a regulator of DDK activity to allow the activation of the MCM2–7 helicase complex in response to mild replicative stress. In contrast, under high replicative stress, FANCI is phosphorylated by ATR. This phosphorylated form of FANCI negatively regulates dormant origin firing and activates replication fork restart/DNA repair that is FA-dependent. In this context, FA complementation group D2 (FANCD2), which is known as a close partner of FANCI, acts as a negative regulator of dormant origin firing [142].
Finally, FANCD2 was shown to facilitate replication of repeat-rich genomic regions such as CFSs by decreasing DNA–RNA hybrid accumulation, thus reducing the need for dormant origin firing [143].
3.2.4. Rap1-Interacting Factor 1 (RIF1) Orchestrates Origins and Replication Timing
RIF1 (Rap1-interacting factor 1) was first discovered in budding yeast as a telomeric chromatin-interacting protein required for the regulation of telomere length via its interaction with Rap1 [144,145]. It was then demonstrated in S. cerevisiae that RIF1 inhibits activation of the DNA damage checkpoint close to telomeres [146,147] and affects telomere replication timing [148]. Although the RIF1 protein is evolutionarily conserved, in metazoans, it was described not to play a specific role at telomeres, but rather to orchestrate the DNA double-strand break repair pathway and DNA recombination [149,150,151,152,153].
Further studies implicated RIF1 from the fission yeast Schizosaccharomyces pombe and mammalian RIF1 in regulating genome-wide DNA replication. S. pombe RIF1 binds selectively not only to telomeres, but also to specific regions of the genome where it may regulate the choice and timing of origin firing in late-replicating regions of chromosomes [154]. In RIF1-deficient cells, activation of dormant or late origins is concomitant with suppression of some active early-firing origins, indicating that RIF1 is a crucial player in the genome-wide origin activation program in S. pombe. In human cells, depletion of RIF1 results in increased early-S phase initiation events, loss of mid-S phase replication foci, and global changes in replication timing domain structures. Domains that normally replicate in the early S phase are delayed, whereas those that normally replicate in the late S phase are advanced [155]. Thus, replication timing is completely disturbed in the absence of RIF1. Another study observed that, in the absence of RIF1, the distance between origins is greater than in control cells during the normal S phase, and there are fewer dormant origins upon replication stress [156].
Also, RIF1 binds tightly to insoluble nuclear structures in late mitosis and the early G1 phase, and regulates chromatin-loop size [155]. Interestingly, RIF1 binding to consensus G-quadruplex-like sequences in fission yeast was identified [157]. These sequences tend to be near dormant origins, and the binding of RIF1 on these sites would allow their repression over a great distance. Overall, these findings indicate that RIF1, through its role in organizing higher-order chromatin architecture, is an essential regulator of replication timing.
Thus, the accumulating data suggest that, through its interaction with chromatin and nuclear structures, RIF1 plays an important role in the regulation of dormant origin availability not only in response to replicative stress, but also in normal conditions.
3.2.5. Chromatin Loop Size Correlates with Dormant Origin Activation
The fluorescent DNA halo technique was essential for establishing the link between chromatin loops and replicon size [158], and for describing the importance of replicon remodeling events in Xenopus embryonic development [159]. Basically, the technique relies on cell permeabilization and soluble protein extraction, allowing supercoiled DNA loops to unroll around an insoluble scaffold, the nuclear matrix. Those structures called DNA “halos” can be visualized by 4′,6-diamidino-2-phenylindole (DAPI) fluorescent staining. Active origins are in or near the nuclear matrix, whereas dormant/inactive origins are in the DNA loops [160] (Figure 2C).
Using the fluorescent DNA halo technique, one study [62] observed a strict correlation between dormant origin activation at a given S phase and reduced chromatin loop size in the next G1/S phase. Combining the DNA halo experiment with fluorescent in situ hybridization (FISH) using a probe targeting the highly amplified adenosine monophosphate deaminase 2 (AMPD2)-specific locus in CHO cells, they demonstrated that, in response to replication stress, activation of dormant origins relocates this locus toward the nuclear matrix.
Cohesin also influences the size of interphase chromatin loops since its absence results in longer chromatin loops due to a limited origin usage [43], showing that, independently of the effect of cohesin acetylation on replication fork progression [161], this structural protein is present at origins and impacts their activity. Finally, chromatin loop size increases in RIF1-depleted cells [155], suggesting that the RIF1 protein is required for proper chromatin loop formation, as already mentioned above.
4. Dormant Origin Deficiency, Genome Stability, and Pathologies
4.1. MCM Mutants and Dormant Origins in Mice
Homozygosity for a null allele of any of the six Mcm genes in mice (Mcm2–7) causes embryonic lethality [162,163,164], consistent with the evidence that these Mcm genes are essential for DNA replication. Only hypomorphic alleles such as Mcm4Chaos3 and Mcm2IRES-CreERT2 (IRES, internal ribosome entry site; ERT2, estrogen receptor 2) result in mice that are viable into adulthood. The Mcm2IRES-CreERT2 allele expresses a tamoxifen-inducible Cre recombinase (CreERT2) inserted into the 3′ untranslated region (UTR) of the endogenous Mcm2 locus, which reduces the expression of MCM2 by 65% when compared to wild-type cells [165]. The Mcm4Chaos3 allele produces an MCM4 protein with a Phe345Ile mutation, which does not affect the helicase activity of the MCM complex in vitro, but does reduce the efficiency of its assembly [164].
Surprisingly, mouse embryonic fibroblasts (MEFs) from Mcm4Chaos3 mice also have a reduced MCM7 protein level in addition to MCM4 [164]. Moreover, immortalized homozygous Mcm4Chaos3 cells display less stable association of MCM2–7 at replication forks compared to wild-type cells [166]. Finally, Mcm4Chaos3/Chaos3 MEFs exhibit about a half reduction in chromatin bound MCM2–7 that causes a lower ability to activate dormant origins in response to treatment with low doses of APH [162,167].
Mice with only one-third of the normal MCM2 level were shown to develop lymphomas at a very young age, and have diverse stem cell proliferation defects. Similarly to Mcm4Chaos3, these mice also have 27% less MCM7 protein than wild-type mice. Moreover, Mcm2IRES-CreERT2 cells exhibit decreased replication origin usage due to lower dormant origin availability even in the presence of HU, as demonstrated by DNA combing experiments [165,168].
Hence, these two mouse models are close phenotypically, showing dormant origin deficiency due to reduced levels of loaded MCM onto the chromatin. Even in an unchallenged S phase, the inability to activate dormant origins leads to accumulation of stalled replication forks that reach mitosis and interfere with chromosome segregation. Both phenotypes lead to improper chromosome stability and premature tumorigenesis, with several differences in the latency of disease development [165,166,168,169].
