FKBP8 Enhances Protein Stability of the CLC-1 Chloride Channel at the Plasma Membrane

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
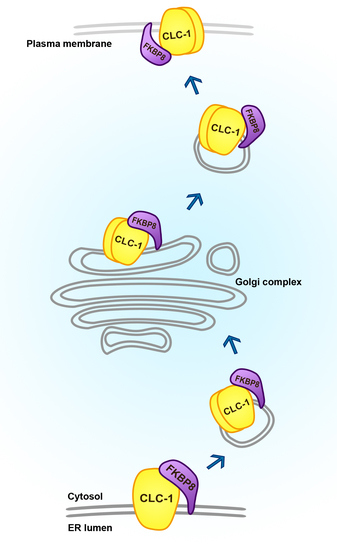
1. Introduction
2. Results
2.1. Surface Biotinylation of FKBP8
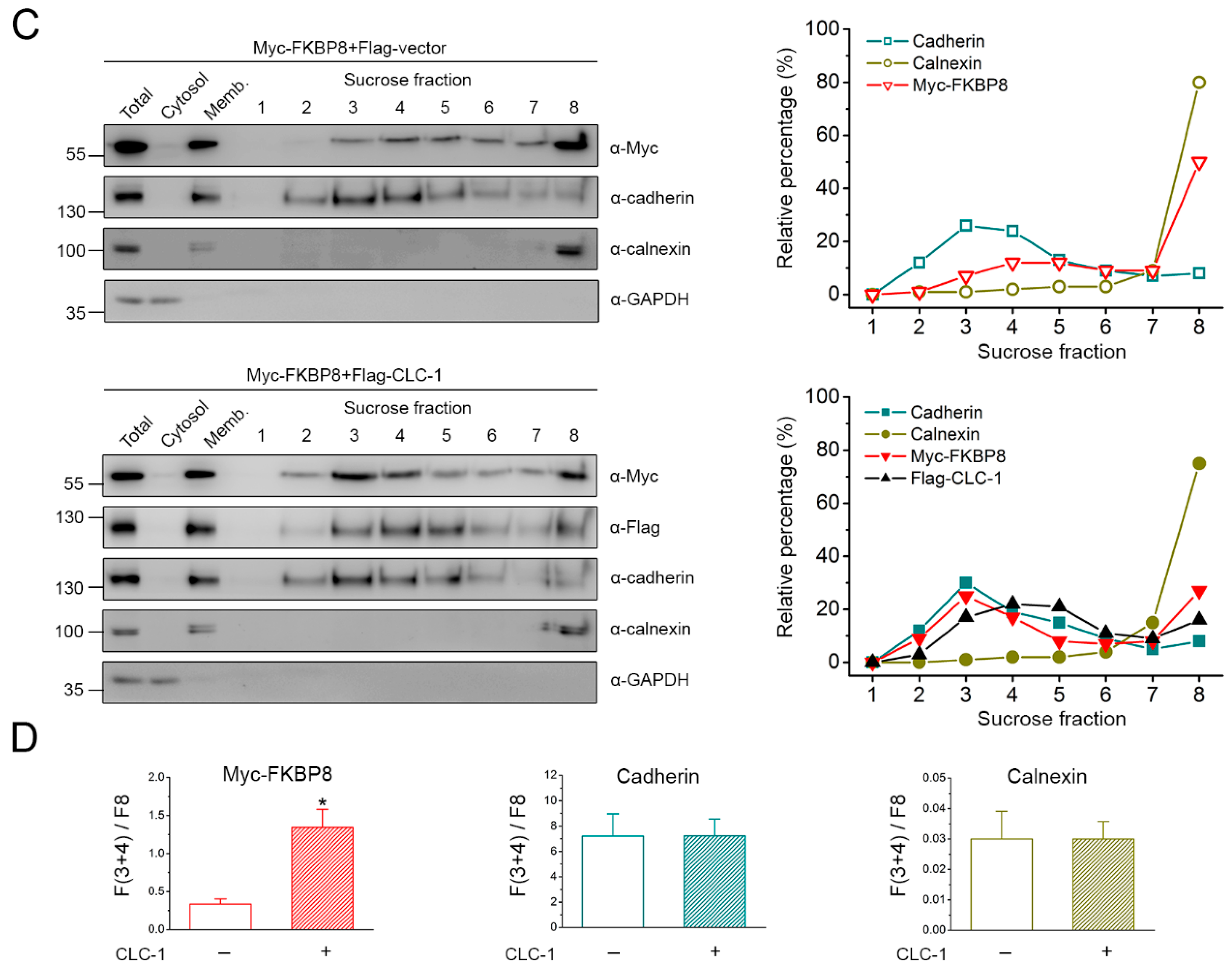
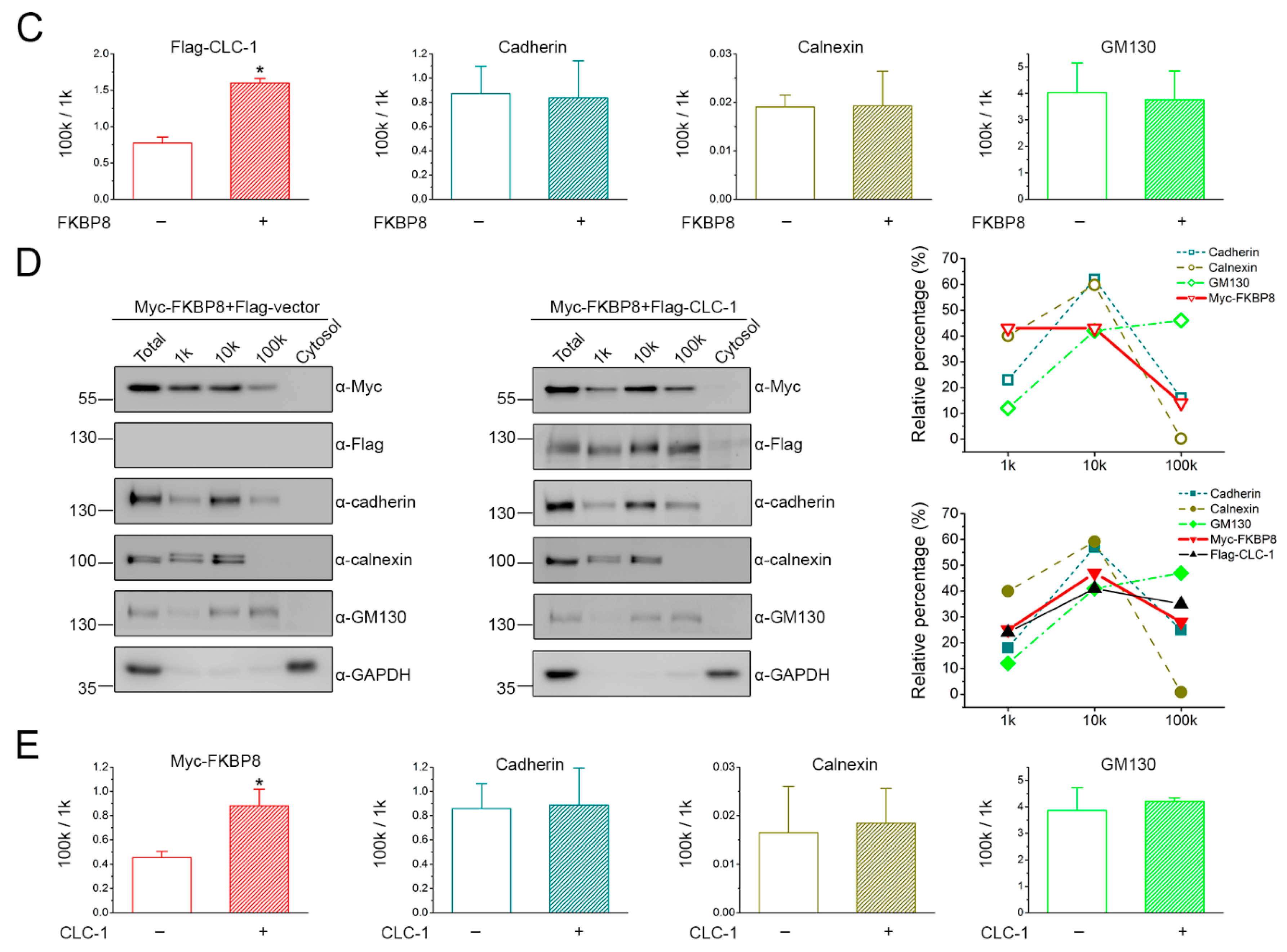
2.2. Localization of FKBP8 at the Plasma Membrane and the Golgi Complex by Subcellular Fractionation
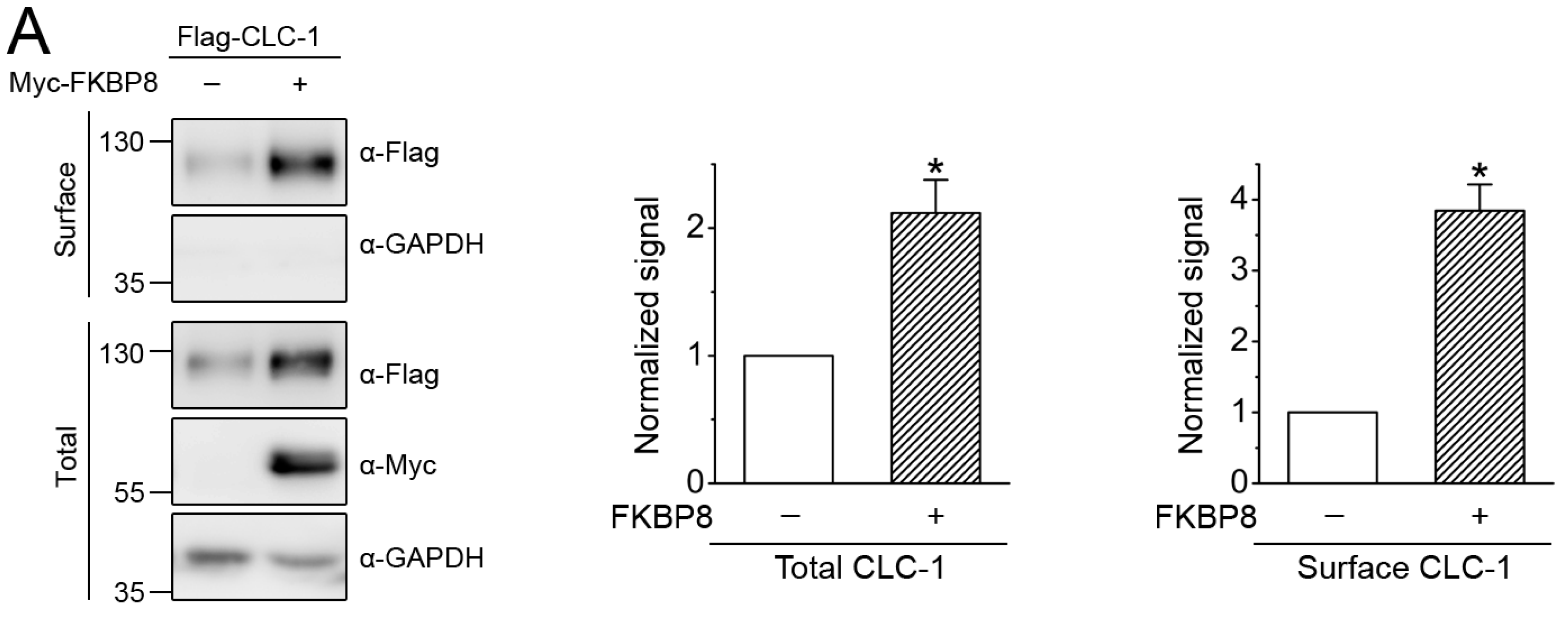
2.3. Regulation of Surface CLC-1 Stability by FKBP8
3. Discussion
4. Materials and Methods
4.1. cDNA Constructs
4.2. Cell Culture and DNA Transfection
4.3. Immunoblotting
4.4. Cell Surface Biotinylation
4.5. Subcellular Fractionation Analyses
4.6. Isolation of Plasma Membrane Proteins from Rat Skeletal Muscle
4.7. Statistical Analyses
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aha1 | activator of Hsp90 ATPase homolog 1 |
| BFA | brefeldin A |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| Cl- | chloride |
| DMEM | Dulbecco’s modified Eagle’s medium |
| D-PBS | Dulbecco’s phosphate buffered saline |
| DTT | dithiothreitol |
| ER | endoplasmic reticulum |
| FKBP8 | FK506-binding protein 8 |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GST | glutathione S-transferase |
| HEK | human embryonic kidney |
| HOP | Hsp70/Hsp90 organizing protein |
| Hsc70 | heat shock cognate protein 70 |
| Hsc70 | heat shock cognate protein 70 |
| Hsp90β | heat shock protein 90β |
| PBS | phosphate buffered saline |
| PMSF | phenylmethylsulfonyl fluoride |
| SR | sarcoplasmic reticulum |
| TBS | Tris buffered saline |
| TGN38 | trans-Golgi network 38 |
References
- Bretag, A.H. Muscle chloride channels. Physiol. Rev. 1987, 67, 618–724. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.H.; Morales-Aguilera, A. Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. J. Physiol. 1971, 219, 367–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulhunty, A.F. Distribution of potassium and chloride permeability over the surface and T-tubule membranes of mammalian skeletal muscle. J. Membr. Biol. 1979, 45, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Lossin, C.; George, A.L., Jr. Myotonia congenita. Adv. Genet. 2008, 63, 25–55. [Google Scholar] [PubMed]
