Stroke-Like Episodes and Cerebellar Syndrome in Phosphomannomutase Deficiency (PMM2-CDG): Evidence for Hypoglycosylation-Driven Channelopathy

 , , , and
, , , and 
Abstract
:
1. Introduction
2. Results
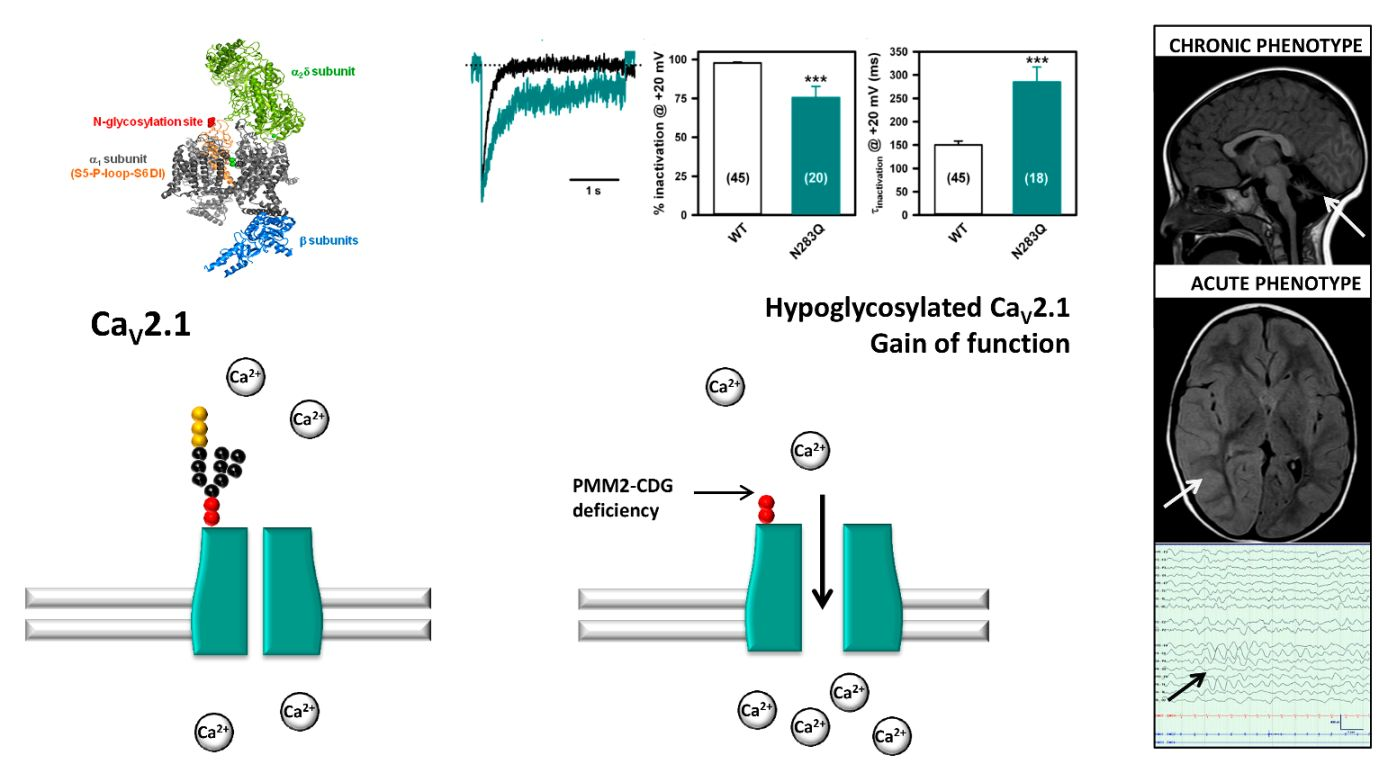
2.1. Stroke-Like Episodes
2.2. SLE Versus SLE Negative Group
2.3. Literature Review Comparing Clinical Presentations between Patients with PMM2-CDG and Patients with CACNA1A Mutations
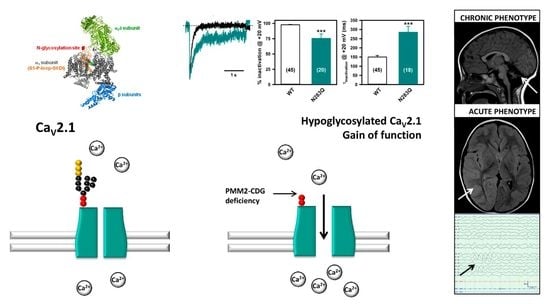
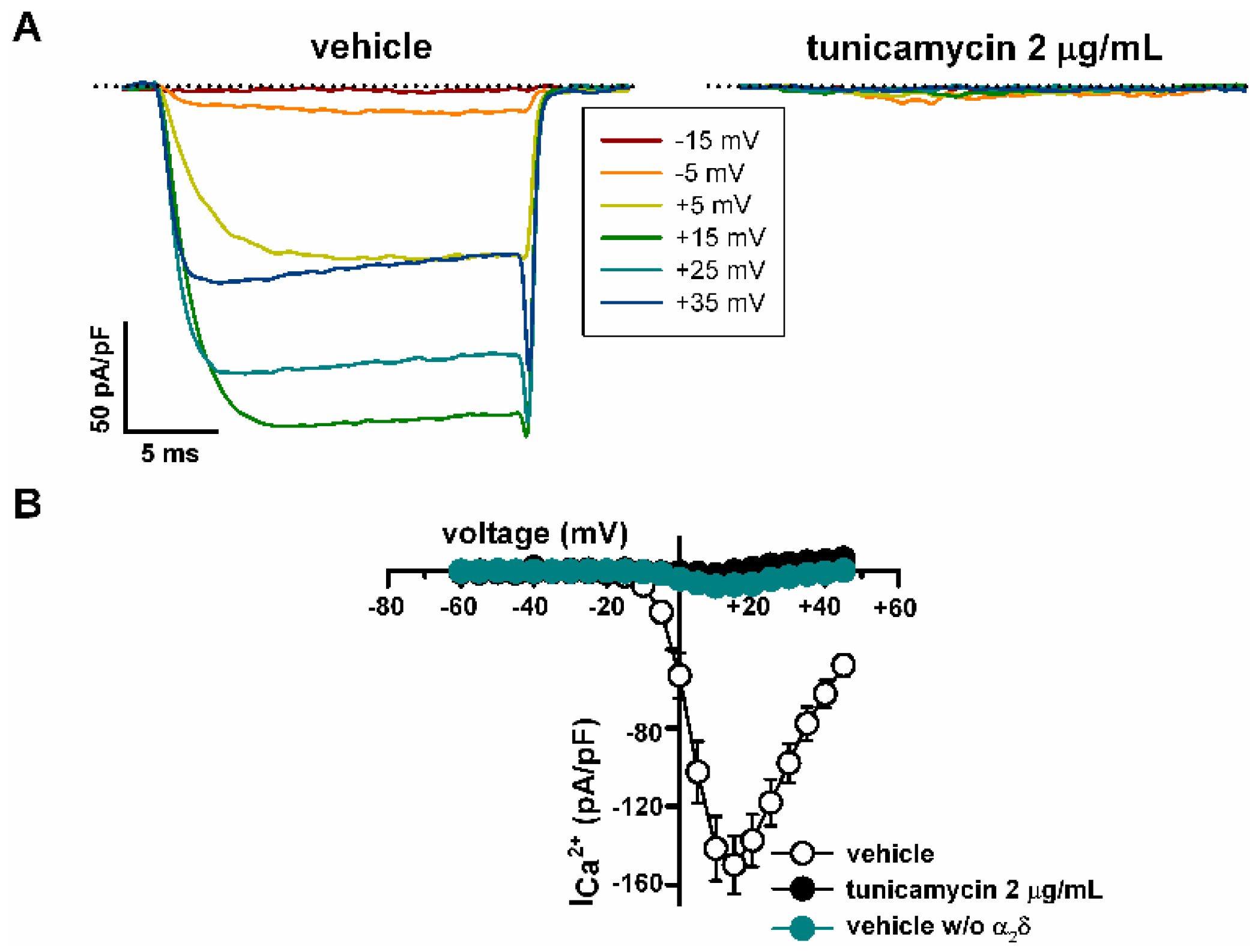
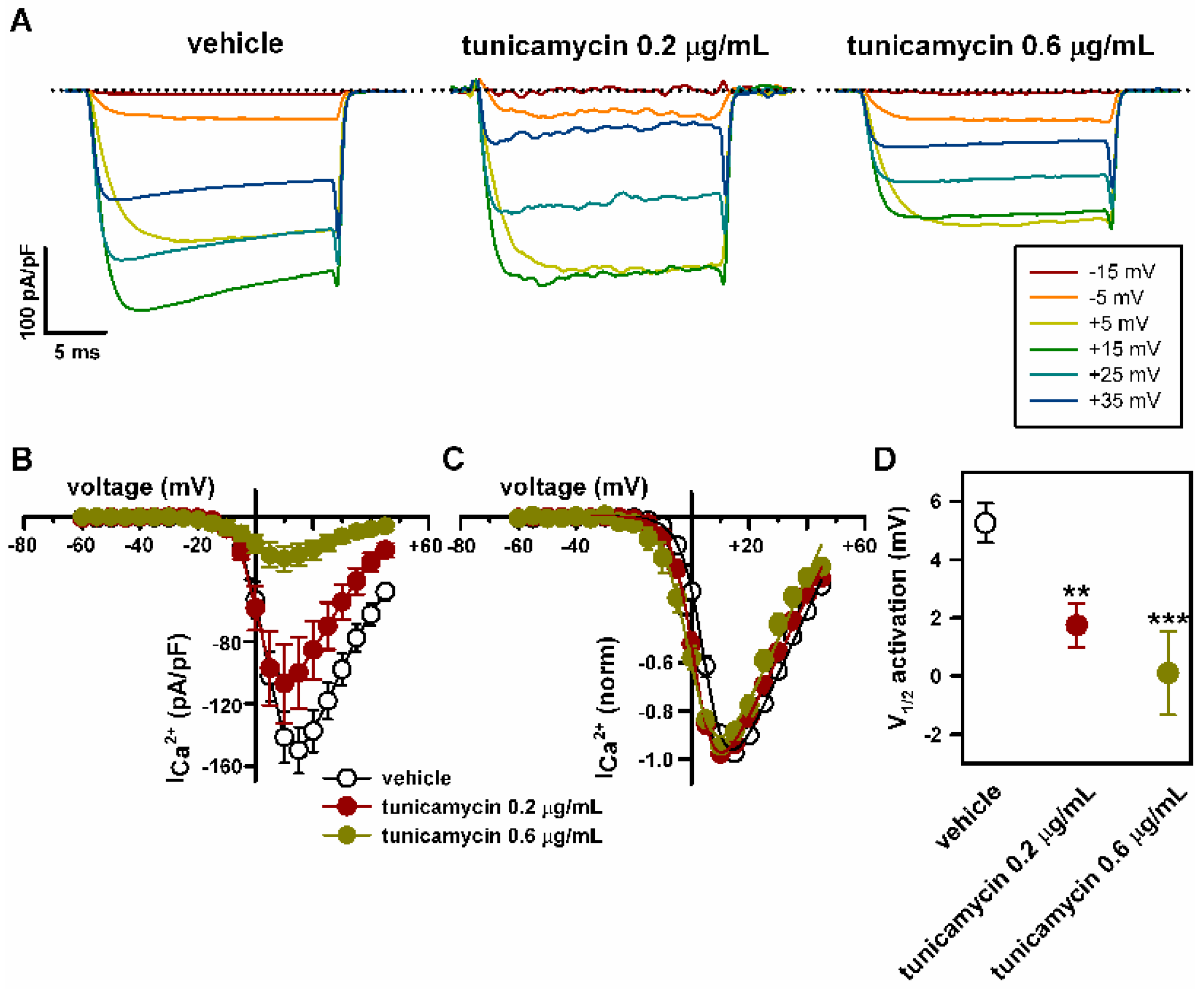
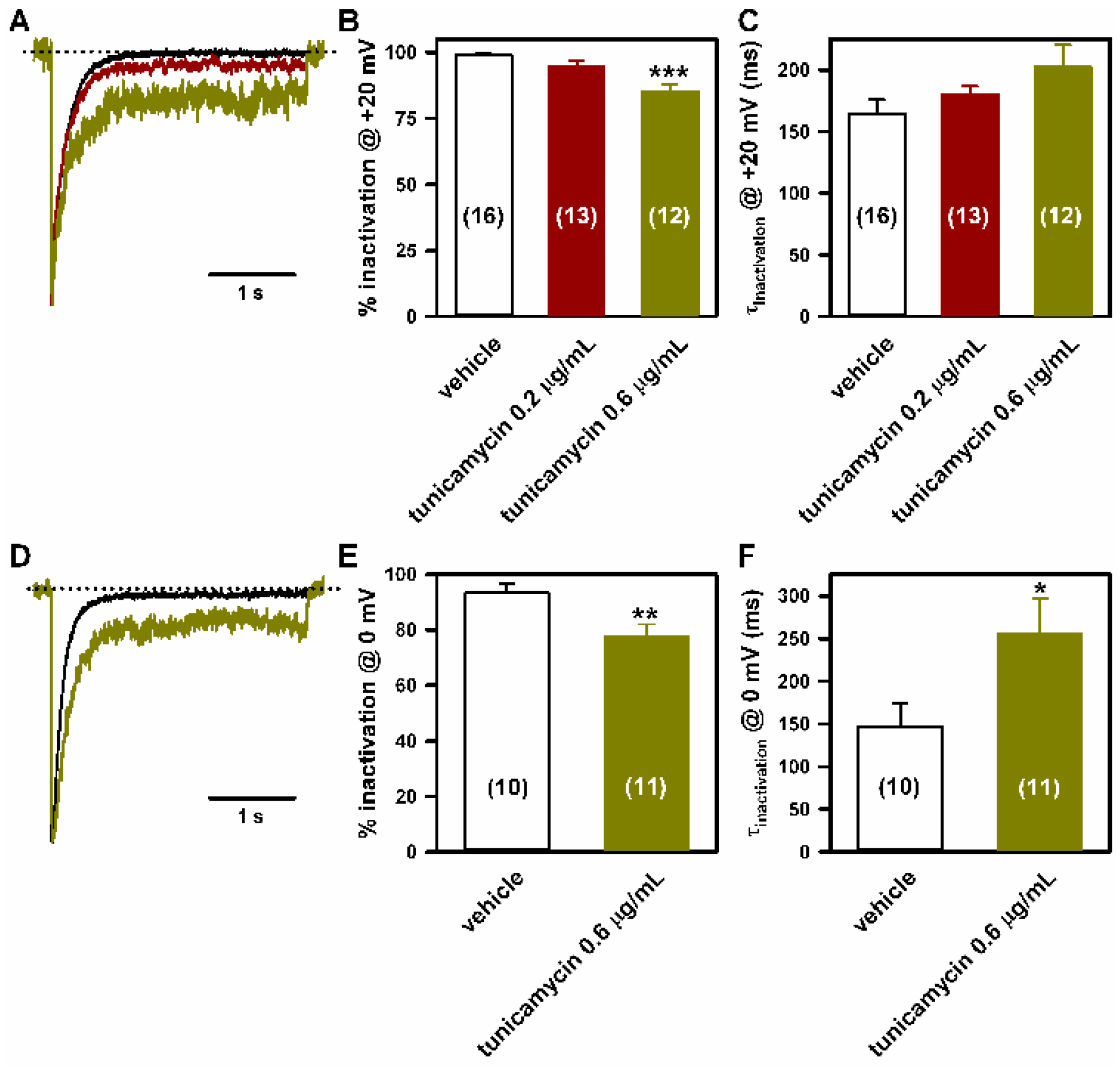
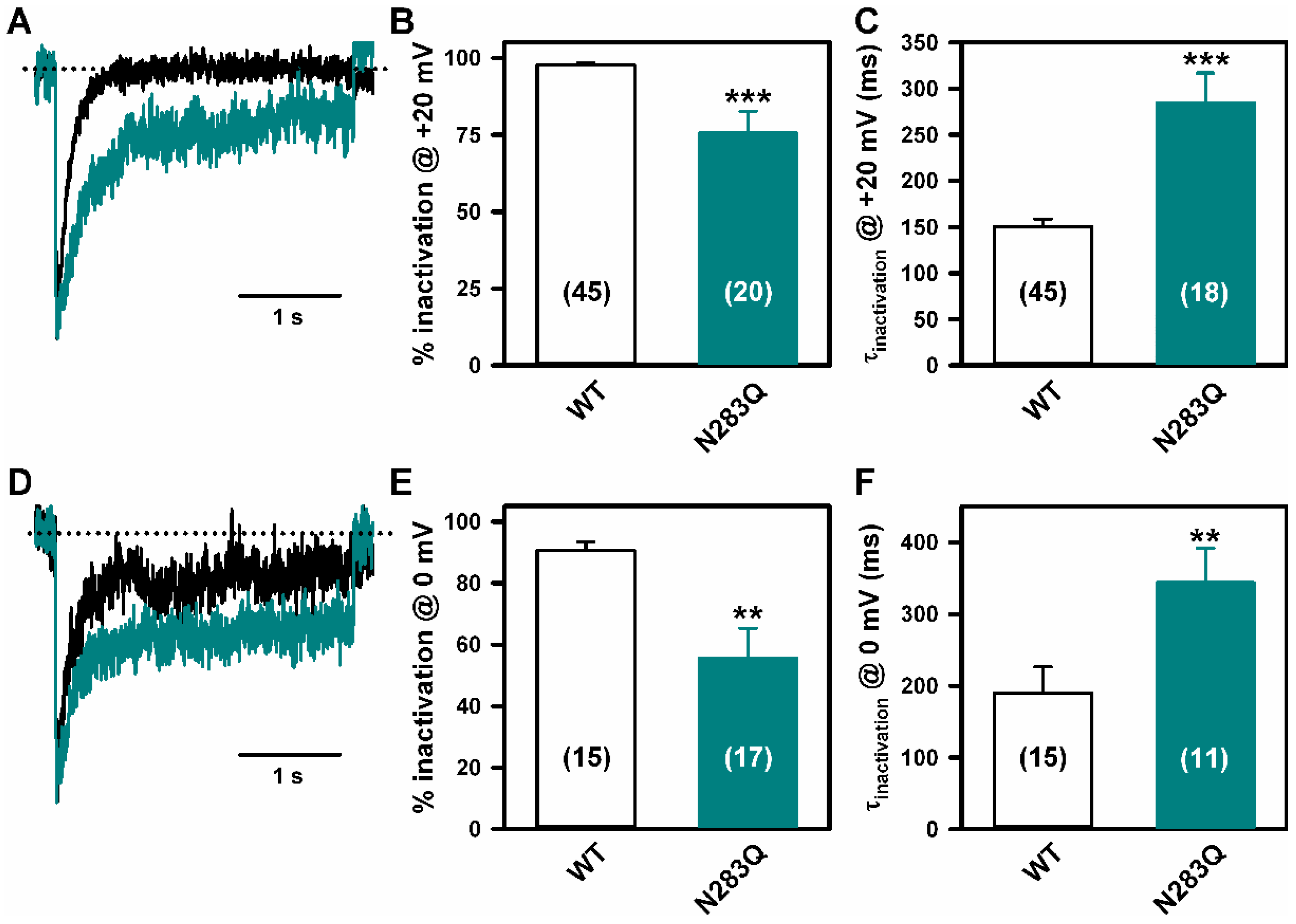
2.4. Deficient N-Glycosylation Alters CaV2.1 Functional Expression, Activation, and Inactivation
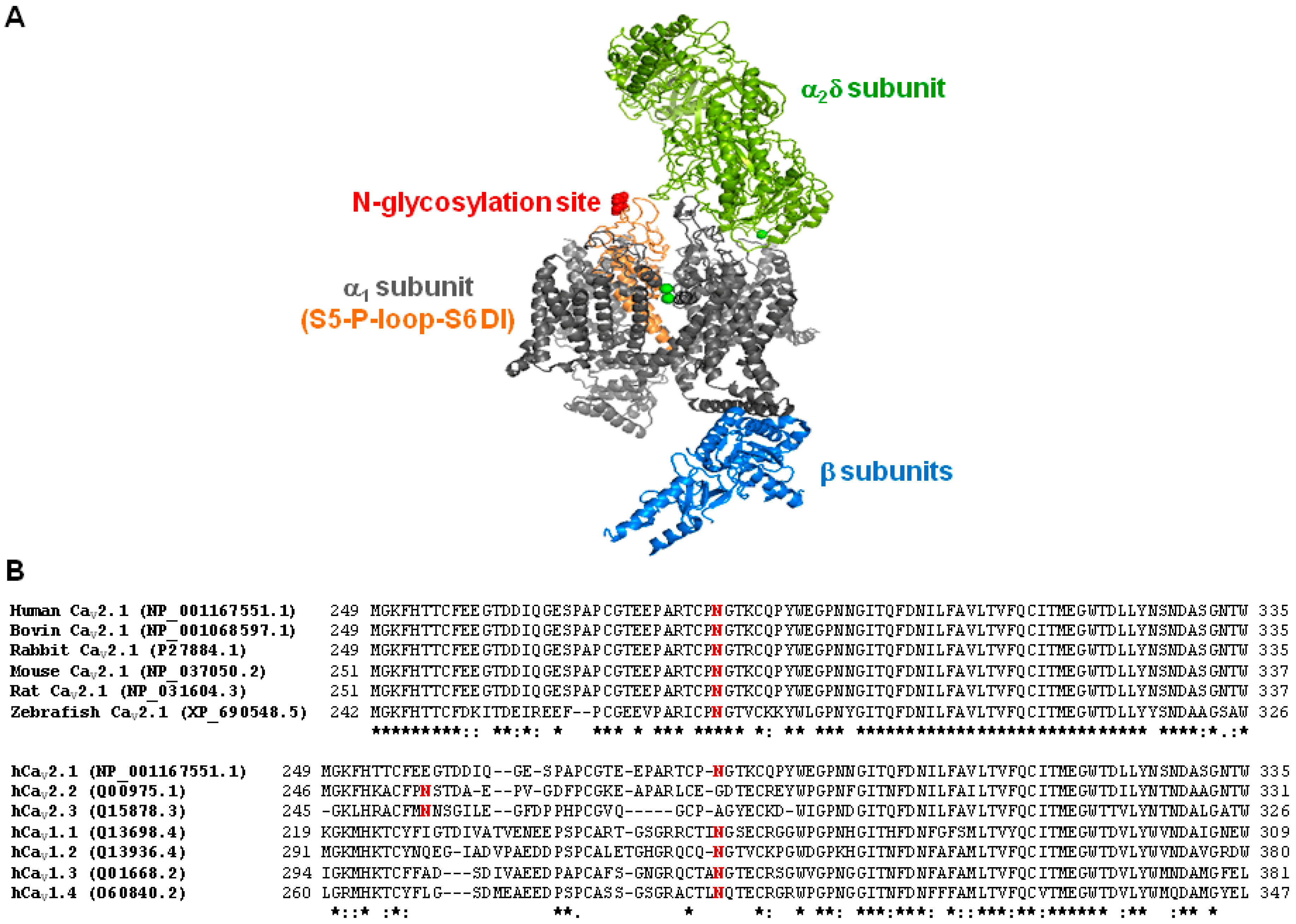
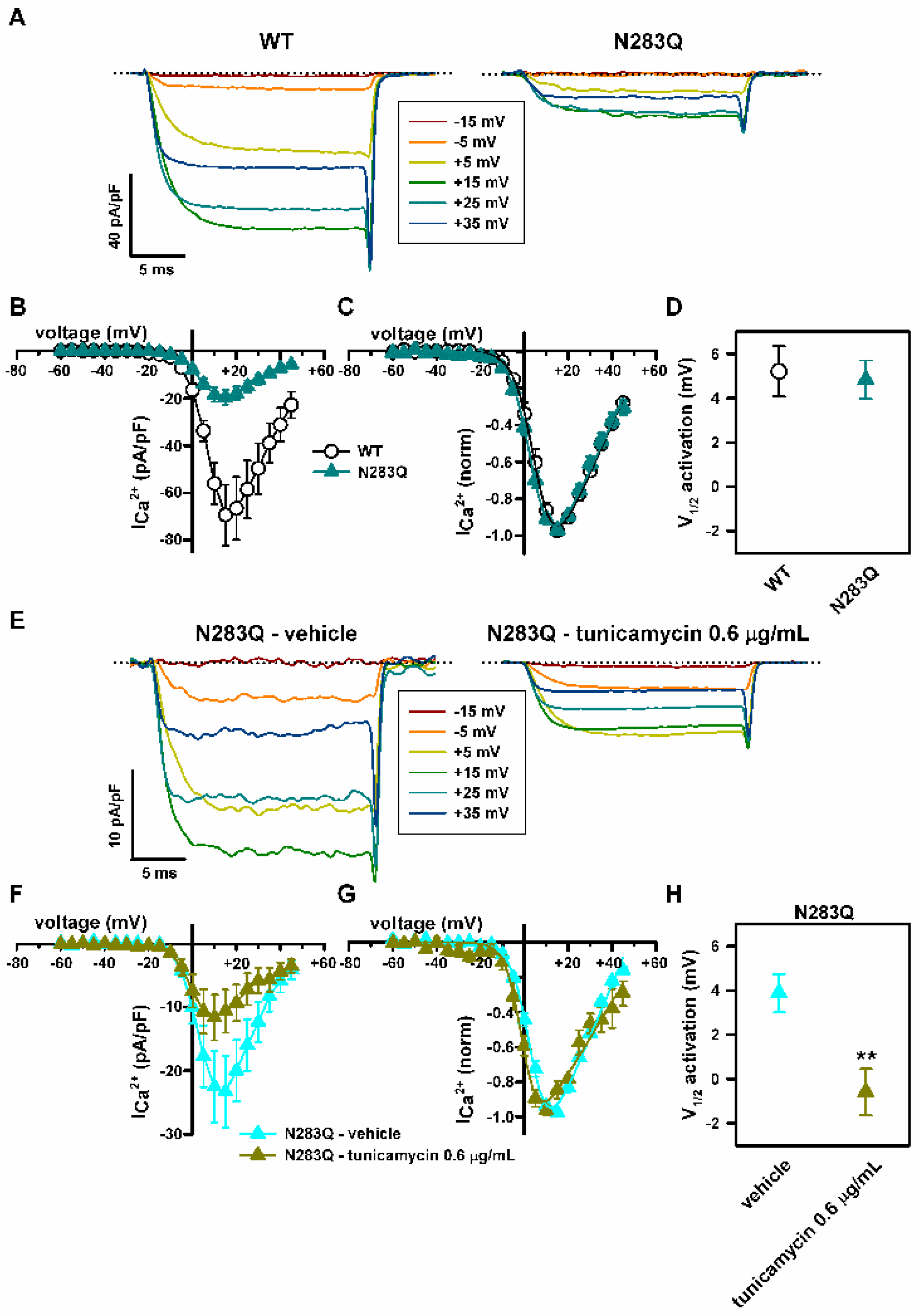
2.5. Mutation of α1A Potential Glycosylation Site Mimics Tunicamycin Effect on CaV2.1 Inactivation
3. Materials and Methods
3.1. Patients
3.2. Literature Review
3.3. cDNA Constructs
3.4. Heterologous Expression
3.5. Inhibition of N-Glycosylation in Live Cells and Western Blot
3.6. Electrophysiology
3.7. Ethics Statement
3.8. Statistical Analysis
4. Discussion and Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Freeze, H.H.; Eklund, E.A.; Ng, B.G.; Patterson, M.C. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012, 11, 453–466. [Google Scholar] [CrossRef]
- Grünewald, S.; Matthijs, G.; Jaeken, J. Congenital disorders of glycosylation: A review. Pediatr. Res. 2002, 52, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Feraco, P.; Mirabelli-Badenier, M.; Severino, M.; Alpigiani, M.G.; Di Rocco, M.; Biancheri, R.; Rossi, A. The shrunken, bright cerebellum: A characteristic MRI finding in congenital disorders of glycosylation type 1a. AJNR Am. J. Neuroradiol. 2012, 33, 2062–2067. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Dueñas, B.; García-Cazorla, A.; Pineda, M.; Poo, P.; Campistol, J.; Cusí, V.; Schollen, E.; Matthijs, G.; Grunewald, S.; Briones, P.; et al. Long-term evolution of eight Spanish patients with CDG type Ia: Typical and atypical manifestations. Eur. J. Paediatr. Neurol. 2009, 13, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Carrozzi, M.; Parini, R.; Battini, R.; Martinelli, D.; Elia, M.; Spada, M.; Lilliu, F.; Ciana, G.; Burlina, A.; et al. A nationwide survey of PMM2-CDG in Italy: High frequency of a mild neurological variant associated with the L32R mutation. J. Neurol. 2015, 262, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Pearl, P.L.; Krasnewich, D. Neurologic course of congenital disorders of glycosylation. J. Child Neurol. 2001, 16, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, S.; Schwartz, M.; Skovby, F. Congenital disorder of glycosylation type Ia (CDG-Ia): Phenotypic spectrum of the R141H/F119L genotype. Arch. Dis. Child. 