4.2. MCM Mutants and Dormant Origins in Stem/Progenitor Cells
The fact that Mcm2 expression has a global effect on cell proliferation within many tissues might explain why the majority of Mcm2IRES-CreERT2 mice develop tumors and display a range of additional hallmarks of age-related disorders. A study that set out to determine the effect of Mcm2 deficiency observed an approximately threefold reduction in the level of neurogenesis within the sub-ventricular zone in Mcm2IRES-CreERT2 mouse brains [165], fewer stem cells in intestinal crypts and in skeletal muscle, and a modest increase in DNA damage.
Consistent with the conclusion that Mcm mutants affect stem cells, neural stem-cell progenitors in Mcm4Chaos3/Chaos3 mouse embryos display a high level of Chk1 activation, increased phosphorylated H2A histone family X (γH2AX) and p53-binding protein 1 (53BP1) foci, an accumulation in the G2–M phase, and more apoptosis, resulting in a reduced ability to form neurospheres in vitro [170]. The renewal of stem cells in the brain appears to be normal, but their ability to differentiate into intermediate progenitors is highly reduced due to an increase of apoptotic cells in the sub-ventricular and intermediate zones [170].
These observations suggest that normal expression of MCM complex proteins is essential for stem/progenitor cell function by reducing the risk of replication-associated genome instability, an idea that was supported by two other studies. One demonstrated that human embryonic stem cells, which have a remarkably short G1 phase, load MCM onto chromatin very rapidly when compared to differentiated cells, in order to have a similar total amount of loaded MCM at the G1–S phase transition [171]. In the second study, hypomorphic expression of the origin licensing factor MCM3 in mouse reduced the number of licensed origins and affected the function of hematopoietic stem cells, as well as the differentiation of highly proliferative erythrocyte precursors, thus demonstrating that the rate of MCM loading is crucial for correct organism development [163]. These observations suggest that hematopoietic progenitors are exceptionally sensitive to replication stress, and that they must license an excess of origins to ensure their correct differentiation and function.
Intriguingly, aging hematopoietic stem cells suffer from replication stress even in wild-type mice. This might be due to the fact that old stem cells have reduced expression of MCM complex proteins, resulting in reduced numbers of dormant origins and, as a consequence, more chromosome instability and cell-cycle defects [172].
4.3. Consequences of Limited Licensing and Firing in Humans
A mutation in the Mcm4 gene, which results in a truncated form of this protein lacking the N-terminal serine/threonine-rich domain, was identified in a group of patients with a syndrome including growth delay, natural killer cell deficiency, adrenal insufficiency, and genome instability [173,174,175]. This truncated form of MCM4 does not affect MCM complex loading [174]. Nevertheless, immortalized fibroblasts from these patients have a high level of chromosome breakage and defects in cell-cycle progression, and they are sensitive to low doses of APH [174], suggesting that the N-terminal amino acids of MCM4 protein are involved in the maintenance of genome integrity during replication. Further studies will be necessary to elucidate the mechanism via which normal MCM4 ensures genome maintenance. One possibility is the role of MCM4 phosphorylation in the checkpoint response, where it was shown that the N-terminal domain of MCM4 has a crucial role. In unperturbed replication, this domain exerts an inhibitory effect on replication initiation, and this inhibitory effect is relieved upon its phosphorylation by DDK. However, in the context of replication stress, this N-terminal phosphorylation by DDK becomes a prerequisite for proper checkpoint activation [176].
Another disease that appears to involve defective replication origin licensing is Meier–Gorlin syndrome (MGS), an autosomal recessive primordial dwarfism syndrome characterized by pre- and post-natal impaired growth. Several studies identified marked locus heterogeneity in this syndrome including mutations in five genes encoding components of the Pre-RC: Orc1, Orc4, Orc6, Cdt1, and Cdc6 [177,178]. The molecular and cellular phenotypes include impaired licensing, altered S phase progression, and proliferation defects, which partially overlap with the phenotypes due to MCM mutations, except for chromosomal instability, and an increased predisposition to cancer. Nonetheless, MGS mutations in Orc1 and Orc6 can cause quite a significant reduction in MCM loading and replication origin licensing [177,179,180].
Mice and human phenotypes caused by mutations in the licensing system illustrate our limited understanding of what happens to cells when the DNA replication program is compromised. For example, the threshold value for the number of licensed origins needed to activate the licensing checkpoint is still not known, nor whether this value varies between cell types.
5. Conclusion and Prospects
Dormant origins are now recognized as an important safeguard against under-replication of the genome, thus ensuring genome maintenance. Activation of dormant origins plays a central role in the rescue of stalled forks in the context of replicative stress, contributing to the complete replication of the DNA. The interactions between dormant origins and other fork restart mechanisms (such as TLS) are mostly unknown, even though some links with DNA damage checkpoint or FA pathways are becoming evident. What determines whether the cell activates dormant origins or induces these other mechanisms in response to fork stalling still remains to be investigated.
How replicon clusters are activated at the molecular level remains unclear, although we know that origin activation is regulated by both Chk1 and CDKs. The RIF1 protein might be the most interesting factor in this process since it is present both at the replication fork and at replication origins, where it plays a role in the DNA damage response, as well as in replication timing.
The study that found a direct correlation between origin activation and chromatin loop size [62] also reported that origins located near the anchorage sites of chromatin loops are preferentially activated in the S phase of the following cell generation (Figure 4). This suggests that cells respond to changes in fork dynamics by adapting origin usage in the next cell cycle, in addition to their rapid response of origin activation. It appears that cells can adapt to grow under conditions of fork slowing by increasing the efficiency of some origins that are usually dormant in normal growth conditions.
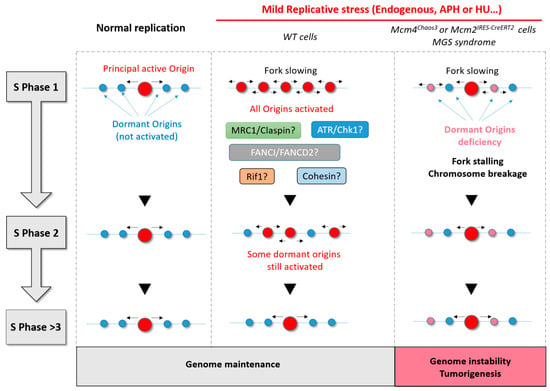
Figure 4.
