Ion Channel Disorders and Sudden Cardiac Death


1
Group of Biomedical Research in Heart Diseases, Hospital del Mar Medical Research Institute (IMIM), C/Doctor Aiguader 88, 08003 Barcelona, Spain
2
Cardiology Department, Hospital del Mar, Passeig Marítim 25-29, 08003 Barcelona, Spain
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(3), 692; https://doi.org/10.3390/ijms19030692
Submission received: 5 February 2018
/
Revised: 22 February 2018
/
Accepted: 23 February 2018
/
Published: 28 February 2018
(This article belongs to the Special Issue Ion Transporters and Channels in Physiology and Pathophysiology)
Abstract
:Long QT syndrome, short QT syndrome, Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia are inherited primary electrical disorders that predispose to sudden cardiac death in the absence of structural heart disease. Also known as cardiac channelopathies, primary electrical disorders respond to mutations in genes encoding cardiac ion channels and/or their regulatory proteins, which result in modifications in the cardiac action potential or in the intracellular calcium handling that lead to electrical instability and life-threatening ventricular arrhythmias. These disorders may have low penetrance and expressivity, making clinical diagnosis often challenging. However, because sudden cardiac death might be the first presenting symptom of the disease, early diagnosis becomes essential. Genetic testing might be helpful in this regard, providing a definite diagnosis in some patients. Yet important limitations still exist, with a significant proportion of patients remaining with no causative mutation identifiable after genetic testing. This review aims to provide the latest knowledge on the genetic basis of cardiac channelopathies and discuss the role of the affected proteins in the pathophysiology of each one of these diseases.
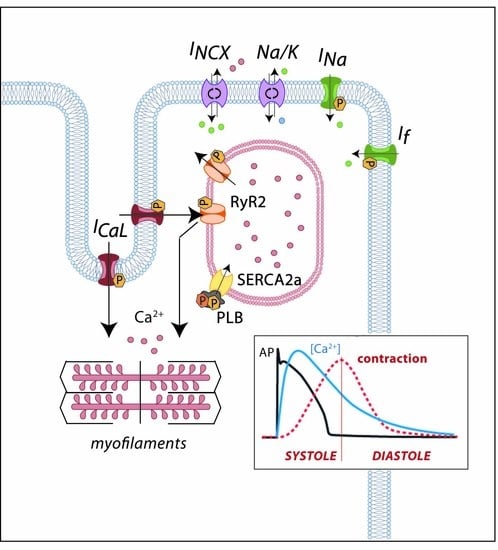
1. Introduction
Sudden cardiac death (SCD) is an unexpected death defined as that occurring within the first hour of the onset of symptoms [1]. It is responsible for 250,000–300,000 deaths annually in the US, with an estimated annual incidence of 50–100/100,000 individuals in industrialized countries [2]. It is therefore a major public health problem, with a tremendous social impact that becomes particularly devastating when affecting young people [3]. Ventricular arrhythmias, especially ventricular fibrillation, are the major underlying rhythms that lead to SCD. In individuals over 35, SCD most commonly occurs in patients with coronary heart disease with or without heart failure, whereas inherited cardiomyopathies and primary electrical disorders prevail in younger SCD victims [4]. Hereditary primary electrical disorders may account for up to 30% of all SCD in the young [5], and primarily include the long QT syndrome (LQTS), the short QT syndrome (SQTS), the Brugada syndrome (BrS) and the catecholaminergic polymorphic ventricular tachycardia (CPVT). These disorders, also known as cardiac channelopathies, most commonly respond to a mutation(s) in a gene(s) encoding cardiac ion channels or receptors and/or their regulatory proteins, the consequence in all cases being a modification in the cardiac action potential or in the intracellular calcium handling that leads to electrical instability and predisposition to life-threatening ventricular arrhythmias [6]. Recently, a disorder termed “early repolarization syndrome” has emerged as a new potential channelopathy. Although defined by a characteristic electrocardiogram (ECG) pattern that has been shown to be more common among patients experiencing SCD [7], most of the potential genetic variants associated with the disorder lack functional or biological validation, so it is still an unresolved issue whether this is a true primary electrical disorder [8]. For this reason, this review will leave out this syndrome and focus on the four above-mentioned channelopathies.
Considering that cardiac channelopathies supervene in patients without structural heart disease that are usually unaware of their disorder and that SCD might be the first presenting symptom, early diagnosis of genetic carriers is warranted. With very few exceptions, cardiac channelopathies are autosomal dominant disorders with incomplete penetrance (meaning that some individuals will not express the trait even though they carry the mutated allele) and variable expressivity (meaning that the level of phenotypic expression will be diverse for individuals with the same genotype) [9]. This represents a significant limitation when the diagnosis is based exclusively on clinical findings. Genetic testing may (1) offer a definite confirmation of a particular electrical disorder, which becomes particularly useful in patients with inconclusive clinical data; (2) confirm or exclude the presence of a disease-causing mutation in family members of an index case; and (3) help personalize treatment recommendations and management of a patient’s specific disorder [9]. Therefore, in recent years genetic testing has been progressively introduced in clinical practice and is currently advised (with different levels of recommendation) in most cardiac channelopathies [10]. However, genetic screening of mutations in genes known to cause cardiac channelopathies might result unsuccessful, providing negative results in around 20% of patients with LQTS, 40% of patients with CPVT, and 80% of patients with SQTS or BrS [9]. This indicates that there is still a long way to go in the field of genetics in primary electrical disorders. This review aims to provide the latest knowledge on the genetic basis of cardiac channelopathies and discuss the role of the affected proteins in the pathophysiology of each one of these diseases. Of note, because of the ongoing description of novel mutations associated with these disorders, and as suggested elsewhere [11], for the sake of simplicity, we will avoid naming these entities by a numerical subtype, except for the major susceptibility genes (i.e., LQT1, LQT2, and LQT3; SQT1; BrS1; and CPVT1).
2. Cardiac Electrical Physiology: Role of Ion Channels and Receptors
Cardiac cells are excitable cells with the ability of generating and propagating an action potential (AP), an electrical signal that will translate into cardiomyocyte contraction. The cardiac AP is generated by a set of ion movements across the cell membrane that take the cell from the resting state to an activated state (by depolarization) and back to the resting membrane potential (repolarization). All phases are the consequence of a synergistic activation and inactivation of several voltage-dependent ion channels. In contractile myocytes (Figure 1A), APs are triggered by the acute entrance of sodium ions (Na+) inside the cell, resulting in an inward current (INa) that shifts the membrane potential from its resting state (−90 mV) to a depolarization state (+20 mV). This phase is followed by the efflux of potassium (K+) ions through an outward current named Ito, which initiates cell repolarization. Consecutively comes the plateau phase, a short period of constant membrane potential due to the balance between inward calcium (Ca2+) currents (ICaL) through the L-type voltage-dependent calcium channels (LTCC) and time-dependent delayed-rectifier outward K+ currents (mainly slow delayed-rectifier IKs and rapid delayed-rectifier IKr). At this point, the Ca2+ entry through LTCC triggers a much larger release of Ca2+ from the sarcoplasmic reticulum (SR) stores through the ryanodine receptor channel type 2 (RyR2), producing a systolic increase in intracellular Ca2+ needed for cell contraction (Figure 1B). Upon inactivation of the LTCC, the net outward K+ currents repolarize the cell and bring the membrane potential to its resting state. The balance between Ca2+ and K+ currents, therefore, determines the AP duration. The basal and acetylcholine-dependent inward-rectifier K+ currents (IK1 and IKACh) control final repolarization and determine the resting membrane potential (Figure 1A). Ca2+ is then extruded from the cell through the Na+/Ca2+ exchanger (NCX) type 1 and taken back into the SR through the SR Ca2+-ATPase type 2a (SERCA2a). This restores low intracellular Ca2+ levels, allowing cell relaxation during diastole (Figure 1B).
As shown in Figure 1, all these processes are tightly mediated by multiple ion channels and regulatory proteins. The slightest modification in the functioning of one of these players may modify the AP and/or the intracellular Ca2+ dynamics, potentially favoring an arrhythmogenic substrate. In the following lines we will discuss how the electrical substrate of cardiac channelopathies can develop depending on the affected gene and the effect of a particular mutation (loss- or gain-of-function) on the channels, receptors and regulators that participate in this process.
3. Long QT Syndrome
The LQTS is a hereditary electrical disorder characterized by an abnormally prolonged QT interval on the ECG (Figure 2A), which reflects a prolonged repolarization in cardiac cells secondary to changes in ion currents participating in the AP (see below). In LQTS, prolonged repolarization allows time for LTCC to recover from inactivation and facilitate new Ca2+ entry that can potentially generate a new depolarization (early afterdepolarization), a mechanism known to promote arrhythmogenesis (Figure 3A) [6]. As such, LQTS patients may present ventricular arrhythmias such as torsades de pointes and SCD.
3.1. Clinical Features
The estimated prevalence of the LQTS is thought to be as high as 1/2,000 individuals [12]. The diagnosis of LQTS is currently established in [13]: (1) the presence of a QT interval corrected for heart rate (QTc) ≥ 480 ms in repeated 12-lead ECGs; (2) the presence of a confirmed pathogenic LQTS mutation, irrespective to the QT duration; (3) a LQTS risk score, based on symptoms, family history and ECG findings [14], >3 in the absence of a secondary cause for QT prolongation.