2001, 85, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Dinopoulos, A.; Mohamed, I.; Jones, B.; Rao, S.; Franz, D.; deGrauw, T. Radiologic and neurophysiologic aspects of stroke-like episodes in children with congenital disorders of glycosylation type-1a. Pediatrics 2007, 119, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Sproule, D.M.; Kaufmann, P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: Basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann. N. Y. Acad. Sci. 2008, 1142, 133–158. [Google Scholar] [CrossRef] [PubMed]
- Van Baalen, A.; Stephani, U.; Rohr, A. Increased brain lactate during stroke-like episode in a patient with congenital disorder of glycosylation type Ia. Brain Dev. 2009, 31, 183. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Tajima, G.; Ono, H.; Kobayashi, M. Different neuroradiological findings during two stroke-like episodes in a patient with a congenital disorder of glycosylation type Ia. Brain Dev. 2009, 31, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.L.; Coleman, L.T.; Smith, L.J.; Shield, L.K. Hemiconvulsion-hemiplegia-epilepsy syndrome: Characteristic early magnetic resonance imaging findings. J. Child Neurol. 2002, 17, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Hart, A.R.; Trinick, R.; Connolly, D.J.; Mordekar, S.R. Profound encephalopathy with complete recovery in three children with familial hemiplegic migraine. J. Paediatr. Child Health 2009, 45, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Blumkin, L.; Michelson, M.; Leshinsky-Silver, E.; Kivity, S.; Lev, D.; Lerman-Sagie, T. Congenital ataxia, mental retardation, and dyskinesia associated with a novel CACNA1A mutation. J. Child Neurol. 2010, 25, 892–987. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Gurnett, C.A.; de Waard, M.; Campbell, K.P. Dual function of the voltage-dependent Ca2+ channel α2δ subunit in current stimulation and subunit interaction. Neuron 1996, 16, 431–440. [Google Scholar] [CrossRef]
- Sandoval, A.; Oviedo, N.; Andrade, A.; Felix, R. Glycosylation of asparagines 136 and 184 is necessary for the α2δ subunit-mediated regulation of voltage-gated Ca2+ channels. FEBS Lett. 2004, 576, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Tétreault, M.P.; Bourdin, B.; Briot, J.; Segura, E.; Lesage, S.; Fiset, C.; Parent, L. Identification of glycosylation sites essential for surface expression of the CaVα2δ1 subunit and modulation of the cardiac CaV1.2 channel activity. J. Biol. Chem. 2016, 291, 4826–4843. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Todorov, B.; Barrett, C.F.; van Dorp, S.; Ferrari, M.D.; van den Maagdenberg, A.M.; De Zeeuw, C.I.; Hoebeek, F.E. Cerebellar ataxia by enhanced CaV2.1 currents is alleviated by Ca2+-dependent K+-channel activators in Cacna1a S218L mutant mice. J. Neurosci. 2012, 32, 15533–15546. [Google Scholar] [CrossRef] [PubMed]