Summary diagram showing the importance of dormant origin activation in response to replicative stress. During normal replication, only the principal origin is activated. If there is no replicative stress, this same principal origin is also activated in the next S phase. Under conditions of mild replicative stress, adjacent or dormant origins fire to compensate for fork slowing and to allow complete replication on time. Many proteins (ATR/Chk1, mannose receptor C-type 1 (Mrc1)/Claspin, Fanconi anemia complementation group 1 (FANCI)/ Fanconi anemia complementation group D2 (FANCD2), and Rap1-interacting factor 1 (RIF1)) are thought to be involved in the regulation of dormant origins under mild replicative stress. RIF1 and Cohesin are two good candidates to explain the persistence of some origin activation in the next S phase. Finally, when cells have few origins or a deficiency in dormant origins, replicative stress leads inevitably to fork stalling, DNA breaks, and genomic instability with a consequent risk of tumorigenesis.
Perhaps most exciting is the prospect that the regulation of dormant origins might be different in cancer cells to that in normal cells. MCM complex proteins are often misregulated at the early stage of cancer [18,181,182], and tumor cells are more sensitive to replicative stress when they have a reduced origin licensing capacity [183]. Mice hypomorphic for Mcm gene expression demonstrate the real importance of dormant origins, but any link with spontaneous cancer development remains to be determined to see whether this information can be useful to deal with anti-cancer molecules more accurately.
Author Contributions
L.C. wrote the manuscript, J.-S.H. proofread it, and V.B designed and proofread it.
Funding
Work in J.-S.H.’s laboratory is supported by funding from the Institut National du Cancer (PLBIO 2016), the Agence Nationale de la Recherche (ANR PRC 2016), Labex TOUCAN, and La Ligue contre le Cancer (Equipe labellisée 2017).
Acknowledgments
We kindly thank Dana Hodroj for proofreading and precious comments.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| 53BP1 | p53-Binding protein 1 |
| APH | Aphidicolin |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| BLM | Bloom syndrome RecQ like helicase |
| BRCA2 | Breast cancer 2 |
| Cdc45 | Cell division cycle protein 45 |
| Cdc6/7 | Cell division cycle 6/7 |
| CDK | Cyclin-dependent kinase |
| Cdt1 | Chromatin licensing and DNA replication factor 1 |
| CFS | Common fragile site |
| Chk1 | Checkpoint kinase 1 |
| Chk2 | Checkpoint kinase 2 |
| CHO | Chinese hamster ovary |
| c-Myc | Myelocytomatosis |
| DDK | Dbf4-dependent kinase |
| DNA | Deoxyribonucleic acid |
| dNTP | Deoxyribonucleotides |
| ERFS | Early-replicating fragile site |
| FANCI/D2 | Fanconi anemia complementation group I/D2 |
| FISH | Fluorescence in situ hybridization |
| GINS | Go-ichi-ni-san |
| HR | Homologous recombination |
| HRAS | Harvey rat sarcoma |
| HU | Hydroxyurea |
| ICL | Interstrand cross-link |
| MCM | Minichromosome maintenance |
| MGS | Meier–Glorin syndrome |
| MMEJ | Micro-homology mediated end-joining |
| Mrc1 | Mannose receptor C-type 1 |
| NHEJ | Non-homologous end-joining |
| ODP | Origin decision point |
| ORC | Origin recognition complex |
| PCNA | Proliferating cell nuclear antigen |
| Pre-IC | Pre-initiation complex |
| Pre-RC | Pre-recognition complex |
| RIF1 | Rap1-interacting factor 1 |
| RNA | Ribonucleic acid |
| RT | Replication timing |
| SNS | Short nascent strand |
| SVZ | Sub-ventricular zone |
| TDP | Timing decision point |
| TOPBP1 | Topoisomerase 2-binding protein 1 |
| TLS | Translesion synthesis |
| TSS | Transcription start sites |
References
- Berezney, R.; Dubey, D.D.; Huberman, J.A. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 2000, 108, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, S.; Ogata, M.; Okumura, K. Anatomy of Mammalian Replication Domains. Genes 2017, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Boulos, R.E.; Drillon, G.; Argoul, F.; Arneodo, A.; Audit, B. Structural organization of human replication timing domains. FEBS Lett. 2015, 589, 2944–2957. [Google Scholar] [CrossRef] [PubMed]
- Löb, D.; Lengert, N.; Chagin, V.O.; Reinhart, M.; Casas-Delucchi, C.S.; Cardoso, M.C.; Drossel, B. 3D replicon distributions arise from stochastic initiation and domino-like DNA replication progression. Nat. Commun. 2016, 7, 11207. [Google Scholar] [CrossRef] [PubMed]
- Hyrien, O. Peaks cloaked in the mist: The landscape of mammalian replication origins. J. Cell Biol. 2015, 208, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Hyrien, O. How MCM loading and spreading specify eukaryotic DNA replication initiation sites. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Karnani, N.; Taylor, C.M.; Malhotra, A.; Dutta, A. Genomic Study of Replication Initiation in Human Chromosomes Reveals the Influence of Transcription Regulation and Chromatin Structure on Origin Selection. Mol. Biol. Cell 2010, 21, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Méchali, M.; Yoshida, K.; Coulombe, P.; Pasero, P. Genetic and epigenetic determinants of DNA replication origins, position and activation. Curr. Opin. Genet. Dev. 2013, 23, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.M.; Ryan, M.; Kim, R.; Zakas, A.L.; Fu, H.; Lin, C.M.; Reinhold, W.C.; Davis, S.R.; Bilke, S.; Liu, H.; et al. Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res. 2011, 21, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Bartholdy, B.; Mukhopadhyay, R.; Lajugie, J.; Aladjem, M.I.; Bouhassira, E.E. Allele-specific analysis of DNA replication origins in mammalian cells. Nat. Commun. 2015, 6, 7051. [Google Scholar] [CrossRef] [PubMed]