Although most patients remain asymptomatic throughout life, up to 13% may experience SCD and 36% may experience syncope before age 40 if untreated [15]. SCD and syncope are the clinical manifestations of sustained and non-sustained ventricular arrhythmias, respectively. The mean age at the time of the first symptom is around 14 [13]. Symptoms in LQTS are characteristically triggered by different circumstances that emphasize the underlying electrical disorder (see below), such as exercise (in LQT1), emotions (in LQT2) or sleep (in LQT3) [16]. Several factors have been shown to be related to the occurrence of arrhythmias in LQTS patients, and therefore must be actively assessed in clinical practice: the presence of prior SCD or syncope, a QTc interval in any ECG longer than 500 ms despite treatment with beta-blockers, and female sex in LQT2 and LQT3 patients [13,15,17]. Certain genetic findings have also shown prognostic significance (see below).
First-line treatment of all LQTS patients at the moment of diagnosis comprises the implementation of certain lifestyle changes (like avoidance of QT-prolonging drugs and genotype-specific triggers for arrhythmias) and treatment with β-blockers [13]. Additionally, an implantable cardioverter defibrillator (ICD) is indicated in patients considered at high risk of fatal arrhythmias, such as in survivor of a previous cardiac arrest, those with syncope or ventricular tachycardias despite treatment with β-blockers, and those with LQT2 and QTc > 500 ms persistently [13]. Genetic testing, which provides conclusive results in up to 80% of patients, particularly directed to the three most prevalent LQTS-susceptible genes (KCNQ1, KCNH2, and SCN5A) is also recommended in all patients with diagnosis of LQTS [10].
3.2. Genetic Bases
As mentioned earlier, LQTS is characterized by prolonged AP duration, which can be consequence of an increase in inward currents (mainly INa and ICaL) or a decrease in outward K+ currents (mainly IKs, IKr, IK1, Figure 3A). Mutations in 20 different genes encoding direct or indirect mediators of these currents have been found in one or several families with LQTS. Table 1 summarizes the genes, their corresponding proteins, and the effect of the LQTS-associated mutations. Most of the described pathogenic variants are inherited in an autosomal dominant pattern, with the exception of those embraced in the Jervell and Lange–Nielsen syndrome (a rare syndrome combining extremely prolonged QT interval and congenital deafness associated to mutations in KCNQ1 and KCNE1) [13] and a reported family with a mutation in TRDN [18], with autosomal recessive inheritance pattern. Autosomal dominant forms LQTS include LQT types 1–13, of which LQT1, LQT2 and LQT3 represent 85% of all genetically-diagnosed cases [19].
LQT1, accounting for >40% of all LQTS, is caused by mutations in the KCNQ1 gene encoding KV7.1, the α-subunit of the voltage-dependent K+ channel that mediates the slow component of the delayed rectifying IKs current. LQT1-related mutations in this gene cause a loss-of-function of the channel and less IKs, explaining the prolonged AP duration and the QT interval on the ECG [20,21]. Since IKs currents respond to progressive adrenergic stimulation such as that present during exercise, LQT1 patients show a marked trend to present arrhythmia-related symptoms during sport practice [16]. The effect of different mutations over the final Kv7.1 function may have prognostic implications: mutations found in the cytoplasmic-loop transmembrane domains, involved in adrenergic channel regulation, are associated with a greater risk of arrhythmias triggered by exercise but not with those during rest or sleep [22,23].
LQT2 (representing 30% of all LQTS) is caused by mutations in the KCNH2 gene encoding KV11.1 (or hERG), the α-subunit of the voltage-dependent K+ channel mediator of the rapid component of the delayed rectifying IKr current. LQT2-related mutations entail a loss of function of KCNH2, in some instances by altering its traffic to the cell membrane, with a consequent reduction in IKr [24,25,26]. Since IKr plays an important role in brisk increases in heart rate, LQT2 patients are prone to present arrhythmia-related symptoms in stress or emotional circumstances [16]. Location of LQT2-related mutations may be relevant to predict a greater reduction of IKr and thus a more aggressive phenotype: as such, patients with mutations affecting the pore region (S5-loop-S6), a determinant structure of the channel, have been reported to have higher risk of suffering life-threatening cardiac events than patients with mutations in other locations [27,28].
Finally, LQT3 is caused by mutations in SCN5A encoding NaV1.5, the α-subunit of the voltage-dependent Na+ channel and mediator of the depolarizing INa current. Gain-of-function mutations in this gene, present in 10% of genetically-diagnosed LQTS patients, prolong AP duration by increasing late depolarizing currents [29]. In LQT3 patients, the defect in INa becomes more evident with slow heart rates, so it is common that these patients develop arrhythmia-related symptoms in circumstances of bradycardia and typically during sleep [16]. Although some particular mutations (NaV1.5-E1784K and NaV1.5-D1790G, also associated with Brugada phenotype) have been related to a better prognosis in LQT3 patients, at present there is not enough evidence supporting that the type and/or location of SCN5A mutations in LQT3 patients have effects on the final phenotype [30].
Mutations in other genes have also been reported in LQTS patients, mainly anecdotically, representing each of them <1% of global cases. As mentioned, all of them lead to a decrease in outward currents or an increase in inward currents directly or indirectly (Table 1), the effect in all cases being a prolongation of AP duration and repolarization:
- -
- Uncommon LQTS mutations causing a decrease in outward currents. Two genes encoding regulatory β-subunits of K+ channels have been associated with the LQTS: mutations in KCNE1, encoding minK, the β1-subunit of voltage-dependent K+ channels, have been reported to interfere with the traffic of KV7.1 and lead to a reduction of IKs current [31,32]; on the other hand, mutations in KCNE2, encoding MiRP1 (minK-related peptide 1), have been reported to impair KV11.1 kinetics (slower activation, faster deactivation and increased drug sensitivity), inducing a decreased IKr [33]. Among the inward-rectifying K+ channel family, two members have also been associated to LQTS, KCNJ2 and KCNJ5. KCNJ2 encodes Kir2.1, the inward rectifying K+ channel that mediates the IK1 current [34]. Loss-of-function mutations in this gene lead not only to LQTS and susceptibility to arrhythmias, but also to periodic paralysis and developmental abnormalities, a condition known as the Andersen–Tawil syndrome [35]. KCNJ5 encodes Kir3.4, the inward rectifying K+ channel mediator of the acetylcholine/adenosine-induced IK,Ach current. Loss-of-function mutations in this gene lead to LQTS by altering the traffic of the channel [36,37]. Finally, mutations in AKAP, a gene that encodes an auxiliary protein (AKAP9, A-kinase anchor protein-9) and not an ion channel, may also cause LQTS by reducing outward currents. AKAP9 is a scaffolding protein between KV7.1 and PKA. Mutations in AKAP have been shown to reduce the KV7.1/PKA interaction, resulting in a net decrease of IKs [38].
- -
- Uncommon LQTS mutations causing an increase in inward currents. Mutations in two different β-subunits regulators of the NaV1.5 channel have been found in families with LQTS: the β1 (encoded by the SCN1B gene) and the β4 (encoded by the SCN4B gene), inducing in both cases an increased INa [39,40]. Increased ICaL currents are seen in LQTS-associated mutations on the CACNA1C gene encoding CaV1.2, the α1C-subunit of the LTCC [41]. Specifically, mutations in this gene have also been associated with the Timothy syndrome, characterized by long QT and associated serious developmental and physical disorders such as autism or immune deficiencies [42]. This particular subtype (also known as LQT8) is considered the most severe variant of LQTS, with the highest mortality rate, generally caused by extracardiac complications [19]. Other genes encoding auxiliary proteins have been associated with LQTS. ANK2 encodes ankyrin-2, a protein in charge of the assembly of the Na+/K+ exchanger, the Na+/Ca2+ exchanger and the inositol triphosphate receptor (IP3R), among others. Loss-of-function mutations in ANK2 increase ICaL by decreasing the amount of Na+/Ca2+ exchanger in the membrane leading to an abnormal restoration of the initial ion state [43,44]. All three calmodulin-encoding genes (CALM1, CALM2, and CALM3) have also been linked to LQTS. Calmodulin is an essential intracellular Ca2+ sensor that acts as a signal-transducing protein and modulates CaV1.2 (and others). Mutations in one of these genes, even in heterozygosis (meaning that only 1/6 alleles is affected), is sufficient to lead to an early and severe form of LQTS with extreme long QTc interval secondary to an impaired CaV1.2 inactivation and increased ICaL currents [45,46,47,48,49]. Mutations in the SNTA1 gene has also been associated to LQTS [50]. This gene encodes α1-syntrophin, a scaffolding protein that associates NaV1.5 channels with the nitric oxide synthase-ATPase plasma membrane Ca2+-transporting four-protein complex (NOS-PMCA4b). SNTA1 mutations disrupt this interaction and increase late INa currents [51,52]. Finally, mutations in the TRDN gene that encodes the triadin, a regulator of RyR, have been associated with the LQTS by putatively decreasing CaV1.2 inactivation and increasing ICaL currents [18].