- Arnoux, J.B.; Boddaertm, N.; Valayannopoulos, V.; Romano, S.; Bahi-Buisson, N.; Desguerre, I.; de Keyzer, Y.; Munnich, A.; Brunelle, F.; Seta, N.; et al. Risk assessment of acute vascular events in congenital disorder of glycosylation type Ia. Mol. Genet. Metab. 2008, 93, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Barros, J.; Damásio, J.; Tuna, A.; Alves, I.; Silveira, I.; Pereira-Monteiro, J.; Sequeiros, J.; Alonso, I.; Sousa, A.; Coutinho, P. Cerebellar ataxia, hemiplegic migraine, and related phenotypes due to a CACNA1A missense mutation: 12-year follow-up of a large Portuguese family. JAMA Neurol. 2013, 70, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Knierim, E.; Leisle, L.; Wagner, C.; Weschke, B.; Lucke, B.; Bohner, G.; Dreier, J.P.; Schuelke, M. Recurrent stroke due to a novel voltage sensor mutation in CaV2.1 responds to verapamil. Stroke 2011, 42, e14–e17. [Google Scholar] [CrossRef] [PubMed]
- García Segarra, N.; Gautschi, I.; Mittaz-Crettol, L.; Kallay Zetchi, C.; Al-Qusairi, L.; Van Bemmelen, M.X.; Maeder, P.; Bonafé, L.; Schild, L.; Roulet-Perez, E. Congenital ataxia and hemiplegic migraine with cerebral edema associated with a novel gain of function mutation in the calcium channel CACNA1A. J. Neurol. Sci. 2014, 342, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Malpas, T.J.; Riant, F.; Tournier-Lasserve, E.; Vahedi, K.; Neville, B.G. Sporadic hemiplegic migraine and delayed cerebral oedema after minor head trauma: A novel de novo CACNA1A gene mutation. Dev. Med. Child Neurol. 2010, 52, 103–104. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; de Diego, V.; Muchart, J.; Cuadras, D.; Felipe, A.; Macaya, A.; Velázquez, R.; Poo, M.P.; Fons, C.; O’Callaghan, M.M.; et al. Phosphomannomutase deficiency (PMM2-CDG): Ataxia and cerebellar assessment. Orphanet J. Rare Dis. 2015, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Douglas, L.; Davies, A.; Wratten, J.; Dolphin, A.C. Do voltage-gated calcium channel α2δ subunits require proteolytic processing into α2 and δ to be functional? Biochem. Soc. Trans. 2006, 34, 894–898. [Google Scholar] [CrossRef] [PubMed]
- De Diego, V.; Martínez-Monseny, A.F.; Muchart, J.; Cuadras, D.; Montero, R.; Artuch, R.; Pérez-Cerdá, C.; Pérez, B.; Pérez-Dueñas, B.; Poretti, A.; Serrano, M. Collaborators of the CDG Spanish-Consortium. Longitudinal volumetric and 2D assessment of cerebellar atrophy in a large cohort of children with phosphomannomutase deficiency (PMM2-CDG). J. Inherit. Metab. Dis. 2017, 40, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.A.; Fernàndez-Castillo, N.; Macaya, A.; Cormand, B.; Valverde, M.A.; Fernández-Fernández, J.M. The Hemiplegic Migraine associated Y1245C mutation in CACNA1A results in a gain of channel function due to its effect on the voltage sensor and G-protein mediated inhibition. Pflügers Arch. 2009, 458, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Van der Knaap, M.S.; Barth, P.G.; Gabreëls, F.J.; Franzoni, E.; Begeer, J.H.; Stroink, H.; Rotteveel, J.J.; Valk, J. A new leukoencephalopathy with vanishing white matter. Neurology 1997, 48, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Kölker, S.; Sauer, S.W.; Hoffmann, G.F.; Müller, I.; Morath, M.A.; Okun, J.G. Pathogenesis of CNS involvement in disorders of amino and organic acid metabolism. J. Inherit. Metab. Dis. 2008, 31, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Iida, S.; Nakamura, M.; Asayama, S.; Kunieda, T.; Kaneko, S.; Osaka, H.; Kusaka, H. Rapidly progressive psychotic symptoms triggered by infection in a patient with methylenetetrahydrofolate reductase deficiency: A case report. BMC Neurol. 2017, 17, 47. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Rebollo, M.; Depienne, C.; Rastetter, A.; Fernández-Álvarez, E.; Muchart, J.; Martorell, L.; Artuch, R.; Obeso, J.A.; Pérez-Dueñas, B. Reversible generalized dystonia and encephalopathy from thiamine transporter 2 deficiency. Mov. Disord. 2012, 27, 1295–1298. [Google Scholar] [CrossRef] [PubMed]
- Tantsis, E.M.; Gill, D.; Griffiths, L.; Gupta, S.; Lawson, J.; Maksemous, N.; Ouvrier, R.; Riant, F.; Smith, R.; Troedson, C.; et al. Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev. Med. Child Neurol. 2016, 58, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Asghar, S.J.; Milesi-Hallé, A.; Kaushik, C.; Glasier, C.; Sharp, G.B. Variable manifestations of familial hemiplegic migraine associated with reversible cerebral edema in children. Pediatr. Neurol. 2012, 47, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Plomp, J.J.; van den Maagdenberg, A.M.; Molenaar, P.C.; Frants, R.R.; Ferrari, M.D. Mutant P/Q-type calcium channel electrophysiology and migraine. Curr. Opin. Investig. Drugs 2001, 2, 1250–1260. [Google Scholar] [PubMed]
- Van den Maagdenberg, A.M.; Pietrobon, D.; Pizzorusso, T.; Kaja, S.; Broos, L.A.; Cesetti, T.; van de Ven, R.C.; Tottene, A.; van der Kaa, J.; Plomp, J.J.; et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 2004, 41, 701–710. [Google Scholar] [CrossRef]
- Tottene, A.; Conti, R.; Fabbro, A.; Vecchia, D.; Shapovalova, M.; Santello, M.; van den Maagdenberg, A.M.; Ferrari, M.D.; Pietrobon, D. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in CaV2.1 knockin migraine mice. Neuron 2009, 61, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, D. CaV2.1 channelopathies. Pflügers Arch. 2010, 460, 375–393. [Google Scholar] [CrossRef] [PubMed]
- Di Guilmi, M.N.; Wang, T.; Inchauspe, C.G.; Forsythe, I.D.; Ferrari, M.D.; van den Maagdenberg, A.M.; Borst, J.G.; Uchitel, O.D. Synaptic gain-of-function effects of mutant CaV2.1 channels in a mouse model of familial hemiplegic migraine are due to increased basal [Ca2+]i. J. Neurosci. 2014, 34, 7047–7058. [Google Scholar] [CrossRef] [PubMed]