- Besnard, E.; Babled, A.; Lapasset, L.; Milhavet, O.; Parrinello, H.; Dantec, C.; Marin, J.-M.; Lemaitre, J.-M. Unraveling cell type—Specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012, 19, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Cayrou, C.; Ballester, B.; Peiffer, I.; Fenouil, R.; Coulombe, P.; Andrau, J.-C.; van Helden, J.; Méchali, M. The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res. 2015, 25, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Coster, G.; Diffley, J.F.X. Bidirectional eukaryotic DNA replication is established by quasi-symmetrical helicase loading. Science 2017, 357, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, Y.; Yuan, Z.; Bai, L.; Schneider, S.; Zhao, G.; Stillman, B.; Speck, C.; Li, H. Cryo-EM structure of Mcm2-7 double hexamer on DNA suggests a lagging-strand DNA extrusion model. Proc. Natl. Acad. Sci. USA 2017, 114, E9529–E9538. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.E.; Ali, F.A.; Costa, A.; Diffley, J.F.X. The mechanism of eukaryotic CMG helicase activation. Nature 2018, 555, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Ge, X.Q. Replication forks, chromatin loops and dormant replication origins. Genome Biol. 2008, 9, 244. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, S.; Búa, S.; Rodríguez-Acebes, S.; Megías, D.; Ortega, S.; de Martino, A.; Méndez, J. In Vivo DNA Re-replication Elicits Lethal Tissue Dysplasias. Cell Rep. 2017, 19, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Zanini, I.M.Y.; Mijic, S.; Herrador, R.; Zellweger, R.; Chaudhuri, A.R.; Creavin, K.D.; Blow, J.J.; Lopes, M. Deregulated origin licensing leads to chromosomal breaks by rereplication of a gapped DNA template. Genes Dev. 2013, 27, 2537–2542. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Carrington, J.T.; Albergante, L.; Al Mamun, M.; Haagensen, E.J.; Komseli, E.-S.; Gorgoulis, V.G.; Newman, T.J.; Blow, J.J. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5757–E5764. [Google Scholar] [CrossRef] [PubMed]
- Syljuåsen, R.G.; Sørensen, C.S.; Hansen, L.T.; Fugger, K.; Lundin, C.; Johansson, F.; Helleday, T.; Sehested, M.; Lukas, J.; Bartek, J. Inhibition of Human Chk1 Causes Increased Initiation of DNA Replication, Phosphorylation of ATR Targets, and DNA Breakage. Mol. Cell. Biol. 2005, 25, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Sequeira-Mendes, J.; Díaz-Uriarte, R.; Apedaile, A.; Huntley, D.; Brockdorff, N.; Gómez, M. Transcription Initiation Activity Sets Replication Origin Efficiency in Mammalian Cells. PLOS Genet. 2009, 5, e1000446. [Google Scholar] [CrossRef] [PubMed]
- Cadoret, J.-C.; Meisch, F.; Hassan-Zadeh, V.; Luyten, I.; Guillet, C.; Duret, L.; Quesneville, H.; Prioleau, M.-N. Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 15837–15842. [Google Scholar] [CrossRef] [PubMed]
- Prioleau, M.-N.; MacAlpine, D.M. DNA replication origins—Where do we begin? Genes Dev. 2016, 30, 1683–1697. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Maehara, K.; Yoshida, K.; Ohkawa, Y.; Fujita, M. Genome-wide analysis of the spatiotemporal regulation of firing and dormant replication origins in human cells. Nucleic Acids Res. 2018, 46, 6683–6696. [Google Scholar] [CrossRef] [PubMed]
- Lucas, I.; Palakodeti, A.; Jiang, Y.; Young, D.J.; Jiang, N.; Fernald, A.A.; Beau, M.M.L. High-throughput mapping of origins of replication in human cells. EMBO Rep. 2007, 8, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Mesner, L.D.; Valsakumar, V.; Cieślik, M.; Pickin, R.; Hamlin, J.L.; Bekiranov, S. Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res. 2013, 23, 1774–1788. [Google Scholar] [CrossRef] [PubMed]
- Das, S.P.; Borrman, T.; Liu, V.W.T.; Yang, S.C.-H.; Bechhoefer, J.; Rhind, N. Replication timing is regulated by the number of MCMs loaded at origins. Genome Res. 2015, 25, 1886–1892. [Google Scholar] [CrossRef] [PubMed]
- Smith, O.K.; Kim, R.; Fu, H.; Martin, M.M.; Lin, C.M.; Utani, K.; Zhang, Y.; Marks, A.B.; Lalande, M.; Chamberlain, S.; et al. Distinct epigenetic features of differentiation-regulated replication origins. Epigenet. Chromatin 2016, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Oda, M.; Ramos, M.-P.; Pascual, M.; Lau, K.; Stasiek, E.; Agyiri, F.; Thompson, R.F.; Glass, J.L.; Jing, Q.; et al. Late-replicating heterochromatin is characterized by decreased cytosine methylation in the human genome. Genome Res 2011, 21, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Casas-Delucchi, C.S.; van Bemmel, J.G.; Haase, S.; Herce, H.D.; Nowak, D.; Meilinger, D.; Stear, J.H.; Leonhardt, H.; Cardoso, M.C. Histone hypoacetylation is required to maintain late replication timing of constitutive heterochromatin. Nucleic Acids Res. 2012, 40, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Gilbert, D.M. Replication timing and transcriptional control: Beyond cause and effect—Part III. Curr. Opin. Cell Biol. 2016, 40, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, D.S.; Berezney, R. The spatio-temporal organization of DNA replication sites is identical in primary, immortalized and transformed mammalian cells. J. Cell Sci. 2002, 115, 4037–4051. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, D.M. Cell fate transitions and the replication timing decision point. J. Cell Biol. 2010, 191, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Anachkova, B.; Djeliova, V.; Russev, G. Nuclear matrix support of DNA replication. J. Cell. Biochem. 2005, 96, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.H.C.; Coverley, D. Relationship between DNA replication and the nuclear matrix. Genes Cells 2013, 18, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Djeliova, V.; Russev, G.; Anachkova, B. Dynamics of association of origins of DNA replication with the nuclear matrix during the cell cycle. Nucleic Acids Res. 2001, 29, 3181–3187. [Google Scholar] [CrossRef] [PubMed]
- Radichev, I.; Parashkevova, A.; Anachkova, B. Initiation of DNA replication at a nuclear matrix-attached chromatin fraction. J. Cell. Physiol. 2005, 203, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Debatisse, M.; Toledo, F.; Anglana, M. Replication initiation in Mammalian Cells: Changing Preferences. Cell Cycle 2004, 3, 18–20. [Google Scholar] [CrossRef]
- Guillou, E.; Ibarra, A.; Coulon, V.; Casado-Vela, J.; Rico, D.; Casal, I.; Schwob, E.; Losada, A.; Méndez, J. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. 2010, 24, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Hand, R. Deoxyribonucleic acid fiber autoradiography as a technique for studying the replication of the mammalian chromosome. J. Histochem. Cytochem. 1975, 23, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J. The bacterial chromosome and its manner of replication as seen by autoradiography. J. Mol. Biol. 1963, 6, 208–213. [Google Scholar] [CrossRef]
- Jackson, D.A.; Pombo, A. Replicon Clusters Are Stable Units of Chromosome Structure: Evidence That Nuclear Organization Contributes to the Efficient Activation and Propagation of S Phase in Human Cells. J. Cell Biol. 1998, 140, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Bielinsky, A.K.; Gerbi, S.A. Discrete start sites for DNA synthesis in the yeast ARS1 origin. Science 1998, 279, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Cayrou, C.; Coulombe, P.; Puy, A.; Rialle, S.; Kaplan, N.; Segal, E.; Méchali, M. New insights into replication origin characteristics in metazoans. Cell Cycle 2012, 11, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Petryk, N.; Kahli, M.; d’Aubenton-Carafa, Y.; Jaszczyszyn, Y.; Shen, Y.; Silvain, M.; Thermes, C.; Chen, C.-L.; Hyrien, O. Replication landscape of the human genome. Nat. Commun. 2016, 7, 10208. [Google Scholar] [CrossRef] [PubMed]