- Colding-Jorgensen, E. Phenotypic variability in myotonia congenita. Muscle Nerve 2005, 32, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Pusch, M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum. Mutat. 2002, 19, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Jurkat-Rott, K.; Lerche, H.; Lehmann-Horn, F. Skeletal muscle channelopathies. J. Neurol. 2002, 249, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Kubisch, C.; Schmidt-Rose, T.; Fontaine, B.; Bretag, A.H.; Jentsch, T.J. ClC-1 chloride channel mutations in myotonia congenita: Variable penetrance of mutations shifting the voltage dependence. Hum. Mol. Genet. 1998, 7, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Saviane, C.; Conti, F.; Pusch, M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 1999, 113, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Duffield, M.; Rychkov, G.; Bretag, A.; Roberts, M. Involvement of helices at the dimer interface in ClC-1 common gating. J. Gen. Physiol. 2003, 121, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberger, S.; Wojciechowski, D.; Sternberg, D.; Lehmann-Horn, F.; Jurkat-Rott, K.; Becher, T.; Begemann, B.; Fahlke, C.; Fischer, M. Disease-causing mutations C277R and C277Y modify gating of human ClC-1 chloride channels in myotonia congenita. J. Physiol. 2012, 590, 3449–3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papponen, H.; Nissinen, M.; Kaisto, T.; Myllyla, V.V.; Myllyla, R.; Metsikko, K. F413C and A531V but not R894X myotonia congenita mutations cause defective endoplasmic reticulum export of the muscle-specific chloride channel CLC-1. Muscle Nerve 2008, 37, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.T.; Zhang, X.D.; Chuang, C.C.; Chen, J.J.; Chen, Y.A.; Chen, S.C.; Chen, T.Y.; Tang, C.Y. Myotonia congenita mutation enhances the degradation of human CLC-1 chloride channels. PLoS ONE 2013, 8, e55930. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.J.; Teijido, O.; Zifarelli, G.; Martin, P.; Ramirez-Espain, X.; Zorzano, A.; Palacin, M.; Pusch, M.; Estevez, R. Myotonia-related mutations in the distal C-terminus of ClC-1 and ClC-0 chloride channels affect the structure of a poly-proline helix. Biochem. J. 2007, 403, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Ronstedt, K.; Sternberg, D.; Detro-Dassen, S.; Gramkow, T.; Begemann, B.; Becher, T.; Kilian, P.; Grieschat, M.; Machtens, J.P.; Schmalzing, G.; et al. Impaired surface membrane insertion of homo- and heterodimeric human muscle chloride channels carrying amino-terminal myotonia-causing mutations. Sci. Rep. 2015, 5, 15382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papponen, H.; Toppinen, T.; Baumann, P.; Myllyla, V.; Leisti, J.; Kuivaniemi, H.; Tromp, G.; Myllyla, R. Founder mutations and the high prevalence of myotonia congenita in northern Finland. Neurology 1999, 53, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Tranebjaerg, L.; Torbergsen, T.; Holmgren, G.; Van Ghelue, M. Spectrum of CLCN1 mutations in patients with myotonia congenita in Northern Scandinavia. Eur. J. Hum. Genet. 2001, 9, 903–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.A.; Peng, Y.J.; Hu, M.C.; Huang, J.J.; Chien, Y.C.; Wu, J.T.; Chen, T.Y.; Tang, C.Y. The Cullin 4A/B-DDB1-Cereblon E3 Ubiquitin Ligase Complex Mediates the Degradation of CLC-1 Chloride Channels. Sci. Rep. 2015, 5, 10667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.J.; Huang, J.J.; Wu, H.H.; Hsieh, H.Y.; Wu, C.Y.; Chen, S.C.; Chen, T.Y.; Tang, C.Y. Regulation of CLC-1 chloride channel biosynthesis by FKBP8 and Hsp90beta. Sci. Rep. 2016, 6, 32444. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Banasavadi-Siddegowda, Y.K.; Mai, J.; Fan, Y.; Bhattacharya, S.; Giovannucci, D.R.; Sanchez, E.R.; Fischer, G.; Wang, X. FKBP38 peptidylprolyl isomerase promotes the folding of cystic fibrosis transmembrane conductance regulator in the endoplasmic reticulum. J. Biol. Chem. 2011, 286, 43071–43080. [Google Scholar] [CrossRef] [PubMed]
- Hutt, D.M.; Roth, D.M.; Chalfant, M.A.; Youker, R.T.; Matteson, J.; Brodsky, J.L.; Balch, W.E. FK506 binding protein 8 peptidylprolyl isomerase activity manages a late stage of cystic fibrosis transmembrane conductance regulator (CFTR) folding and stability. J. Biol. Chem. 2012, 287, 21914–21925. [Google Scholar] [CrossRef] [PubMed]
- Koulov, A.V.; LaPointe, P.; Lu, B.; Razvi, A.; Coppinger, J.; Dong, M.Q.; Matteson, J.; Laister, R.; Arrowsmith, C.; Yates, J.R., 3rd; et al. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 2010, 21, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Walker, V.E.; Atanasiu, R.; Lam, H.; Shrier, A. Co-chaperone FKBP38 promotes HERG trafficking. J. Biol. Chem. 2007, 282, 23509–23516. [Google Scholar] [CrossRef] [PubMed]