Other rare mutations have been related to the LQTS, with less established mechanisms. Mutations in CAV3 may lead to LQTS by increasing INa or by decreasing IK1 currents. CAV3 encodes caveolin-3 (Cav3), an anchoring protein that regulates membrane expression of multiple ion channels. Mutations in CAV3 have been associated with loss of Kir2.1 (and a consequent decrease in IK1) [53] or a gain of NaV1.5 (increasing late INa currents) [54], whereas other groups have found no association between these previously described CAV3 mutations and LQTS [55]. A loss-of-function mutation in the TRPM4 gene, encoding the transient receptor potential melastatin 4 (TRPM4), has been found in one family with LQTS history, with no established mechanism to explain such association [56]. Lastly, mutations in the RYR2 gene encoding the RyR type 2 have also been reported in LQTS patients. Considering the strong implication of this gene in CPVT (see below) and the potential phenotypical overlap between both diseases, there is some controversy whether RYR2 is a true LQTS-susceptible gene [57,58] or a CPVT gene putatively diagnosed as LQTS [59].
4. Short QT Syndrome
The short QT syndrome was first described in 2000 [60] and since then around 200 cases from 50 different families have been reported world-wide [8]. This highly-lethal rare disease is characterized by the presence of an abnormally short QTc interval on the ECG (Figure 2B) and a predisposition to develop life-threatening ventricular arrhythmias [60,61]. The short QT interval on the ECG reflects a short AP duration at the cellular level, caused by faster repolarization rates (and shorter refractory periods) that typically are heterogeneous along the cardiac tissue, favouring the occurrence of ventricular arrhythmias (Figure 3B) [6].
4.1. Clinical Features
The SQTS is diagnosed in the presence of a QTc ≤ 340 ms [13]. The diagnosis should also be considered in the presence of a QTc ≤ 360 ms together with the presence of a confirmed pathogenic mutation, a family history of SQTS or SCD at age <40, or a documented VT/VF episode [13]. Initial descriptions pointed to very aggressive clinical manifestations in SQTS patients, with up to 60% experiencing syncope and 31% SCD in follow-up, frequently at young age [62,63]. More recent publications highlight a great variability of phenotypes, with more than half patients remaining asymptomatic despite showing continuous short QTc on the ECG [64]. Other arrhythmias, such as atrial fibrillation, are common in SQTS patients [65].
Considering the low number of cases described to date, the optimal strategy for prevention of SCD is unclear [13]. Little is known about patient risk stratification [65]: neither the length of the QTc interval, the history of previous syncope, the gender or the genetic background have been shown to be predictors of the occurrence of arrhythmias [13]. Current guidelines recommend ICD implant in patients with SQTS who are survivors of a previous cardiac arrest and in those with documented sustained VT. Quinidine or sotalol (which prolong cardiac repolarization and therefore QT interval primarily due to inhibition of repolarizing currents) may be used in asymptomatic SQTS patients or in those with contraindications for ICD [10,13]. Given the limited yield of genetic testing in SQTS, recommendations in this regard are feeble [10,13].
4.2. Genetic Bases
Although SQTS is considered an inherited channelopathy whose clinical manifestations are linked to mutations in cardiac ion channels, only in 15% of cases is a responsible mutation found [8,65]. To date, six genes have been described in families with SQTS (Table 2). All associated mutations have been reported in genes exclusively encoding ion channel subunits and found to be inherited in an autosomal dominant pattern with high penetrance. The consequence of all SQTS-related mutations is a shortening of the cardiac AP that can be due either to an increase in repolarizing outward currents or a decrease in inward currents (Figure 3B).
KCNH2 (associated with the so-called SQT1) seems to be the major SQTS-susceptible gene described so far [66]. Gain-of-function mutations in KCNH2 encoding KV11.1 lead to an increase in IKr by slowing the inactivation of the channel [67,68,69]. Gain-of-function mutations in the KCNQ1 gene have also been related to SQTS, inducing an increase in IKs either by accelerating the activation kinetics of KV7.1 [70] or by decreasing its inactivation due to an impaired regulation of minK, the β1-subunit of voltage-dependent K+ channels [71]. Finally, mutations in the KCNJ2 gene have been linked to a shortening of the AP by shifting the voltage-dependency and increasing peak currents through Kir2.1 [72,73] or by enhancing the membrane expression of the channel [74].
Several loss-of-function mutations in genes encoding different subunits of the LTCC have been associated with the SQTS. Mutations in CACNA1C, encoding CaV1.2, the α1C-subunit of the voltage-dependent LTCC, have been identified to shorten the AP by reducing the α1C-subunit traffic to the membrane [75]. On the other hand, a mutation in CACNB2b, encoding the β2-subunit of the LTCC, was shown to reduce dramatically ICaL without affecting traffic [75]. Both mutations in CACNA1C and CACNB2b, by decreasing inward currents at early phases of cell repolarization, also induce transmural and epicardial dispersion of repolarization, leading to a combined phenotype of BrS (with the characteristic ST elevation on the ECG) and SQTS (with short QTc interval) [75]. Finally, a mutation in CACNA2D1 gene, encoding the α2δ1-subunit of LTCC, seemed to be related to a case of SQTS. Although the proposed mechanism was a decrease in ICaL currents through CaV1.2 [76], genotype-positive relatives of the index case failed to display ECG features of SQTS, so at present it is not clear whether CACNA2D1 is a real SQTS-causal gene or not [8].
5. Brugada Syndrome
First described in 1992, the BrS is characterized by a typical ECG pattern (coved-type ST-segment elevation in right precordial leads, Figure 2C) and a susceptibility to develop ventricular arrhythmias and SCD [77]. From experimental studies we now know that the characteristic ECG pattern responds to an imbalance between inward and outward currents in early phases of repolarization (phase 1 of the AP), created either by a decrease in inward (mainly INa, and less importantly ICaL) or an increase in outward (Ito, IKs, or IKr) currents (Figure 3C) [78]. This imbalance sets also the basis for the development of ventricular arrhythmias by a mechanism of phase 2 re-entry, initiated when voltages in phase 1 reach approximately −30 mV, potentially leading to a premature repolarization that could be affecting only a subset of cells, and not others. The heterogeneous repolarization across the cardiac tissue may facilitate then re-entrant arrhythmias [78]. However, the arrhythmogenic mechanism underlying the BrS has been and still is under extensive debate, with other authors considering the disease a primary depolarization disorder [79]. Taking into account the amount of genes associated with the BrS, with different outcomes in the AP (see below), one could speculate that the BrS is a heterogeneous disease potentially explained by more than one arrhythmogenic mechanism.
5.1. Clinical Features
The BrS is currently diagnosed in patients with a characteristic pattern of ST-segment elevation (defined as coved-type or type 1) ≥2 mm in ≥1 leads from V1 to V2 positioned in the second, third, or fourth intercostal space [13]. The ECG may be observed either spontaneously or after being unmasked by a provocative drug test with a sodium-channel blocker (Figure 2C) [13]. Sodium-channel blockers are antiarrhythmic agents that, by inhibiting INa, increase the imbalance between inward and outward currents in early phases of the AP, and therefore may exacerbate the phenotypic expression of the BrS [80]. Of note, the BrS must be distinguished from other conditions, globally known as Brugada phenocopies, that may induce a Brugada-like ECG pattern in patients without the syndrome, among which metabolic and electrolyte disturbances, mechanical compression of the right ventricle, and acute ischemic or pericardial diseases are the most common [81].
The prevalence of the BrS is highly variable in different geographical areas, but it has been estimated in 5/10,000 inhabitants, although it could be higher given that many patients present concealed forms of the disease [78]. According to previous reports, the BrS could be responsible for 4–12% of all SCD and for up to 20% of SCD in subjects without structural heart disease [82]. Patients with BrS usually remain asymptomatic, but syncope or SCD due to ventricular arrhythmias have been described in 17–42% of diagnosed individuals [83,84]. Age at presentation is around the third-fourth decade of life [80]. For SCN5A-mutation carriers (the gene most commonly affected in BrS patients, see below), like in the case of LQT3 patients, symptoms typically appear during rest or sleep [80]. Gender differences have been reported, with the BrS being 8–10 times more prevalent in men, in whom the syndrome entails a worse prognosis [85]. Besides gender, other factors have been associated with an increased risk of life-threatening arrhythmias in patients with BrS. After extensive debate for more than a decade, referral authors have come to agree that a history of previous syncope, a spontaneous (not-induced) type-1 ECG and the inducibility of ventricular arrhythmias during programmed electrical stimulation (a catheter-based invasive test to test arrhythmia susceptibility) are all predictors of future SCD in BrS patients [84,86].
To date, ICD continues to be the only proven effective treatment to prevent SCD in BrS patients. Current guidelines recommend ICD implantation in all patients with previous arrhythmia-related symptoms [10,13]. ICD should also be considered in asymptomatic patients who develop ventricular arrhythmias during programmed ventricular stimulation [10,13]. Quinidine, a drug with blocking effects on the Ito current (and thus the potential to decrease the ionic imbalance in phase 1 of the AP), has been proven useful in patients with contraindication for ICD, multiple ICD shocks, arrhythmic storms or in high-risk children [10,13,87]. Recently, catheter ablation of the underlying substrate in regions that are supposed to display the highest ionic imbalance (such as the epicardium of the right ventricular outflow tract) has become available in specialized centers and can be offered to high-risk patients with multiple arrhythmic episodes. Interestingly, this technique not only reduces the incidence of arrhythmias but also eliminates the ECG pattern in BrS patients [88].