- Vecchia, D.; Tottene, A.; van den Maagdenberg, A.M.; Pietrobon, D. Mechanism underlying unaltered cortical inhibitory synaptic transmission in contrast with enhanced excitatory transmission in CaV2.1 knockin migraine mice. Neurobiol. Dis. 2014, 69, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Bahamonde, M.I.; Serra, S.A.; Drechsel, O.; Rahman, R.; Marcé-Grau, A.; Prieto, M.; Ossowski, S.; Macaya, A.; Fernández-Fernández, J.M. A Single amino acid deletion (ΔF1502) in the S6 segment of CaV2.1 domain III associated with congenital ataxia increases channel activity and promotes Ca2+ influx. PLoS ONE 2015, 10, e0146035. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimons, R.B.; Wolfenden, W.H. Migraine coma: Meningitic migraine with cerebral oedema associated with a new form of autosomal dominant cerebellar ataxia. Brain 1985, 108, 555–577. [Google Scholar] [CrossRef] [PubMed]
- Ducros, A.; Denier, C.; Joutel, A.; Vahedi, K.; Michel, A.; Darcel, F.; Madigand, M.; Guerouaou, D.; Tison, F.; Julien, J.; et al. Recurrence of the T666M calcium channel CACNA1A gene mutation in familial hemiplegic migraine with progressive cerebellar ataxia. Am. J. Hum. Genet. 1999, 64, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Kors, E.E.; Terwindt, G.M.; Vermeulen, F.L.; Fitzsimons, R.B.; Jardine, P.E.; Heywood, P.; Love, S.; van den Maagdenberg, A.M.; Haan, J.; Frants, R.R.; et al. Delayed cerebral edema and fatal coma after minor head trauma: Role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann. Neurol. 2001, 49, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Bolay, H.; Reuter, U.; Dunn, A.K.; Huang, Z.; Boas, D.A.; Moskowitz, M.A. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat. Med. 2002, 8, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, D.; Striessnig, J. Neurobiology of migraine. Nat. Rev. Neurosci. 2003, 4, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, N.; Stam, A.H.; Haan, J.; Ferrari, M.D.; Terwindt, G.M. Familial and sporadic hemiplegic migraine: Diagnosis and treatment. Curr. Treat. Options Neurol. 2013, 15, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, D.; Moskowitz, M.A. Pathophysiology of migraine. Annu. Rev. Physiol. 2013, 75, 365–391. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Black, S.A.G.; Bladen, C.; Chen, L.; Zamponi, G.W. Surface expression and function of CaV3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch. 2013, 465, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Orestes, P.; Osuru, H.P.; McIntire, W.E.; Jacus, M.O.; Salajegheh, R.; Jagodic, M.M.; Choe, W.; Lee, J.; Lee, S.S.; Rose, K.E.; et al. Reversal of neuropathic pain in diabetes by targeting glycosylation of CaV3.2 T-Type calcium channels. Diabetes 2013, 62, 3828–3838. [Google Scholar] [CrossRef] [PubMed]
- Felix, R.; Gurnett, C.A.; De Waard, M.; Campbell, K.P. Dissection of functional domains of the voltage-dependent Ca2+ channel α2δ subunit. J. Neurosci. 1997, 17, 6884–6891. [Google Scholar] [PubMed]
- Platano, D.; Qin, N.; Noceti, F.; Birnbaumer, L.; Stefani, E.; Olcese, R. Expression of the α2δ subunit interferes with prepulse facilitation in cardiac L-type calcium channels. Biophys. J. 2000, 78, 2959–2972. [Google Scholar] [CrossRef]