- Walker, V.E.; Wong, M.J.; Atanasiu, R.; Hantouche, C.; Young, J.C.; Shrier, A. Hsp40 chaperones promote degradation of the HERG potassium channel. J. Biol. Chem. 2009, 285, 3319–3329. [Google Scholar] [CrossRef] [PubMed]
- Shirane, M.; Nakayama, K.I. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 2003, 5, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.B.; Hong, Y.; Dhe-Paganon, S.; Yoon, H.S. FKBP family proteins: Immunophilins with versatile biological functions. Neurosignals 2008, 16, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Miyakoshi, L.M.; Marques-Coelho, D.; De Souza, L.E.R.; Lima, F.R.S.; Martins, V.R.; Zanata, S.M.; Hedin-Pereira, C. Evidence of a Cell Surface Role for Hsp90 Complex Proteins Mediating Neuroblast Migration in the Subventricular Zone. Front. Cell. Neurosci. 2017, 11, 138. [Google Scholar] [CrossRef] [PubMed]
- Stangl, S.; Gehrmann, M.; Riegger, J.; Kuhs, K.; Riederer, I.; Sievert, W.; Hube, K.; Mocikat, R.; Dressel, R.; Kremmer, E.; et al. Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc. Natl. Acad. Sci. USA 2011, 108, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Sidera, K.; Samiotaki, M.; Yfanti, E.; Panayotou, G.; Patsavoudi, E. Involvement of cell surface HSP90 in cell migration reveals a novel role in the developing nervous system. J. Biol. Chem. 2004, 279, 45379–45388. [Google Scholar] [CrossRef] [PubMed]
- Broquet, A.H.; Thomas, G.; Masliah, J.; Trugnan, G.; Bachelet, M. Expression of the molecular chaperone Hsp70 in detergent-resistant microdomains correlates with its membrane delivery and release. J. Biol. Chem. 2003, 278, 21601–21606. [Google Scholar] [CrossRef] [PubMed]
- Lynes, E.M.; Bui, M.; Yap, M.C.; Benson, M.D.; Schneider, B.; Ellgaard, L.; Berthiaume, L.G.; Simmen, T. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 2012, 31, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Myhill, N.; Lynes, E.M.; Nanji, J.A.; Blagoveshchenskaya, A.D.; Fei, H.; Carmine Simmen, K.; Cooper, T.J.; Thomas, G.; Simmen, T. The subcellular distribution of calnexin is mediated by PACS-2. Mol. Biol. Cell 2008, 19, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Gilady, S.Y.; Bui, M.; Lynes, E.M.; Benson, M.D.; Watts, R.; Vance, J.E.; Simmen, T. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones 2010, 15, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Saita, S.; Shirane, M.; Nakayama, K.I. Selective escape of proteins from the mitochondria during mitophagy. Nat. Commun. 2013, 4, 1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.Q.; Nakaya, Y.; Du, Z.; Yamane, T.; Shirane, M.; Kudo, T.; Takeda, M.; Takebayashi, K.; Noda, Y.; Nakayama, K.I.; et al. Interaction of presenilins with FKBP38 promotes apoptosis by reducing mitochondrial Bcl-2. Hum. Mol. Genet. 2005, 14, 1889–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edlich, F.; Weiwad, M.; Erdmann, F.; Fanghanel, J.; Jarczowski, F.; Rahfeld, J.U.; Fischer, G. Bcl-2 regulator FKBP38 is activated by Ca2+/calmodulin. EMBO J. 2005, 24, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Melville, D.; Zhang, M.; Schekman, R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife 2013, 2, e00947. [Google Scholar] [CrossRef] [PubMed]
- Prior, K.K.; Wittig, I.; Leisegang, M.S.; Groenendyk, J.; Weissmann, N.; Michalak, M.; Jansen-Durr, P.; Shah, A.M.; Brandes, R.P. The Endoplasmic Reticulum Chaperone Calnexin Is a NADPH Oxidase NOX4 Interacting Protein. J. Biol. Chem. 2016, 291, 7045–7059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, K.; Wraight, C.; Stanley, K.K. TGN38 is maintained in the trans-Golgi network by a tyrosine-containing motif in the cytoplasmic domain. EMBO J. 1993, 12, 2219–2228. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Rabouille, C.; Watson, R.; Nilsson, T.; Hui, N.; Slusarewicz, P.; Kreis, T.E.; Warren, G. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 1995, 131, 1715–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzini-Armstrong, C. The relationship between form and function throughout the history of excitation-contraction coupling. J. Gen. Physiol. 2018, 150, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Volpe, P.; Villa, A.; Podini, P.; Martini, A.; Nori, A.; Panzeri, M.C.; Meldolesi, J. The endoplasmic reticulum-sarcoplasmic reticulum connection: Distribution of endoplasmic reticulum markers in the sarcoplasmic reticulum of skeletal muscle fibers. Proc. Natl. Acad. Sci. USA 1992, 89, 6142–6146. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Characterisation of the dystrophin-related protein utrophin in highly purified skeletal muscle sarcolemma vesicles. Biochim. Biophys. Acta 1996, 1283, 215–222. [Google Scholar] [CrossRef]