5.2. Genetic Bases
The BrS is a genetically heterogeneous disease, with only 20–30% of all diagnosed patients having a known causal mutation and most associated genes having been described in anecdotic cases. This manifests a big gap of knowledge in the genetics of this disease [89]. Therefore, genetic testing is modestly recommended in BrS patients [10]. In general, all BrS-susceptible genes are inherited following an autosomal-dominant pattern, with the exception of KCNE5, which presents an X-linked inheritance. Nevertheless, considering the low disease penetrance observed in some families, some authors suggest that some cases of the syndrome could be associated with a much more complex pattern of inheritance [90].
To date, mutations in 25 different genes have been linked to the BrS, 18 of which encoding ion channel subunits and 7 encoding regulatory proteins (Table 3). With some exceptions, the two main molecular mechanisms that have been proposed to explain the BrS pattern are either a decrease in the INa currents or an increase in the Ito currents during early repolarization (Figure 3D). The major BrS-susceptible gene is SCN5A (BrS1), which encodes the α-subunit of the Na+ channel, NaV1.2, and is responsible of 25% of all genetically-diagnosed patients [91]. More than 300 different mutations in SCN5A have been associated with the disease, most leading to a loss of function either by impairing traffic or by modifying the channels’ gating properties [92]. The exact molecular mechanism depends on the location of the mutation within the channel, which can also determine the clinical severity of the disease [93]. Two different groups have demonstrated that, among BrS patients, those with mutations leading to a truncated protein, presumably causing greater INa decrease, have more severe phenotypes [93,94]. Mutations affecting any other regulatory subunit of the Na+ channel are also candidates for BrS. Indeed, mutations in genes encoding 3 β-subunits that regulate NaV1.2 traffic and function (SCN1B, SCN2B and SCN3B) and consequently reduce INa currents have been described in BrS patients [95,96,97,98]. The SCN10A gene, encoding NaV1.8, the neuronal Na+ channel also present in the intracardiac terminal neurons, has also been linked to the BrS [99]. Some authors have proposed this as a major-susceptibility gene, given that in some series SCN10A mutations have been found in up to 10% of all BrS patients [100]. However other authors have failed to reproduce these findings [101].
Reduction of inward currents other than INa could potentially induce the Brugada pattern, by creating a similar imbalance in early phases of repolarization. As such, reduced ICaL currents, secondary to loss-of-function mutations affecting CaV1.2 or the regulatory subunits β2 and α2δ1 (encoded by CACNA1C, CACNB2b and CACNA2D1 respectively), have also been reported as causative of BrS [75,102,103]. Interestingly, because ICaL also contributes to later phases of repolarization, mutations in these genes have also been associated with a reduction in the AP duration and a short QT interval on the ECG, leading to a combined phenotype of BrS and SQTS [75,102,103].
Mutations in several subunits that mediate the Ito currents have also been associated with the BrS. Gain-of-function mutations in KCND3, which encodes KV4.3, the α-subunit of the voltage-dependent K+ channel mediator of the transient Ito current, induce BrS by direct increase in Ito currents [104]. Similarly, gain-of-function mutations in the KCNE3 gene, encoding the minK-related peptide 2, the β-subunit that interacts with KV4.3 and regulates its current density, lead also to BrS [105,106,107]. Other sporadic mutations indirectly affecting Ito have also been described in BrS patients. A mutation in the KCNAB2 gene encoding the β2-subunit that regulates KV4.3 traffic to the membrane was found to increase Ito currents by enhancing the membrane expression of the channel and induce BrS [108]. Mutations in KCND2, which encodes KV4.2, another contributor to the Ito current, have also been found in BrS patients, who show a 50%-increased peak in Ito [109]. Mutations in the KCNE5 gene, encoding the minK-related peptide 4, a β-subunit that regulates KV7.1, increase currents through KV4.3 without affecting KV7.1, suggesting that minK-related peptide 4 is also a modulator of the Ito current and a putative BrS-susceptible gene [110].
By a mechanism similar to that proposed for mutations causing an increase in Ito currents, mutations inducing increases in other repolarizing currents may predispose to BrS. Increases in the IK-ATP currents have been seen in gain-of-function mutations of KCNJ8 and ABCC9, encoding respectively Kir6.1 and its modulator SUR2, and explain some BrS cases [111,112,113]. Similarly, several mutations in KCNH2 that affect the N or C-terminal tails of KV11.1, the hERG channel, have shown to result in increased IKr and potentially induce BrS [68,114,115].
Less is known about the mechanisms of the mutations found in other genes encoding ion channels that have been found in BrS patients. Loss-of-function mutations in HCN4 gene, which encodes the pacemaker channel (hyperpolarization-activated, cyclic nucleotide-gated ion channel 4), mediator of the If current and responsible for the spontaneous activity of the sinoatrial node (SAN), have been reported in two different works [116,117]. Finally, mutations in the TRPM4 channel, which, like HCN4, seems to participate in the diastolic depolarization that gives rise to the AP in the SAN, have been found in BrS patients and proposed as related to the conduction disorders that may appear in the disease [118,119]. Strikingly, both gain-of-function and loss-of-function TRPM4 mutations have been described in BrS patients, reflecting that much more investigation needs to be done in this regard [120].
Among the seven genes that have been associated with the BrS that encode regulatory proteins and not ion channels, five of them interact with INa currents. FGF12 gene encodes the fibroblast growth homologous factor 12, a potent regulator of NaV1.5 traffic and function [121]. Mutations in this gene associated with BrS were found to strongly reduce Na+ but not Ca2+ currents [122]. Severe traffic defects of NaV1.5 with the consequent reduction in INa currents have also been reported in patients with mutations in the GPD1L gene, encoding the glycerol-3-phosphate dehydrogenase 1-like protein, and the SLMAP gene, encoding the sarcolemma associated protein [123,124,125]. A dominant negative mutant of MOG1 (encoded by the RANGRF gene) was described to also decrease NaV1.5 traffic to the membrane in BrS patients [126]. Nevertheless, the same mutation was found in non-affected relatives and complete knock-down of MOG-1 was tolerated without any sign of disease, questioning whether this is a true BrS-susceptible gene [127,128]. Moreover, mutations that affect the expression of the desmosomal protein plakophillin-2 (encoded by PKP2) also have been shown to reduce the number of NaV1.5 channels at the intercalated disc, possibly explaining the molecular substrate for BrS in these patients [129]. The last two regulatory proteins associated with BrS interact with Ito currents. SEMA3A gene encodes for semaphorin-3A, a protein that binds to KV4.3 and reduces peak current densities without perturbing cell surface expression. In the context of BrS, loss-of-function mutations of SEMA3A lead to an increase in Ito currents [130]. Finally, a recent genome-wide association study (GWAS) identified a strong association between a region near the HEY2 gene and BrS [131]. This gene encodes a transcription factor that regulates electric patterning across the ventricular wall and affects cardiac ion channel gene expression. Although no BrS-associated mutation has been described so far, genome-wide co-expression analysis postulated KCNIP2, the β-subunit regulator of the Ito current, as one of the regulated genes by HEY2 and potentially associated with BrS [132].
6. Catecholaminergic Polymorphic Ventricular Tachycardia
Although the earliest clinical reference goes back to 1975 [133], the first series of catecholamine-induced tachycardia or CPVT was not described until 20 years later [134]. CPVT, although not a pure channelopathy, was included among the primary electrical disorders much later [59]. This rare but extremely severe disease is characterized by the presence of a normal ECG at baseline but the predisposition to adrenergic-induced ventricular tachycardias that typically are bidirectional or polymorphic (Figure 2D) [135] and can induce SCD. Mechanistic studies have elucidated the molecular basis of CPVT, which relies on an abnormal release of Ca2+ from the SR in response to adrenergic stimulation. Excess Ca2+ is handled by the cell membrane Na+/Ca2+-exchanger, which transports three Na+ ions into the cell per single Ca2+ ion extruded, creating a net depolarizing current that can lead to arrhythmogenesis by a mechanism called delayed afterdepolarizations (Figure 3D) [136,137].
6.1. Clinical Features
According to the 2015 European Society of Cardiology guidelines, CPVT can be currently diagnosed (class I recommendation): (1) in the presence of a structurally normal heart, normal ECG, and exercise- or emotion-induced bidirectional or polymorphic ventricular tachycardia; (2) in patients who are carriers of a pathogenic mutation in RYR2 or CASQ2 genes [13]. However, it is important to note that mutations in other genes have recently been identified in patients with clinical features of CPVT (see below). With an estimated prevalence of 1 in 10,000 [138], CPVT is an inherited disorder with both autosomal dominant and recessive patterns of transmission. Although an incomplete penetrance has been reported (around 15% of all patients are silent carriers), CPVT is usually an aggressive disorder, with symptoms likely appearing during childhood and a high incidence of cardiac events in follow-up (around 80% of untreated patients will experiment an arrhythmia, and up to 30% SCD) [139]. Due to the particular mechanism of this condition, arrhythmia-related symptoms such as syncope typically occur in adrenergically mediated circumstances such as exercise or emotional stress [135]. Male-sex seems to be a risk factor in patients carrying mutations in RYR2 [135]. No other predictors of risk have been identified to date [13].
Besides avoidance of competitive sports or strenuous exercise and stressful environments, all CPVT patients should receive treatment with β-blockers upon diagnosis to prevent SCD [13]. However, up to 45% of patients may still experience symptoms under treatment. In these cases, an ICD is recommended although additional flecainide may be useful [10,13,140,141,142]. There is still some debate regarding the mechanism by which flecainide reduces the incidence of arrhythmias, with some authors postulating that flecainide modulates RyR2 thus reducing Ca2+ leak, and some others supporting that it blocks NaV1.2, raising the threshold for delayed afterdepolarizations [143,144]. For survivors of a previous cardiac arrest, an ICD is warranted [13]. Genetic testing in CPVT might be reasonable, although its diagnostic yield is modest, with approximately 60% having a recognized causative mutation [10].