- Qin, N.; Olcese, R.; Stefani, E.; Birnbaumer, L. Modulation of human neuronal α1E-type calcium channel by α2δ-subunit. Am. J. Physiol. 1998, 274, 1324–1331. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient/Episode | Sex/Age | Molecular Findings | ICARS | Trigger | Free Symptom Period | Initial Clinical Presentation | Body Temperature (°C) | C-reactive Protein/PCT | EEG | Neuroimaging (<72 h) | Treatment | Duration of Symptoms (h) | Recovery |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M 3 yr 11 mo | p.V44A p.Q33Stop | NA | Head trauma | 3 h | Lethargy, epileptic seizures, left hemiparesis | 38.5 | Normal | Asymmetric slow background, right hemisphere FIRDA epileptic activity | MRI: Asymmetric hemispheric vasogenic edema | LEV, PHE, MDZ | 96 | Tonic upgaze some days after discharge Left hemiparesis until 4 months later |
| 2 | M 15 yr | p. V44A p.R123G | NA | Head trauma | 24 h | Somnolence, vomiting | 37.4 | No data available | Normal | MRI: No significant changes | No | 48 | Complete recovery |
| 3 | F 5 yr | p.T237M IVS7-9T > G | 46 | Head trauma | 4 h | Somnolence, vomiting | 37.5 | Normal | Normal | No significant changes | No | 7 | Complete recovery |
| 4-I | F 3 yr 6 mo | p.L32R IVS-1G > C | 8 | Head trauma | 1 h | Irritability and lethargy | 38.0 | Normal | Normal | No significant changes | No | 3 | Complete recovery |
| 4-II | F 14 yr 5 mo | Enterovirus | NA | Dysarthria, dysphasia, irritability, left hemiparesis | 38.0 | Normal | Normal | CT angiography: No significant changes | LEV | 36 | Complete recovery | ||
| 5-I | F 7 yr 4 mo | p.D65Y IVS7-9T > G | 56 | Head trauma | 12 h | Irritability, lethargy, aphasia and dysphagia | 38.6 | No data available | Normal | MRI: No significant changes | Risperidone | 168 | Complete recovery |
| 5-II | F 10 yr 2 mo | Head trauma | 2 h | Irritability and lethargy | 38.5 | Normal | Normal | MRI: No significant changes | MDZ | 24 | Complete recovery | ||
| 6 | F 3 yr 3 mo | p.P113L p.F207S | NA | Influenza virus infection | NA | Epileptic seizures, irritability and lethargy | 38.9 | 38.0 mg/L 1.04 ng/mL | Asymmetric slow background, epileptic activity | CT: No significant changes | LEV, PHE, MDZ | 264 | Tonic upgaze deviation and irritability until 1 month later. Fully recovered 7 months later. |
| 7 | M 5 yr 8 mo | p.D65Y p.R141H | 65 | Upper respiratory viral infection | NA | Lethargy, epileptic seizures, left arm monoparesis | 38.5 | 11.4 mg/L 24.7 ng/mL | Asymmetric slow background, no epileptic activity | MRI: No significant changes | VPA, LEV, DZP, MDZ | 144 | Distal weakness left arm, fully recovered in 2 weeks |
| Mean values/SD | 7 yr 7 mo/ 4 yr 10 mo | 6 out of 9 Head trauma | 13.0/8.4 | 38.2/0.5 | 3 out of 9 Abnormal EEG | 1/9 Hemispheric edema | 87.8 h/86.9 h | ||||||
| Range | 3 yr 3 mo/15 yr | 3–24 h | 37.4–38.9 | 3–264 h |
| SLE Positive (SD) | SLE Negative (SD) | p-Value | ||
|---|---|---|---|---|