- Schindler, J.; Nothwang, H.G. Aqueous polymer two-phase systems: Effective tools for plasma membrane proteomics. Proteomics 2006, 6, 5409–5417. [Google Scholar] [CrossRef] [PubMed]
- Morre, D.M.; Morre, D.J. Aqueous two-phase partition applied to the isolation of plasma membranes and Golgi apparatus from cultured mammalian cells. J. Chromatogr. B Biomed. Sci. Appl. 2000, 743, 377–387. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Yuan, L.C.; Bonifacino, J.S.; Klausner, R.D. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: Evidence for membrane cycling from Golgi to ER. Cell 1989, 56, 801–813. [Google Scholar] [CrossRef]
- Hsu, P.H.; Ma, Y.T.; Fang, Y.C.; Huang, J.J.; Gan, Y.L.; Chang, P.T.; Jow, G.M.; Tang, C.Y.; Jeng, C.J. Cullin 7 mediates proteasomal and lysosomal degradations of rat Eag1 potassium channels. Sci. Rep. 2017, 7, 40825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.; Martin, M.; Wiederrecht, G. Isolation of a cDNA encoding a novel human FK506-binding protein homolog containing leucine zipper and tetratricopeptide repeat motifs. Gene 1995, 160, 297–302. [Google Scholar] [CrossRef]
- Babst, M. Quality control: Quality control at the plasma membrane: One mechanism does not fit all. J. Cell Biol 2014, 205, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacGurn, J.A. Garbage on, garbage off: New insights into plasma membrane protein quality control. Curr. Opin. Cell Biol. 2014, 29, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Apaja, P.M.; Lukacs, G.L. Protein homeostasis at the plasma membrane. Physiology 2014, 29, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [Google Scholar] [CrossRef] [PubMed]