6.2. Genetic Bases
CPVT is the only arrhythmogenic disorder that does not affect directly the cardiac AP. As mentioned before, the molecular mechanism leading to delayed depolarizations and arrhythmias is an impaired regulation of the SR Ca2+ handling. All the described CPVT-related genes thus far affect in one way or another RyR2, the channel responsible of Ca2+ release from the SR.
Although seven genes have been associated with CPVT (Table 4), 60% of all patients carry a mutation in RYR2 gene [145]. Mutations in this protein, inherited in an autosomal dominant pattern, concentrate in three regions: surrounding the Ca2+-binding site, in the transmembrane segments and in the FKBP12.6-binding domain, a protein that regulates RyR2 gating [146]. In all cases, RYR2 mutations impair normal RyR2 functioning, causing opening and Ca2+ leak during diastole [137]. Similarly, mutations in the CASQ2 gene, present in 5% of all genetically-diagnosed CPVT patients, also lead to an abnormal Ca2+ leak through RyR2 by decreasing the expression of its encoded protein, calsequestrin 2 [147]. Calsequestrin 2 is the main Ca2+-buffering protein in the SR. If its expression is reduced, free-Ca2+ in the SR increases leading to a RyR2 leak [148]. These mutations, inherited in a dominant or recessive manner, are associated to a higher rate of sudden death [149,150,151]. The physical link between RyR2 and calsequestrin 2 is found in triadin, a protein encoded by TRDN [152]. Mutations in this protein, recessively inherited, result in its complete degradation and SR leak due to impaired regulation of the complex calsequestrin-triadin-RyR2 [153].
Additionally, mutations in the three calmodulin-encoding genes have also been associated to CPVT. In this context, CaM is a regulator of Ca2+ handling that avoids Ca2+ leak during diastole. CALM1 mutations disrupt the interaction between CaM and the RyR2 favoring the Ca2+ leak [154]. CALM2 and CALM3 mutations do not disrupt the interaction but lower the CaM-Ca2+-binding affinity, prompting spontaneous Ca2+ waves and spark activity, phenotype that mimics an increase in RyR2 function [46,155]. Nevertheless, mutations in CALM2 have been found in patients with a double diagnosis for LQTS and CPVT, and considering the overlapping features of both diseases, one should be cautious in establishing CALM2 as a unique CPVT-susceptible gene.
Recently, the TECLR gene was also identified as a CPVT-susceptible gene. Recessively-inherited mutations in the gene encoding for the trans-2,3-enoyl-CoA reductase-like protein were found in three different non-related families [156]. This protein, strongly expressed in the heart and localized in the SR, is thought to participate in the synthesis of fatty acids. Mutations in this gene correlated with a reduction in RyR2 and calsequestrin-2 protein levels and a consequent impairment in calcium handling
Mutations in ANK2 and KCNJ2 genes have been reported in patients with exercise-induced bi-directional ventricular tachycardia, thus mimicking the clinical phenotype of CPVT. However, it is now believed that these mutations, previously linked to the LQT4 and LQT7-Andersen–Tawil syndrome, respectively, are clinical phenocopies of CPVT (Priori ESC guidelines) and should be included in the differential diagnosis of this entity. In the case of ANK2 mutations, the clinical phenotype might be characterized by a normal ECG at rest and polymorphic ventricular tachycardias induced by exercise, with an overall better outcome than with the CPVT [157]. These disorders have recently been proposed as a distinct entity known as the Ankyrin B syndrome [158]. KCNJ2 mutations responsible for the Andersen–Tawil syndrome typically manifest with the phenotypic triad of ventricular arrhythmias, commonly induced by exercise, periodic paralysis, and facial and limb dysmorphism, but these latter features might be variably present, which, together with a mild QTc prolongation, may very much mimic CPVT [159]. However, distinction with true CPVT is important because patients with Andersen–Tawil syndrome show a much more benign course [35,159].
7. Conclusions
The normal electric function of the heart is exquisitely orchestrated by macromolecular complexes with ion channels in its core. These complexes include proteins regulating functions from transcription to function or degradation of such channels. Any disruption of this gear assembly may generate electrical instability and a substrate capable of inducing ventricular arrhythmias and SCD [160]. Therefore, it is extremely important to gain knowledge on the electrophysiological mechanisms that control ion channel functions. At present, there are still many unanswered questions, such as why mutations in the same gene and with the same general output can cause different arrhythmogenic disorders, or why patients carrying the same mutation may present a diverse phenotype, from remaining asymptomatic throughout life to experiencing SCD at an early age. The most probable explanation to this puzzle is that, apart from the identified mutations, we are missing modifying factors that modulate the outcome in a mutation- and patient-specific manner. Understanding these diverse mechanisms may guide us to design directed therapies for inherited arrhythmogenic syndromes in the future.
Acknowledgments
This study was supported by research grants from the Instituto de Salud Carlos III (Spanish Ministry of Economy, Industry and Competitiveness)—FEDER (Fondo Europeo de Desarrollo Regional) PI13/01830, awarded to Begoña Benito, and by a grant from the Daniel Bravo Foundation awarded to Anna Garcia-Elias. Authors also want to thank the Juan de la Cierva program from the Spanish Ministry of Economy, Industry and Competitiveness.
Authors Contribution
Anna Garcia-Elias, Begoña Benito designed and wrote the manuscript. Both read and approved the final version of the work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zipes, D.P.; Wellens, H.J.J. Clinical Cardiology: New Frontiers Sudden Cardiac Death. Circulation 1998, 98, 2334–2351. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.I.; Chugh, S.; DiMarco, J.; Albert, C.; Anderson, M.; Bonow, R.; Buxton, A.; Chen, P.-S.; Estes, M.; Jouven, X.; et al. Sudden Cardiac Death Prediction and Prevention Report from a National Heart, Lung, and Blood Institute and Heart Rhythm Society Workshop. Circulation 2010, 122, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Van Der Werf, C.; van Langen, I.M.; Wilde, A.A.M. Sudden death in the young: What do we know about it and how to prevent? Circ. Arrhythm. Electrophysiol. 2010, 3, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Eckart, R.E.; Shry, E.A.; Burke, A.P.; McNear, J.A.; Appel, D.A.; Castillo-Rojas, L.M.; Avedissian, L.; Pearse, L.A.; Potter, R.N.; Tremaine, L.; et al. Sudden death in young adults: An autopsy-based series of a population undergoing active surveillance. J. Am. Coll. Cardiol. 2011, 58, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Puranik, R.; Chow, C.K.; Duflou, J.A.; Kilborn, M.J.; McGuire, M.A. Sudden death in the young. Heart Rhythm 2005, 2, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Benito, B. Sudden death in patients without structural heart disease. Rev. Española Cardiol. 2013, 13, 14–23. [Google Scholar]
- Cheng, Y.; Lin, X.; Ji, C.; Chen, X.; Liu, L.; Tang, K.; Wu, S. Role of Early Repolarization Pattern in Increasing Risk of Death. J. Am. Heart Assoc. 2016, 5, e003375. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Underwood, K.; Nevelev, D.; Kofman, S.; Priori, S.G. The new kids on the block of arrhythmogenic disorders: Short QT syndrome and early repolarization. J. Cardiovasc. Electrophysiol. 2017, 28, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Tester, D.J.; Ackerman, M.J. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation 2011, 123, 1021–1037. [Google Scholar] [CrossRef] [PubMed]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Gillis, A.M.; Bryant, W.J.; Hlatky, M.A.; Callans, D.J.; Granger, C.B.; Curtis, A.B.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: Executive Summary. Heart Rhythm 2017, 24390. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Ackerman, M.J. Exercise extreme caution when calling rare genetic variants novel arrhythmia syndrome susceptibility mutations. Heart Rhythm 2010, 7, 1883–1885. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; Mosca, F.; et al. Prevalence of the congenital long-qt syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Moss, A.J.; Vincent, G.M.; Crampton, R.S. Current Perspectives Diagnostic Criteria for the Long QT Syndrome An Update. Circulation 1993, 782–785. [Google Scholar] [CrossRef]