| Number of patients/episodes | 7/9 | 32 | ||
| Age (years) | 13.7 (11.6) * | 16.2 (9.6) | 0.46 | |
| Sex (Male:Female) | 3:4 | 21:11 | 0.19 | |
| Liver function | AST (UI/mL) Normal values 2–34 UI/mL | 378 (317.3) | 172 (235.5) | 0.61 |
| ALT (UI/mL) Normal values 2–36 UI/mL | 277 (220.6) | 216 (327.1) | 0.70 | |
| Coagulation | PT (%) Normal values 80–120% | 95.4 (13.1) | 95.9(24.0) | 0.82 |
| aPTT (seconds) Normal values 23–35 seconds | 33.3 (4.6) | 31.5 (6.0) | 0.53 | |
| F IX (%) Normal values 50–120% | 49.5 (14.9) | 75.2 (25.0) | 0.17 | |
| F XI (%) Normal values 50–120% | 35.9 (15.1) | 59.3 (35.5) | 0.28 | |
| AT III (%) Normal values 60–120% | 43.0 (14.1) | 51.2 (26.8) | 0.82 | |
| Protein C (%) Normal values 60–140% | 38.6 (17.2) | 83.4 (129.9) | 0.46 | |
| Protein S (%) Normal values 60–140% | 52.9 (22.6) | 63.5 (17.5) | 0.31 | |
| Vascular events | Positive personal history | 1/7 | 3/32 | 0.51 |
| Positive familial history | 1/7 | 2/32 | 0.41 | |
| Vermis Midsagittal Relative Diameter (MRI) | 0.48 | 0.41 | 0.44 | |
| Seizures | Febrile seizures | 2/7 | 4/32 | 0.23 |
| Epilepsy | 2/7 | 6/32 | 0.58 | |
| Gene | CACNA1A | PMM2 |
|---|---|---|
| Acute: stroke-like episodes | ||
| Prevalence | 21% [8], >70 cases reported [14]. | 18 to 40–50%, >49 cases reported [20]. |
| Age at onset | At any age between 1 to 73 years old [21]. | Any age, frequently between 3 to 6 years old [5,8,11]. |
| Trigger/Onset of encephalopathy after trigger | Minor head trauma [22], infections, diagnostic procedures (cerebral or coronary angiography), physical activity [21]; Immediately or 2–3 h after the trigger [14]. | Infections, head trauma, angiography, alcohol ingestion [5]; Minutes to hours after the trigger. |
| Prodromi | Severe headache, yawning, truncal unsteadiness [22]. | None |
| Focal deficits | Hemiparesis, dysphasia, nystagmus, dyskinetic limb movement [12,13,14]. | Hemiparesis, dysphasia, dysphagia, conjugated eye deviation, blindness [5,8,11]. |
| Epilepsy/EEG findings | Hemi-clonic or hemi-tonic convulsions/Globally slow background activity, spikes, spike-and-slow wave complexes, slow wave bursts, photoparoxysmal response [14]. | Clonic convulsions/Low voltage pattern in affected areas, asymmetric slow background activity with moderately low frontal intermittent rhythmic delta activity (FIRDA) in the affected hemisphere [8]. |
| Autonomic signs | High fever, recurrent vomiting [14]. | High fever, recurrent vomiting [11]. |
| MRI findings during the episode | Normal, ischemic lesion with prominent perifocal edema, or panhemispheric edema [14,23]. | Normal or asymmetric hemispheric cytotoxic edema [8,11]. |
| Duration/number of episodes | Few minutes to 10 days/1–11 episodes [14,21]. | 1h to several months/1–2 episodes [5,11]. |
| Recovery | 24 h to months. Complete recovery in the majority of the patients, but residual hemiplegia possible [14,22]. | 1h to several months. Complete recovery in the majority of the patients, but exceptionally residual motor symptoms may persist [5]. |