- Langley, A.R.; Gräf, S.; Smith, J.C.; Krude, T. Genome-wide identification and characterisation of human DNA replication origins by initiation site sequencing (ini-seq). Nucleic Acids Res. 2016, 44, 10230–10247. [Google Scholar] [CrossRef] [PubMed]
- Cayrou, C.; Coulombe, P.; Vigneron, A.; Stanojcic, S.; Ganier, O.; Peiffer, I.; Rivals, E.; Puy, A.; Laurent-Chabalier, S.; Desprat, R.; et al. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 2011, 21, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Méchali, M. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010, 11, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Lebofsky, R.; Heilig, R.; Sonnleitner, M.; Weissenbach, J.; Bensimon, A.; Matera, A.G. DNA Replication Origin Interference Increases the Spacing between Initiation Events in Human Cells. Mol. Biol. Cell 2006, 17, 5337–5345. [Google Scholar] [CrossRef] [PubMed]
- Lõoke, M.; Reimand, J.; Sedman, T.; Sedman, J.; Järvinen, L.; Värv, S.; Peil, K.; Kristjuhan, K.; Vilo, J.; Kristjuhan, A. Relicensing of Transcriptionally Inactivated Replication Origins in Budding Yeast. J. Biol. Chem. 2010, 285, 40004–40011. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Aladjem, M.I.; Redon, C.E. Order from clutter: Selective interactions at mammalian replication origins. Nat. Rev. Genet. 2017, 18, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.R.; Gilbert, D.M. The replication origin decision point is a mitogen-independent, 2-aminopurine-sensitive, G1-phase event that precedes restriction point control. Mol. Cell. Biol. 1997, 17, 4312–4321. [Google Scholar] [CrossRef] [PubMed]
- Rhind, N. DNA replication timing: Random thoughts about origin firing. Nat. Cell Biol. 2006, 8, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Das, S.P.; Rhind, N. How and Why Multiple MCMs are Loaded at Origins of DNA Replication. Bioessays 2016, 38, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Singh, S.; Pradhan, S.; Narayan, G. MCM Paradox: Abundance of Eukaryotic Replicative Helicases and Genomic Integrity. Mol. Biol. Int. 2014, 2014, 574850. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Lee, L.; Lynch, B.; Tsukiyama, T. Nucleosome occupancy as a novel chromatin parameter for replication origin functions. Genome Res. 2017, 27, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Courbet, S.; Gay, S.; Arnoult, N.; Wronka, G.; Anglana, M.; Brison, O.; Debatisse, M. Replication fork movement sets chromatin loop size and origin choice in mammalian cells. Nature 2008, 455, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; García, P.; Helleday, T.; Petermann, E. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 153. [Google Scholar] [CrossRef] [PubMed]
- Samassekou, O.; Bastien, N.; Lichtensztejn, D.; Yan, J.; Mai, S.; Drouin, R. Different TP53 mutations are associated with specific chromosomal rearrangements, telomere length changes, and remodeling of the nuclear architecture of telomeres. Genes Chromosomes Cancer 2014, 53, 934–950. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, B.; Li, L.; Zhang, H.; Chen, Y.; Cui, X.; Hu, J.; Jiang, J.; Qi, Y.; Li, F. Correlations of telomere length, P53 mutation, and chromosomal translocation in soft tissue sarcomas. Int. J. Clin. Exp. Pathol. 2015, 8, 5666–5673. [Google Scholar] [PubMed]
- Hanel, W.; Moll, U.M. Links Between Mutant p53 and Genomic Instability. J. Cell. Biochem. 2012, 113, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.H. Increase in DNA replication sites in cells held at the beginning of S phase. Chromosoma 1977, 62, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, R.; Schulte, D.; Hu, B.; Musahl, C.; Göhring, F.; Knippers, R. Interactions of Human Nuclear Proteins P1Mcm3 and P1Cdc46. Eur. J. Biochem. 1995, 228, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Kawasaki, Y.; Tye, B.K. Physical interactions among Mcm proteins and effects of Mcm dosage on DNA replication in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 5081–5090. [Google Scholar] [CrossRef] [PubMed]
- Rowles, A.; Chong, J.P.J.; Brown, L.; Howell, M.; Evan, G.I.; Blow, J.J. Interaction between the Origin Recognition Complex and the Replication Licensing Systemin Xenopus. Cell 1996, 87, 287–296. [Google Scholar] [CrossRef]
- Donovan, S.; Harwood, J.; Drury, L.S.; Diffley, J.F.X. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc. Natl. Acad. Sci. USA 1997, 94, 5611–5616. [Google Scholar] [CrossRef] [PubMed]
- Mahbubani, H.M.; Chong, J.P.J.; Chevalier, S.; Thömmes, P.; Blow, J.J. Cell Cycle Regulation of the Replication Licensing System: Involvement of a Cdk-dependent Inhibitor. J. Cell Biol. 1997, 136, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.C.; Tutter, A.V.; Cvetic, C.; Gilbert, C.H.; Prokhorova, T.A.; Walter, J.C. MCM2–7 Complexes Bind Chromatin in a Distributed Pattern Surrounding the Origin Recognition Complex inXenopus Egg Extracts. J. Biol. Chem. 2002, 277, 33049–33057. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.M.; Göhler, T.; Luciani, M.G.; Oehlmann, M.; Ge, X.; Gartner, A.; Jackson, D.A.; Blow, J.J. Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006, 173, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Schwob, E.; Méndez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Blow, J.J. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J. Cell Biol. 2010, 191, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Koundrioukoff, S.; Carignon, S.; Técher, H.; Letessier, A.; Brison, O.; Debatisse, M. Stepwise Activation of the ATR Signaling Pathway upon Increasing Replication Stress Impacts Fragile Site Integrity. PLoS Genet. 2013, 9, e1003643. [Google Scholar] [CrossRef] [PubMed]
- Shechter, D.; Gautier, J. MCM proteins and checkpoint kinases get together at the fork. Proc. Natl. Acad. Sci. USA 2004, 101, 10845–10846. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Suzuki, A.; Sugimura, K.; Okumura, K.; Zineldeen, D.H.; Shimada, M.; Niida, H.; Mizuno, T.; Hanaoka, F.; Nakanishi, M. Cyclin A–Cdk1 regulates the origin firing program in mammalian cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3184–3189. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Petermann, E.; Gillespie, D.A.; Caldecott, K.W.; Jackson, D.A. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J. 2007, 26, 2719–2731. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Woodcock, M.; Helleday, T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 16090–16095. [Google Scholar] [CrossRef] [PubMed]