- Priori, S.G.; Schwartz, P.J.; Napolitano, C.; Bloise, R.; Ronchetti, E.; Grillo, M.; Vicentini, A.; Spazzolini, C.; Nastoli, J.; Bottelli, G.; et al. Risk stratification in the long-QT syndrome. N. Engl. J. Med. 2003, 348, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J. Genotype-phenotype correlation in the long-QT syndrome. Circulation 2001, 103, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Moss, A.J.; Peterson, D.R.; McNitt, S.; Zareba, W.; Andrews, M.L.; Robinson, J.L.; Locati, E.H.; Ackerman, M.J.; Benhorin, J.; et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital Long-QT syndrome. Circulation 2008, 117, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Altmann, H.M.; Tester, D.J.; Will, M.L.; Middha, S.; Evans, J.M.; Eckloff, B.W.; Ackerman, M.J. Homozygous/compound heterozygous triadin mutations associated with autosomal-recessive long-QT syndrome and pediatric sudden cardiac arrest: Elucidation of the triadin knockout syndrome. Circulation 2015, 131, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Lahrouchi, N.; Priori, S.G. Genetics of Sudden Cardiac Death. Circ. Res. 2015, 116, 1919–1936. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Oliveras, A.; Bartolucci, C.; Muñoz, C.; de la Cruz, A.; Peraza, D.A.; Gimeno, J.R.; Martín-Martínez, M.; Severi, S.; Felipe, A.; et al. D242N, a KV7.1 LQTS mutation uncovers a key residue for IKsvoltage dependence. J. Mol. Cell. Cardiol. 2017, 110, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Thottathil, P.; Lopes, C.M.; Moss, A.J.; McNitt, S.; Jin, O.U.; Robinson, J.L.; Zareba, W.; Ackerman, M.J.; Kaufman, E.S.; et al. Trigger-specific ion-channel mechanisms, risk factors, and response to therapy in type 1 long QT syndrome. Heart Rhythm 2012, 9, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Shimizu, W.; Wilde, A.A.M.; Towbin, J.A.; Zareba, W.; Robinson, J.L.; Qi, M.; Vincent, G.M.; Ackerman, M.J.; Kaufman, E.S.; et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation 2007, 115, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincent, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef]
- Furutani, M.; Trudeau, M.; Hagiwara, N.; Seki, A.; Gong, Q.; Zhou, Z.; Imamura, S.; Nagashima, H.; Kasanuki, H.; Takao, A.; et al. Novel Mechanism Associated With an Inherited Cardiac Arrhythmia. Circulation 1999, 99, 2290–2295. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Anderson, C.L.; Burgess, D.E.; Elayi, C.S.; January, C.T.; Delisle, B.P. Molecular pathogenesis of long QT syndrome type 2. J. Arrhythmia 2016, 32, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Zareba, W.; Kaufman, E.S.; Gartman, E.; Peterson, J.; Benhorin, D.R.; Towbin, J.A.; Keating, M.T.; Priori, S.G.; Schwartz, P.J.; et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation 2002, 105, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, W.; Moss, A.J.; Wilde, A.A.M.; Towbin, J.A.; Ackerman, M.J.; January, C.T.; Tester, D.J.; Zareba, W.; Robinson, J.L.; Qi, M.; et al. Genotype-Phenotype Aspects of Type 2 Long QT Syndrome. J. Am. Coll. Cardiol. 2009, 54, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shen, J.; Splawski, I.; Atkinson, D.; Li, Z.; Robinson, J.L.; Moss, A.J.; Towbin, J.A.; Keating, M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995, 80, 805–811. [Google Scholar] [CrossRef]
- Wilde, A.; Moss, A.; Kaufman, E.; Shimizu, W.; Peterson, D.; Benhorin, J.; Lopes, C.; Towbin, J.; Spazzolini, C.; Crotti, L.; et al. Clinical Aspects of Type 3 Long-QT Syndrome: An International Multicenter Study. Circulation 2016, 134, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Krumerman, A.; Gao, X.; Bian, J.; Melman, Y.F.; Kagan, A.; Mcdonald, T.V.; Gao, X.; Bian, J.; Melman, F.; Kagan, A.; et al. An LQT mutant minK alters KvLQT1 trafficking Andrew. Am. J. Physiol. Cell Physiol. 2004, 10461, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Harmer, S.C.; Tinker, A. The role of abnormal trafficking of KCNE1 in long QT syndrome 5. Biochem. Soc. Trans. 2007, 35, 1074–1076. [Google Scholar] [CrossRef] [PubMed]
- Abbott, G.W.; Sesti, F.; Splawski, I.; Buck, M.E.; Lehmann, M.H.; Timothy, K.W.; Keating, M.T.; Goldstein, S.A. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999, 97, 175–187. [Google Scholar] [CrossRef]
- Fodstad, H.; Swan, H.; Auberson, M.; Gautschi, I.; Loffing, J.; Schild, L.; Kontula, K. Loss-of-function mutations of the K+channel gene KCNJ2 constitute a rare cause of long QT syndrome. J. Mol. Cell. Cardiol. 2004, 37, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Tawil, R.; Ptacek, L.J.; Pavlakis, S.G.; DeVivo, D.C.; Penn, A.S.; Özdemir, C.; Griggs, R.C. Andersen’s syndrome: Potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann. Neurol. 1994, 35, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Liang, B.; Liu, J.; Li, J.; Grunnet, M.; Olesen, S.P.; Rasmussen, H.B.; Ellinor, P.T.; Gao, L.; et al. Identification of a Kir3.4 Mutation in Congenital Long QT Syndrome. Am. J. Hum. Genet. 2010, 86, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, J.; Hong, L.; Liang, B.; Graff, C.; Yang, Y.; Christiansen, M.; Olesen, S.P.; Zhang, L.; Kanters, J.K. The phenotype characteristics of type 13 long QT syndrome with mutation in KCNJ5 (Kir3.4-G387R). Heart Rhythm 2013, 10, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Marquardt, M.L.; Tester, D.J.; Sampson, K.J.; Ackerman, M.J.; Kass, R.S. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 20990–20995. [Google Scholar] [CrossRef] [PubMed]
- Riuró, H.; Campuzano, O.; Arbelo, E.; Iglesias, A.; Batlle, M.; Pérez-Villa, F.; Brugada, J.; Pérez, G.J.; Scornik, F.S.; Brugada, R. A missense mutation in the sodium channel β1b subunit reveals SCN1B as a susceptibility gene underlying long QT syndrome. Heart Rhythm 2014, 11, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Medeiros-Domingo, A.; Kaku, T.; Tester, D.J.; Iturralde-Torres, P.; Itty, A.; Ye, B.; Valdivia, C.; Ueda, K.; Canizales-Quinteros, S.; Tusié-Luna, M.T.; et al. SCN4B-encoded sodium channel β4 subunit in congenital long-QT syndrome. Circulation 2007, 116, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Boczek, N.J.; Ye, D.; Miyake, C.Y.; De la Uz, C.M.; Allen, H.D.; Ackerman, M.J.; Kim, J.J. Novel long QT syndrome-associated missense mutation, L762F, in CACNA1C-encoded L-type calcium channel imparts a slower inactivation tau and increased sustained and window current. Int. J. Cardiol. 2016, 220, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Mohler, P.J.; Schott, J.-J.; Gramolini, A.O.; Dilly, K.W.; Guatimoisim, S.; duBell, W.H.; Song, L.-S.; Haurogne, K.; Kyndt, F.; Ali, M.E.; et al. Ankyrin-B mutations causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 2003, 421, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Swayne, L.A.; Murphy, N.P.; Asuri, S.; Chen, L.; Xu, X.; McIntosh, S.; Wang, C.; Lancione, P.J.; Roberts, J.D.; Kerr, C.; et al. Novel Variant in the ANK2 Membrane-Binding Domain Is Associated with Ankyrin-B Syndrome and Structural Heart Disease in a First Nations Population with a High Rate of Long QT Syndrome. Circ. Cardiovasc. Genet. 2017, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Johnson, C.N.; Graf, E.; de Ferrari, G.M.; Cuneo, B.F.; Ovadia, M.; Papagiannis, J.; Feldkamp, M.D.; Rathi, S.G.; Kunic, J.D.; et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 2013, 127, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Makita, N.; Yagihara, N.; Crotti, L.; Johnson, C.N.; Beckmann, B.M.; Roh, M.S.; Shigemizu, D.; Lichtner, P.; Ishikawa, T.; Aiba, T.; et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ. Cardiovasc. Genet. 2014, 7, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.J.; Boczek, N.J.; Etheridge, S.P.; Ackerman, M.J. CALM3 mutation associated with long QT syndrome. Heart Rhythm 2015, 12, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Boczek, N.J.; Gomez-Hurtado, N.; Ye, D.; Calvert, M.L.; Tester, D.J.; Kryshtal, D.O.; Hwang, H.S.; Johnson, C.N.; Chazin, W.J.; Loporcaro, C.G.; et al. Spectrum and prevalence of CALM1-, CALM2-, and CALM3-encoded calmodulin variants in long QT syndrome and functional characterization of a novel long QT syndrome-associated calmodulin missense variant, E141G. Circ. Cardiovasc. Genet. 2016, 9, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, M.; Sala, L.; Dreizehnter, L.; Crotti, L.; Sinnecker, D.; Mura, M.; Pane, L.S.; Altomare, C.; Torre, E.; Mostacciuolo, G.; et al. Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc. Res. 2017, 113, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.I.; Wang, C.; Thomas, M.J.; Pitt, G.S. α1-Syntrophin variant identified in drug-induced long QT syndrome increases late sodium current. PLoS ONE 2016, 11, e0152355. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Valdivia, C.; Medeiros-Domingo, A.; Tester, D.J.; Vatta, M.; Farrugia, G.; Ackerman, M.J.; Makielski, J.C. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc. Natl. Acad. Sci. USA 2008, 105, 9355–9360. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Ai, T.; Kim, J.J.; Mohapatra, B.; Xi, Y.; Li, Z.; Abbasi, S.; Purevjav, E.; Samani, K.; Ackerman, M.J.; et al. Alpha-1-syntrophin mutation and the long-QT syndrome: A disease of sodium channel disruption. Circ. Arrhythm. Electrophysiol. 2008, 1, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, R.; Vega, A.L.; Song, C.; Zhou, Q.; Tan, B.; Berger, S.; Makielski, J.C.; Eckhardt, L.L. The interaction of caveolin 3 protein with the potassium inward rectifier channel Kir2.1: Physiology and pathology related to long QT syndrome 9 (LQT9). J. Biol. Chem. 2013, 288, 17472–17480. [Google Scholar] [CrossRef] [PubMed]