- French, P.J.; van Doorninck, J.H.; Peters, R.H.; Verbeek, E.; Ameen, N.A.; Marino, C.R.; de Jonge, H.R.; Bijman, J.; Scholte, B.J. A delta F508 mutation in mouse cystic fibrosis transmembrane conductance regulator results in a temperature-sensitive processing defect in vivo. J. Clin. Investig. 1996, 98, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Okiyoneda, T.; Barriere, H.; Bagdany, M.; Rabeh, W.M.; Du, K.; Hohfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Villella, V.R.; Esposito, S.; Bruscia, E.M.; Vicinanza, M.; Cenci, S.; Guido, S.; Pettoello-Mantovani, M.; Carnuccio, R.; De Matteis, M.A.; Luini, A.; et al. Disease-relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cell Death Differ. 2013, 20, 1101–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yao, Y.; Tang, X.Q.; Wang, Z.Z. Mouse RIC-3, an endoplasmic reticulum chaperone, promotes assembly of the alpha7 acetylcholine receptor through a cytoplasmic coiled-coil domain. J. Neurosci. 2009, 29, 12625–12635. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Jiang, Q.; Bai, X.; Yang, Y.F.; Ruan, M.Y.; Cai, S.Q. Tetrameric Assembly of K+ Channels Requires ER-Located Chaperone Proteins. Mol. Cell 2017, 65, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.L.; Kelly, J.W. Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: Progress and prognosis. Cold Spring Harb. Perspect. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.F.; Lin, I.W.; Chen, S.C.; Wu, H.H.; Yang, C.S.; Fang, H.Y.; Chiu, M.M.; Jeng, C.J. The subfamily-specific assembly of Eag and Erg K+ channels is determined by both the amino and the carboxyl recognition domains. J. Biol. Chem. 2014, 289, 22815–22834. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Y.-J.; Lee, Y.-C.; Fu, S.-J.; Chien, Y.-C.; Liao, Y.-F.; Chen, T.-Y.; Jeng, C.-J.; Tang, C.-Y. FKBP8 Enhances Protein Stability of the CLC-1 Chloride Channel at the Plasma Membrane. Int. J. Mol. Sci. 2018, 19, 3783. https://doi.org/10.3390/ijms19123783
Peng Y-J, Lee Y-C, Fu S-J, Chien Y-C, Liao Y-F, Chen T-Y, Jeng C-J, Tang C-Y. FKBP8 Enhances Protein Stability of the CLC-1 Chloride Channel at the Plasma Membrane. International Journal of Molecular Sciences. 2018; 19(12):3783. https://doi.org/10.3390/ijms19123783
Chicago/Turabian StylePeng, Yi-Jheng, Yi-Ching Lee, Ssu-Ju Fu, Yun-Chia Chien, Yi-Fan Liao, Tsung-Yu Chen, Chung-Jiuan Jeng, and Chih-Yung Tang. 2018. "FKBP8 Enhances Protein Stability of the CLC-1 Chloride Channel at the Plasma Membrane" International Journal of Molecular Sciences 19, no. 12: 3783. https://doi.org/10.3390/ijms19123783