| Treatment | Analgesia, antiepileptic drugs, sleep, acetazolamide, verapamil [13,22,23,24]. | IV hydration, antiepileptic drugs [5]. |
| Chronic: time between stroke-like episodes | ||
| Ataxia | Episodic attacks of cerebellar dysfunction lasting from 5 minutes to 5 h [23]. | Persistent ataxia [1,2,4,5,11]. |
| Abnormal eye movement | Saccadic eye movements, downgaze nystagmus, strabismus, tonic upgaze episodes [22]. | Strabismus, nystagmus, tonic upgaze episodes [4,20,25]. |
| Other neurologic manifestations | Developmental disability, dysarthria, migraine with aura, motor stereotypies, benign paroxysmal torticollis [14,21]. | Developmental disability, hypotonia, dysarthria [4,20,25]. |
| MRI | Cerebellar vermis atrophy, mild delay in white matter myelination [22]. | Atrophy of the cerebellar hemispheres and vermis [25], pontine hypoplasia [11]. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izquierdo-Serra, M.; Martínez-Monseny, A.F.; López, L.; Carrillo-García, J.; Edo, A.; Ortigoza-Escobar, J.D.; García, Ó.; Cancho-Candela, R.; Carrasco-Marina, M.L.; Gutiérrez-Solana, L.G.; et al. Stroke-Like Episodes and Cerebellar Syndrome in Phosphomannomutase Deficiency (PMM2-CDG): Evidence for Hypoglycosylation-Driven Channelopathy. Int. J. Mol. Sci. 2018, 19, 619. https://doi.org/10.3390/ijms19020619
Izquierdo-Serra M, Martínez-Monseny AF, López L, Carrillo-García J, Edo A, Ortigoza-Escobar JD, García Ó, Cancho-Candela R, Carrasco-Marina ML, Gutiérrez-Solana LG, et al. Stroke-Like Episodes and Cerebellar Syndrome in Phosphomannomutase Deficiency (PMM2-CDG): Evidence for Hypoglycosylation-Driven Channelopathy. International Journal of Molecular Sciences. 2018; 19(2):619. https://doi.org/10.3390/ijms19020619
Chicago/Turabian StyleIzquierdo-Serra, Mercè, Antonio F. Martínez-Monseny, Laura López, Julia Carrillo-García, Albert Edo, Juan Darío Ortigoza-Escobar, Óscar García, Ramón Cancho-Candela, M Llanos Carrasco-Marina, Luis G. Gutiérrez-Solana, and et al. 2018. "Stroke-Like Episodes and Cerebellar Syndrome in Phosphomannomutase Deficiency (PMM2-CDG): Evidence for Hypoglycosylation-Driven Channelopathy" International Journal of Molecular Sciences 19, no. 2: 619. https://doi.org/10.3390/ijms19020619
APA StyleIzquierdo-Serra, M., Martínez-Monseny, A. F., López, L., Carrillo-García, J., Edo, A., Ortigoza-Escobar, J. D., García, Ó., Cancho-Candela, R., Carrasco-Marina, M. L., Gutiérrez-Solana, L. G., Cuadras, D., Muchart, J., Montero, R., Artuch, R., Pérez-Cerdá, C., Pérez, B., Pérez-Dueñas, B., Macaya, A., Fernández-Fernández, J. M., & Serrano, M. (2018). Stroke-Like Episodes and Cerebellar Syndrome in Phosphomannomutase Deficiency (PMM2-CDG): Evidence for Hypoglycosylation-Driven Channelopathy. International Journal of Molecular Sciences, 19(2), 619. https://doi.org/10.3390/ijms19020619