- Vatta, M.; Ackerman, M.J.; Ye, B.; Makielski, J.C.; Ughanze, E.E.; Taylor, E.W.; Tester, D.J.; Balijepalli, R.C.; Foell, J.D.; Li, Z.; et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation 2006, 114, 2104–2112. [Google Scholar] [CrossRef] [PubMed]
- Hedley, P.L.; Kanters, J.K.; Dembic, M.; Jespersen, T.; Skibsbye, L.; Aidt, F.H.; Eschen, O.; Graff, C.; Behr, E.R.; Schlamowitz, S.; et al. The role of CAV3 in long-QT syndrome: Clinical and functional assessment of a caveolin-3/Kv11.1 double heterozygote versus caveolin-3 single heterozygote. Circ. Cardiovasc. Genet. 2013, 6, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Hof, T.; Liu, H.; Salle, L.; Schott, J.; Ducreux, C.; Millat, G.; Chevalier, P.; Probst, V.; Guinamard, R.; Bouvagnet, P.; et al. TRPM4 non-selective cation channel variants in long QT syndrome. BMC Med. Genet. 2017, 18, 31. [Google Scholar] [CrossRef] [PubMed]
- Kauferstein, S.; Kiehne, N.; Erkapic, D.; Schmidt, J.; Hamm, C.W.; Bratzke, H.; Pitschner, H.F.; Kuniss, M.; Neumann, T. A novel mutation in the cardiac ryanodine receptor gene (RyR2) in a patient with an unequivocal LQTS. Int. J. Cardiol. 2011, 146, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, M.; Ohno, S.; Ichikawa, M.; Takayama, K.; Fukumoto, D.; Horie, M. Novel RYR2 mutations causative for long QT syndromes. Eur. Heart J. 2017, 38, 1–28. [Google Scholar] [CrossRef]
- Tester, D.J.; Kopplin, L.J.; Will, M.L.; Ackerman, M.J. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm 2005, 2, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Gussak, I.; Brugada, P.; Brugada, J.; Wright, R.S.; Kopecky, S.L.; Chaitman, B.R.; Bjerregaard, P. Idiopathic Short QT Interval: A New Clinical Syndrome? Cardiology 2000, 94, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Gaita, F.; Giustetto, C.; Bianchi, F.; Wolpert, C.; Schimpf, R.; Riccardi, R.; Grossi, S.; Richiardi, E.; Borggrefe, M. Short QT syndrome: A familial cause of sudden death. Circulation 2003, 108, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Giustetto, C.; Di Monte, F.; Wolpert, C.; Borggrefe, M.; Schimpf, R.; Sbragia, P.; Leone, G.; Maury, P.; Anttonen, O.; Haissaguerre, M.; et al. Short QT syndrome: Clinical findings and diagnostic-therapeutic implications. Eur. Heart J. 2006, 27, 2440–2447. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Kanthan, A.; Monteforte, N.; Memmi, M.; Bloise, R.; Novelli, V.; Miceli, C.; O’Rourke, S.; Borio, G.; Zienciuk-Krajka, A.; et al. Novel insight into the natural history of short QT syndrome. J. Am. Coll. Cardiol. 2014, 63, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Akdis, D.; Saguner, A.M.; Medeiros-Domingo, A.; Schaller, A.; Balmer, C.; Steffel, J.; Brunckhorst, C.; Duru, F. Multiple clinical profiles of families with the short QT syndrome. Europace 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Europace 2011, 13, 1077–1109. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Falgueras, A.; Sarquella-Brugada, G.; Brugada, J.; Brugada, R.; Campuzano, O. Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances. Biology 2017, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Brugada, R.; Hong, K.; Dumaine, R.; Cordeiro, J.; Gaita, F.; Borggrefe, M.; Menendez, T.M.; Brugada, J.; Pollevick, G.D.; Wolpert, C.; et al. Sudden Death Associated with Short-QT Syndrome Linked to Mutations in HERG. Circulation 2004, 109, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Sakaguchi, T.; Ashihara, T.; Ding, W.G.; Nagaoka, I.; Oka, Y.; Nakazawa, Y.; Yao, T.; Jo, H.; Ito, M.; et al. A novel KCNH2 mutation as a modifier for short QT interval. Int. J. Cardiol. 2009, 137, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Quan, X.Q.; Fromme, S.; Cox, R.H.; Zhang, P.; Zhang, L.; Guo, D.; Guo, J.; Patel, C.; Kowey, P.R.; et al. A novel mutation in the KCNH2 gene associated with short QT syndrome. J. Mol. Cell. Cardiol. 2011, 50, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Bellocq, C.; van Ginneken, A.C.G.; Bezzina, C.R.; Alders, M.; Escande, D.; Mannens, M.M.A.M.; Baró, I.; Wilde, A.A.M. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004, 109, 2394–2397. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Oliveras, A.; de La Cruz, A.; Bartolucci, C.; Muñoz, C.; Salar, E.; Gimeno, J.R.; Severi, S.; Comes, N.; Felipe, A.; et al. A new KCNQ1 mutation at the S5 segment that impairs its association with KCNE1 is responsible for short QT syndrome. Cardiovasc. Res. 2015, 107, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Pandit, S.V.; Rivolta, I.; Berenfeld, O.; Ronchetti, E.; Dhamoon, A.; Napolitano, C.; Anumonwo, J.; di Barletta, M.R.; Gudapakkam, S.; et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 2005, 96, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Makiyama, T.; Akao, M.; Ehara, E.; Ohno, S.; Iguchi, M.; Nishio, Y.; Sasaki, K.; Itoh, H.; Yokode, M.; et al. A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc. Res. 2012, 93, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, E.; Sicca, F.; Brignone, M.S.; D’adamo, M.C.; Napolitano, C.; Servettini, I.; Moro, F.; Ruan, Y.; Guglielmi, L.; Pieroni, S.; et al. Genetically induced dysfunctions of Kir2.1 channels: Implications for short QT3 syndrome and autism-epilepsy phenotype. Hum. Mol. Genet. 2014, 23, 4875–4886. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Pollevick, G.D.; Cordeiro, J.M.; Casis, O.; Sanguinetti, M.C.; Aizawa, Y.; Guerchicoff, A.; Pfeiffer, R.; Oliva, A.; Wollnik, B.; et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007, 115, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Templin, C.; Ghadri, J.R.; Rougier, J.S.; Baumer, A.; Kaplan, V.; Albesa, M.; Sticht, H.; Rauch, A.; Puleo, C.; Hu, D.; et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur. Heart J. 2011, 32, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef]
- Benito, B.; Brugada, J.; Brugada, R.; Brugada, P. Brugada Syndrome. Rev. Española Cardiol. 2009, 62, 1297–1315. [Google Scholar] [CrossRef]
- Park, D.S.; Cerrone, M.; Morley, G.; Vasquez, C.; Fowler, S.; Liu, N.; Bernstein, S.A.; Liu, F.Y.; Zhang, J.; Rogers, C.S.; et al. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J. Clin. Investig. 2015, 125, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Benito, B.; Brugada, R.; Brugada, J.; Brugada, P. Brugada Syndrome. Prog. Cardiovasc. Dis. 2008, 51, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Baranchuk, A.; Nguyen, T.; Ryu, M.H.; Femenía, F.; Zareba, W.; Wilde, A.A.M.; Shimizu, W.; Brugada, P.; Pérez-Riera, A.R. Brugada phenocopy: New terminology and proposed classification. Ann. Noninvasive Electrocardiol. 2012, 17, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Brugada, P.; Borggrefe, M.; Brugada, J.; Brugada, R.; Corrado, D.; Gussak, I.; LeMarec, H.; Nademanee, K.; Perez Riera, A.R.; et al. Brugada syndrome: Report of the second consensus conference. Heart Rhythm 2005, 2, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Gasparini, M.; Pappone, C.; Della Bella, P.; Giordano, U.; Bloise, R.; Giustetto, C.; de Nardis, R.; Grillo, M.; et al. Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation 2002, 105, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Brugada, R.; Antzelevitch, C.; Towbin, J.; Nademanee, K.; Brugada, P. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1to V3. Circulation 2002, 105, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Benito, B.; Sarkozy, A.; Mont, L.; Henkens, S.; Berruezo, A.; Tamborero, D.; Arzamendi, D.; Berne, P.; Brugada, R.; Brugada, P.; et al. Gender Differences in Clinical Manifestations of Brugada Syndrome. J. Am. Coll. Cardiol. 2008, 52, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Sroubek, J.; Probst, V.; Mazzanti, A.; Hevia, J.C.; Ohkubo, K.; Zorzi, A.; Kostopoulou, A.; Yin, X.; Napolitano, C.; Milan, D.J.; et al. Programmed Ventricular Stimulation for Risk Stratification in the Brugada Syndrome: A Pooled Analysis. Circulation 2016, 133, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013, 10, 1932–1963. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Pappone, C.; Berruezo, A.; Vicedomini, G.; Manguso, F.; Ciconte, G.; Giannelli, L.; Santinelli, V. Brugada Syndrome Phenotype Elimination by Epicardial Substrate Ablation. Circ. Arrhythm. Electrophysiol. 2015, 8, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Juang, J.M.J.; Horie, M. Genetics of Brugada syndrome. J. Arrhythmia 2016, 32, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Gasparini, M.; Pappone, C.; Della Bella, P.; Brignole, M.; Giordano, U.; Giovannini, T.; Menozzi, C.; Bloise, R.; et al. Clinical and Genetic Heterogeneity of Right Bundle Branch Block and ST-Segment Elevation Syndrome. Circulation 2000, 102, 2509–2515. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Kapplinger, J.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; Kamakura, S.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Meregalli, P.G.; Tan, H.L.; Probst, V.; Koopmann, T.T.; Tanck, M.W.; Bhuiyan, Z.A.; Sacher, F.; Kyndt, F.; Schott, J.J.; Albuisson, J.; et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 2009, 6, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Benito, B.; Campuzano, O.; Ishac, R.; Iglesias, A.; Junttila, M.; Michaud, J.; Brugada, J.; Brugada, P.; Brugada, R. Role of genetic testing in risk stratification of Brugada syndrome. Eur. Heart J. 2009, 30, 5121. [Google Scholar]