- Técher, H.; Koundrioukoff, S.; Carignon, S.; Wilhelm, T.; Millot, G.A.; Lopez, B.S.; Brison, O.; Debatisse, M. Signaling from Mus81-Eme2-Dependent DNA Damage Elicited by Chk1 Deficiency Modulates Replication Fork Speed and Origin Usage. Cell Rep. 2016, 14, 1114–1127. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-Dependent Kinase Suppression by WEE1 Kinase Protects the Genome through Control of Replication Initiation and Nucleotide Consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Kelly, R.; Martín, Y.; Koundrioukoff, S.; Tanenbaum, M.E.; Smits, V.A.J.; Medema, R.H.; Debatisse, M.; Freire, R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 2011, 194, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Chabosseau, P.; Buhagiar-Labarchède, G.; Onclercq-Delic, R.; Lambert, S.; Debatisse, M.; Brison, O.; Amor-Guéret, M. Pyrimidine pool imbalance induced by BLM helicase deficiency contributes to genetic instability in Bloom syndrome. Nat. Commun. 2011, 2, 368. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Helleday, T.; Caldecott, K.W.; Cohen-Fix, O. Claspin Promotes Normal Replication Fork Rates in Human Cells. Mol. Biol. Cell 2008, 19, 2373–2378. [Google Scholar] [CrossRef] [PubMed]
- Scorah, J.; McGowan, C.H. Claspin and Chk1 Regulate Replication Fork Stability by Different Mechanisms. Cell Cycle 2009, 8, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Ragu, S.; Magdalou, I.; Machon, C.; Dardillac, E.; Técher, H.; Guitton, J.; Debatisse, M.; Lopez, B.S. Slow Replication Fork Velocity of Homologous Recombination-Defective Cells Results from Endogenous Oxidative Stress. PLoS Genet. 2016, 12, e1006007. [Google Scholar] [CrossRef] [PubMed]
- Anglana, M.; Apiou, F.; Bensimon, A.; Debatisse, M. Dynamics of DNA Replication in Mammalian Somatic Cells: Nucleotide Pool Modulates Origin Choice and Interorigin Spacing. Cell 2003, 114, 385–394. [Google Scholar] [CrossRef]
- Conti, C.; Saccà, B.; Herrick, J.; Lalou, C.; Pommier, Y.; Bensimon, A.; Matera, A.G. Replication Fork Velocities at Adjacent Replication Origins Are Coordinately Modified during DNA Replication in Human Cells. Mol. Biol. Cell 2007, 18, 3059–3067. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Nellimoottil, T.; Peace, J.M.; Knott, S.R.V.; Villwock, S.K.; Yee, J.M.; Jancuska, J.M.; Rege, S.; Tecklenburg, M.; Sclafani, R.A.; et al. The level of origin firing inversely affects the rate of replication fork progression. J. Cell Biol. 2013, 201, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Acebes, S.; Mourón, S.; Méndez, J. Uncoupling fork speed and origin activity to identify the primary cause of replicative stress phenotypes. J. Biol. Chem. 2018, 293, 12855–12861. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Elder, F.F.; Robinson, T.J. Rodent common fragile sites: Are they conserved? Evidence from mouse and rat. Chromosoma 1989, 97, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Stout-Weider, K.; Hermann, K.; Schrock, E.; Heiden, T. Common fragile sites are conserved features of human and mouse chromosomes and relate to large active genes. Genome Res. 2006, 16, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Koundrioukoff, S.; Wilhelm, T.; Letessier, A.; Brison, O.; Debatisse, M. Updating the mechanisms of common fragile site instability: How to reconcile the different views? Cell. Mol. Life Sci. 2014, 71, 4489–4494. [Google Scholar] [CrossRef] [PubMed]
- Bergoglio, V.; Boyer, A.-S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.W.T.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Freudenreich, C.H. An AT-rich Sequence in Human Common Fragile Site FRA16D Causes Fork Stalling and Chromosome Breakage in S. cerevisiae. Mol. Cell 2007, 27, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Fungtammasan, A.; Walsh, E.; Chiaromonte, F.; Eckert, K.A.; Makova, K.D. A genome-wide analysis of common fragile sites: What features determine chromosomal instability in the human genome? Genome Res. 2012, 22, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.W.; Pierce, L.C.T.; Ng, M.C.Y.; Wang, Y.-H. Role of DNA secondary structures in fragile site breakage along human chromosome 10. Hum. Mol. Genet. 2013, 22, 1443–1456. [Google Scholar] [CrossRef] [PubMed]
- Corbin, S.; Neilly, M.E.; Espinosa, R.; Davis, E.M.; McKeithan, T.W.; Beau, M.M.L. Identification of Unstable Sequences within the Common Fragile Site at 3p14.2: Implications for the Mechanism of Deletions within Fragile Histidine Triad Gene/Common Fragile Site at 3p14.2 in Tumors. Cancer Res. 2002, 62, 3477–3484. [Google Scholar] [PubMed]
- Finnis, M.; Dayan, S.; Hobson, L.; Chenevix-Trench, G.; Friend, K.; Ried, K.; Venter, D.; Woollatt, E.; Baker, E.; Richards, R.I. Common chromosomal fragile site FRA16D mutation in cancer cells. Hum. Mol. Genet. 2005, 14, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, E.; Matricardi, L.; Tosoni, E.; Bensimon, A.; Russo, A. Replication dynamics at common fragile site FRA6E. Chromosoma 2010, 119, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.-M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Ozeri-Galai, E.; Lebofsky, R.; Rahat, A.; Bester, A.C.; Bensimon, A.; Kerem, B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell 2011, 43, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Millot, G.A.; Blin, M.E.; Brison, O.; Dutrillaux, B.; Debatisse, M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 2013, 4, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Debatisse, M.; Le Tallec, B.; Letessier, A.; Dutrillaux, B.; Brison, O. Common fragile sites: Mechanisms of instability revisited. Trends Genet. 2012, 28, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Rosselli, F. The FANC pathway and mitosis: A replication legacy. Cell Cycle 2009, 8, 2907–2912. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Ge, X.Q. A model for DNA replication showing how dormant origins safeguard against replication fork failure. EMBO Rep. 2009, 10, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Melander, F.; Stucki, M.; Falck, J.; Bekker-Jensen, S.; Goldberg, M.; Lerenthal, Y.; Jackson, S.P.; Bartek, J.; Lukas, J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004, 23, 2674–2683. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. The DNA damage response during DNA replication. Curr. Opin. Cell Biol. 2005, 17, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Carr, A.M. Checkpoint responses to replication fork barriers. Biochimie 2005, 87, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.M.; Gillespie, P.J.; Blow, J.J. Replication factory activation can be decoupled from the replication timing program by modulating Cdk levels. J. Cell Biol. 2010, 188, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Treslin Collaborates with TopBP1 in Triggering the Initiation of DNA Replication. Cell 2010, 140, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, Z.; Chen, J. MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J. Cell Biol. 2011, 193, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Sansam, C.L.; Cruz, N.M.; Danielian, P.S.; Amsterdam, A.; Lau, M.L.; Hopkins, N.; Lees, J.A. A vertebrate gene, ticrr, is an essential checkpoint and replication regulator. Genes Dev. 2010, 24, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Direct regulation of Treslin by cyclin-dependent kinase is essential for the onset of DNA replication. J. Cell Biol. 2011, 193, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Kumagai, A.; Schlacher, K.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Interaction of Chk1 with Treslin Negatively Regulates the Initiation of Chromosomal DNA Replication. Mol. Cell 2015, 57, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, T.; Hood, B.; Schamus, S.; O’Connor, M.J.; Conrads, T.P.; Bakkenist, C.J. ATR kinase inhibition induces unscheduled origin firing through a Cdc7-dependent association between GINS and And-1. Nat. Commun. 2017, 8, 1392. [Google Scholar] [CrossRef] [PubMed]
- Sokka, M.; Koalick, D.; Hemmerich, P.; Syväoja, J.; Pospiech, H.; Sokka, M.; Koalick, D.; Hemmerich, P.; Syväoja, J.E.; Pospiech, H. The ATR-Activation Domain of TopBP1 Is Required for the Suppression of Origin Firing during the S Phase. Int. J. Mol. Sci. 2018, 19, 2376. [Google Scholar] [CrossRef] [PubMed]
- Gold, D.A.; Dunphy, W.G. Drf1-dependent Kinase Interacts with Claspin through a Conserved Protein Motif. J. Biol. Chem. 2010, 285, 12638–12646. [Google Scholar] [CrossRef] [PubMed]
- Serçin, Ö.; Kemp, M.G. Characterization of functional domains in human Claspin. Cell Cycle 2011, 10, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Uno, S.; Masai, H. Efficient expression and purification of human replication fork-stabilizing factor, Claspin, from mammalian cells: DNA-binding activity and novel protein interactions. Genes Cells 2011, 16, 842–856. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; de Renty, C.; Li, Y.; Xiao, H.; Kemp, M.G.; Han, Z.; DePamphilis, M.L.; Zhu, W. And-1 coordinates with Claspin for efficient Chk1 activation in response to replication stress. EMBO J. 2015, 34, 2096–2110. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-Y.; Li, K.; Stewart, G.S.; Elledge, S.J. Human Claspin works with BRCA1 to both positively and negatively regulate cell proliferation. Proc. Natl. Acad. Sci. USA 2004, 101, 6484–6489. [Google Scholar] [CrossRef] [PubMed]
- Szyjka, S.J.; Viggiani, C.J.; Aparicio, O.M. Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol. Cell 2005, 19, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-C.; Suzuki, M.; Yamakawa, S.; Uno, S.; Ishii, A.; Yamazaki, S.; Fukatsu, R.; Fujisawa, R.; Sakimura, K.; Tsurimoto, T.; et al. Claspin recruits Cdc7 kinase for initiation of DNA replication in human cells. Nat. Commun. 2016, 7, 12135. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Kanoh, Y.; Shimmoto, M.; Hayano, M.; Ueda, K.; Fukatsu, R.; Kakusho, N.; Masai, H. Checkpoint-Independent Regulation of Origin Firing by Mrc1 through Interaction with Hsk1 Kinase. Mol. Cell. Biol. 2017, 37, e00355-16. [Google Scholar] [CrossRef] [PubMed]
- Knipscheer, P.; Räschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Schärer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand crosslink repair. Science 2009, 326, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anemia and the repair of Watson and Crick crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Räschle, M.; Knipscheer, P.; Enoiu, M.; Angelov, T.; Sun, J.; Griffith, J.D.; Ellenberger, T.E.; Schärer, O.D.; Walter, J.C. Mechanism of Replication-Coupled DNA Interstrand Crosslink Repair. Cell 2008, 134, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Howlett, N.G.; Taniguchi, T.; Durkin, S.G.; D’Andrea, A.D.; Glover, T.W. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet. 2005, 14, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Jones, M.J.K.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J.; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; García-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Hardy, C.F.; Sussel, L.; Shore, D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992, 6, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Bunker, R.D.; Mattarocci, S.; Ribeyre, C.; Faty, M.; Gut, H.; Scrima, A.; Rass, U.; Rubin, S.M.; Shore, D.; et al. Rif1 and Rif2 shape telomere function and architecture through multivalent Rap1 interactions. Cell 2013, 153, 1340–1353. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Rushton, M.D.; Maringele, L. A Novel Checkpoint and RPA Inhibitory Pathway Regulated by Rif1. PLoS Genet. 2011, 7, e1002417. [Google Scholar] [CrossRef] [PubMed]
- Ribeyre, C.; Shore, D. Anticheckpoint pathways at telomeres in yeast. Nat. Struct. Mol. Biol. 2012, 19, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.-Y.; Robertson, E.D.; Hiraga, S.; Alvino, G.M.; Collingwood, D.; McCune, H.J.; Sridhar, A.; Brewer, B.J.; Raghuraman, M.K.; Donaldson, A.D. The effect of Ku on telomere replication time is mediated by telomere length but is independent of histone tail acetylation. Mol. Biol. Cell 2011, 22, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Barral, P.; Vannier, J.-B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol. Cell 2013, 49, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Virgilio, M.D.; Callen, E.; Yamane, A.; Zhang, W.; Jankovic, M.; Gitlin, A.D.; Feldhahn, N.; Resch, W.; Oliveira, T.Y.; Chait, B.T.; et al. Rif1 Prevents Resection of DNA Breaks and Promotes Immunoglobulin Class Switching. Science 2013. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 Regulates DSB Repair Using Rif1 to Control 5′ End Resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Fong, K.-W.; Wang, J.; Wang, W.; Chen, J. RIF1 Counteracts BRCA1-mediated End Resection during DNA Repair. J. Biol. Chem. 2013, 288, 11135–11143. [Google Scholar] [CrossRef] [PubMed]
- Hayano, M.; Kanoh, Y.; Matsumoto, S.; Renard-Guillet, C.; Shirahige, K.; Masai, H. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev. 2012, 26, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Ishii, A.; Kanoh, Y.; Oda, M.; Nishito, Y.; Masai, H. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012, 31, 3667–3677. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, S.; Ly, T.; Garzón, J.; Hořejší, Z.; Ohkubo, Y.; Endo, A.; Obuse, C.; Boulton, S.J.; Lamond, A.I.; Donaldson, A.D. Human RIF1 and protein phosphatase 1 stimulate DNA replication origin licensing but suppress origin activation. EMBO Rep. 2017, 18, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Kanoh, Y.; Matsumoto, S.; Fukatsu, R.; Kakusho, N.; Kono, N.; Renard-Guillet, C.; Masuda, K.; Iida, K.; Nagasawa, K.; Shirahige, K.; et al. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol. 2015, 22, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Buongiorno-Nardelli, M.; Micheli, G.; Carri, M.T.; Marilley, M. A relationship between replicon size and supercoiled loop domains in the eukaryotic genome. Nature 1982, 298, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, J.-M.; Danis, E.; Pasero, P.; Vassetzky, Y.; Méchali, M. Mitotic remodeling of the replicon and chromosome structure. Cell 2005, 123, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Pardoll, D.M.; Coffey, D.S. Supercoiled loops and eucaryotic DNA replicaton. Cell 1980, 22, 79–85. [Google Scholar] [CrossRef]
- Terret, M.-E.; Sherwood, R.; Rahman, S.; Qin, J.; Jallepalli, P.V. Cohesin acetylation speeds the replication fork. Nature 2009, 462, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.-H.; Wallace, M.D.; Abratte, C.; Southard, T.; Schimenti, J.C. Incremental genetic perturbations to MCM2-7 expression and subcellular distribution reveal exquisite sensitivity of mice to DNA replication stress. PLoS Genet. 2010, 6, e1001110. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Díaz, M.; Flach, J.; Rodriguez-Acebes, S.; López-Contreras, A.J.; Martínez, D.; Cañamero, M.; Fernández-Capetillo, O.; Isern, J.; Passegué, E.; et al. Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat. Commun. 2015, 6, 8548. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Alcaraz, A.; Liachko, I.; Buske, T.R.; Andrews, C.A.; Munroe, R.J.; Hartford, S.A.; Tye, B.K.; Schimenti, J.C. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007, 39, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, S.C.; Bailey, K.J.; Freeland, A. Reduced Mcm2 Expression Results in Severe Stem/Progenitor Cell Deficiency and Cancer. Stem Cells 2007, 25, 3121–3132. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Buske, T.R.; Schimenti, J.C. Genetic Screen for Chromosome Instability in Mice: Mcm4 and Breast Cancer. Cell Cycle 2007, 6, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Luebben, S.W.; Yamaguchi, S.; Ilves, I.; Matise, I.; Buske, T.; Botchan, M.R.; Shima, N. Stalled Fork Rescue via Dormant Replication Origins in Unchallenged S Phase Promotes Proper Chromosome Segregation and Tumor Suppression. Mol. Cell 2011, 41, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Kunnev, D.; Rusiniak, M.E.; Kudla, A.; Freeland, A.; Cady, G.K.; Pruitt, S.C. DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene 2010, 29, 3630–3638. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Smolka, M.B.; Schimenti, J.C. Chronic DNA Replication Stress Reduces Replicative Lifespan of Cells by TRP53-Dependent, microRNA-Assisted MCM2-7 Downregulation. PLoS Genet. 2016, 12, e1005787. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Han, J.; Cheng, E.-C.; Yamaguchi, S.; Shima, N.; Thomas, J.-L.; Lin, H. Embryonic Stem Cells License a High Level of Dormant Origins to Protect the Genome against Replication Stress. Stem Cell Rep. 2015, 5, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.P.; Dumitru, R.; Coryell, P.; Baxley, R.M.; Chen, W.; Twaroski, K.; Webber, B.R.; Tolar, J.; Bielinsky, A.-K.; Purvis, J.E.; et al. Rapid DNA replication origin licensing protects stem cell pluripotency. eLife 2017, 6, e30473. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.P.; Nobbs, M.; McGettigan, P.; Lynch, S.; Ennis, S. Recessive mutations in MCM4/PRKDC cause a novel syndrome involving a primary immunodeficiency and a disorder of DNA repair. J. Med. Genet. 2012, 49, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.R.; Guasti, L.; Meimaridou, E.; Chuang, C.-H.; Schimenti, J.C.; King, P.J.; Costigan, C.; Clark, A.J.L.; Metherell, L.A. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J. Clin. Investig. 2012, 122, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Sheu, Y.-J.; Stillman, B. The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 2010, 463, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Bicknell, L.S.; Bongers, E.M.H.F.; Leitch, A.; Brown, S.; Schoots, J.; Harley, M.E.; Aftimos, S.; Al-Aama, J.Y.; Bober, M.; Brown, P.A.J.; et al. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat. Genet. 2011, 43, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Guernsey, D.L.; Matsuoka, M.; Jiang, H.; Evans, S.; Macgillivray, C.; Nightingale, M.; Perry, S.; Ferguson, M.; LeBlanc, M.; Paquette, J.; et al. Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nat. Genet. 2011, 43, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Bleichert, F.; Balasov, M.; Chesnokov, I.; Nogales, E.; Botchan, M.R.; Berger, J.M. A Meier-Gorlin syndrome mutation in a conserved C-terminal helix of Orc6 impedes origin recognition complex formation. eLife 2013, 2, e00882. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; Alagoz, M.; Alcantara, D.; Outwin, E.; Brunner, H.G.; Bongers, E.M.H.F.; O’Driscoll, M.; Jeggo, P.A. Deficiency in origin licensing proteins impairs cilia formation: Implications for the aetiology of Meier-Gorlin syndrome. PLoS Genet. 2013, 9, e1003360. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.A.; Tachibana, K.K.; Laskey, R.A.; Coleman, N. Control of DNA replication and its potential clinical exploitation. Nat. Rev. Cancer 2005, 5, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Gillespie, P.J. Replication Licensing and Cancer—A Fatal Entanglement? Nat. Rev. Cancer 2008, 8, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, K.M.; Jones, R.M.; Petermann, E.; Jeggo, P.A. Diminished origin licensing capacity specifically sensitises tumour cells to replication stress. Mol. Cancer Res. 2013, 11, 370–380. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).