- Watanabe, H.; Koopmann, T.T.; Le Scouarnec, S.; Yang, T.; Ingram, C.R.; Schott, J.; Demolombe, S.; Probst, V.; Anselme, F.; Escande, D.; et al. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. Structure 2008, 118, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Riuró, H.; Beltran-Alvarez, P.; Tarradas, A.; Selga, E.; Campuzano, O.; Vergés, M.; Pagans, S.; Iglesias, A.; Brugada, J.; Brugada, P.; et al. A Missense Mutation in the Sodium Channel β2 Subunit Reveals SCN2B as a New Candidate Gene for Brugada Syndrome. Hum. Mutat. 2013, 34, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martinez, H.; Burashnikov, E.; Springer, M.; Wu, Y.; Varro, A.; Pfeiffer, R.; Koopmann, T.T.; Cordeiro, J.M.; Guerchicoff, A.; et al. A mutation in the β3 subunit of the cardiac sodium channel associated with brugada ECG phenotype. Circ. Cardiovasc. Genet. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Takahashi, N.; Ohno, S.; Sakurada, H.; Nakamura, K.; On, Y.K.; Park, J.E.; Makiyama, T.; Horie, M.; Arimura, T.; et al. Novel SCN3B mutation associated with brugada syndrome affects intracellular trafficking and function of Nav1.5. Circ. J. 2013, 77, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Remme, C.A.; Schumacher, C.A.; Scicluna, B.P.; Wolswinkel, R.; de Jonge, B.; Bezzina, C.R.; Veldkamp, M.W. Functional NaV1.8 channels in intracardiac neurons: The link between SCN10A and cardiac electrophysiology. Circ. Res. 2012, 111, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martínez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A are responsible for a large fraction of cases of brugada syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Behr, E.R.; Savio-Galimberti, E.; Barc, J.; Holst, A.G.; Petropoulou, E.; Prins, B.P.; Jabbari, J.; Torchio, M.; Berthet, M.; Mizusawa, Y.; et al. Role of common and rare variants in SCN10A: Results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc. Res. 2015, 106, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Béziau, D.M.; Barc, J.; O’Hara, T.; Le Gloan, L.; Amarouch, M.Y.; Solnon, A.; Pavin, D.; Lecointe, S.; Bouillet, P.; Gourraud, J.B.; et al. Complex Brugada syndrome inheritance in a family harbouring compound SCN5A and CACNA1C mutations. Basic Res. Cardiol. 2014, 109, 446. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, M.; Ohno, S.; Wang, Q.; Shirayama, T.; Itoh, H.; Horie, M. . Nonsense-mediated mRNA decay due to a CACNA1C splicing mutation in a patient with Brugada syndrome. Heart Rhythm 2014, 11, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Ye, D.; Tester, D.J.; Crotti, L.; Mugione, A.; Nesterenko, V.V.; Albertson, R.M.; Antzelevitch, C.; Schwartz, P.J.; Ackerman, M.J. Transient Outward Current (Ito) Gain-of-Function Mutations in the Transient Outward Current (Ito) KCND3-Encoded Kv4.3 Potassium Channel and Brugada Syndrome. Heart Rhythm 2011, 8, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- You, T.; Mao, W.; Cai, B.; Li, F.; Xu, H. Two novel Brugada syndrome-associated mutations increase KV4.3 membrane expression and function. Int. J. Mol. Med. 2015, 36, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Radicke, S.; Cotella, D.; Graf, E.M.; Banse, U.; Jost, N.; Varró, A.; Tseng, G.N.; Ravens, U.; Wettwer, E. Functional modulation of the transient outward current Itoby KCNE β-subunits and regional distribution in human non-failing and failing hearts. Cardiovasc. Res. 2006, 71, 695–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delpón, E.; Cordeiro, J.M.; Núñez, L.; Thomsen, P.E.B.; Guerchicoff, A.; Pollevick, G.D.; Wu, Y.; Kanters, J.K.; Larsen, C.T.; Hofman-Bang, J.; et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ. Arrhythm. Electrophysiol. 2008, 1, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Portero, V.; Le Scouarnec, S.; Es-Salah-Lamoureux, Z.; Burel, S.; Gourraud, J.B.; Bonnaud, S.; Lindenbaum, P.; Simonet, F.; Violleau, J.; Baron, E.; et al. Dysfunction of the voltage-gated K+channel β2 subunit in a familial case of Brugada syndrome. J. Am. Heart Assoc. 2016, 5, e003122. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.J.; Adler, A.; Green, S.; Al-Zoughool, F.; Doroshenko, P.; Orr, N.; Uppal, S.; Healey, J.S.; Birnie, D.; Sanatani, S.; et al. Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J-wave syndrome associated with sudden cardiac death. Circ. Cardiovasc. Genet. 2014, 7, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Zankov, P.; Ding, W.G.; Itoh, H.; Makiyama, T.; Doi, T.; Shizuta, S.; Hattori, T.; Miyamoto, A.; Naiki, N.; et al. KCNE5 (KCNE1L) variants are novel modulators of brugada syndrome and idiopathic ventricular fibrillation. Circ. Arrhythm. Electrophysiol. 2011, 4, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Medeiros-domingo, A.; Tan, B.; Crotti, L.; Tester, D.J.; Eckhardt, L.; Cuoretti, A.; Kroboth, S.L.; Song, C.; Zhou, Q.; Kopp, D.; et al. Gain-of-Function Mutations, S422L, in the KCNJ8-Encoded Cardiac K-ATP Channel Kir6.1 as a Pathogenic Substrate for J Wave Syndromes. Heart Rhythm 2010, 7, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martínez, H.; Terzic, A.; Park, S.; Pfeiffer, R.; Burashnikov, E.; Wu, Y.; Borggrefe, M.; Veltmann, C.; Schimpf, R.; et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int. J. Cardiol. 2014, 171, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Barajas-Martínez, H.; Hu, D.; Ferrer, T.; Onetti, C.G.; Wu, Y.; Burashnikov, E.; Boyle, M.; Surman, T.; Urrutia, J.; Veltmann, C.; et al. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm 2012, 9, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Wilders, R.; Verkerk, A.O. Role of the R1135H KCNH2 mutation in Brugada syndrome. Int. J. Cardiol. 2010, 144, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ohno, S.; Ding, W.G.; Fukuyama, M.; Miyamoto, A.; Itoh, H.; Makiyama, T.; Wu, J.; Bai, J.; Hasegawa, K.; et al. Gain-of-function KCNH2 mutations in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2014, 25, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Nakamura, K.; Hayashi, T.; Inagaki, N.; Takahashi, M.; Arimura, T.; Morita, H.; Higashiuesato, Y.; Hirano, Y.; Yasunami, M.; et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J. Biol. Chem. 2004, 279, 27194–27198. [Google Scholar] [CrossRef] [PubMed]
- Biel, S.; Aquila, M.; Hertel, B.; Berthold, A.; Neumann, T.; DiFrancesco, D.; Moroni, A.; Thiel, G.; Kauferstein, S. Mutation in S6 domain of HCN4 channel in patient with suspected Brugada syndrome modifies channel function. Pflugers Arch. Eur. J. Physiol. 2016, 468, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; El Zein, L.; Kruse, M.; Guinamard, R.; Beckmann, A.; Bozio, A.; Kurtbay, G.; Mégarbané, A.; Ohmert, I.; Blaysat, G.; et al. Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ. Cardiovasc. Genet. 2010, 3, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chatel, S.; Simard, C.; Syam, N.; Salle, L.; Probst, V.; Morel, J.; Millat, G.; Lopez, M.; Abriel, H.; et al. Molecular Genetics and Functional Anomalies in a Series of 248 Brugada Cases with 11 Mutations in the TRPM4 Channel. PLoS ONE 2013, 8, e54131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinamard, R.; Bouvagnet, P.; Hof, T.; Liu, H.; Simard, C.; Sall, L. TRPM4 in cardiac electrical activity. Cardiovasc. Res. 2015, 108, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hennessey, J.A.; Kirkton, R.D.; Wang, C.; Graham, V.; Puranam, R.S.; Rosenberg, P.B.; Bursac, N.; Pitt, G.S. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ. Res. 2011, 109, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, J.A.; Marcou, C.A.; Wang, C.; Wei, E.Q.; Wang, C.; Tester, D.J.; Torchio, M.; Dagradi, F.; Crotti, L.; Schwartz, P.J.; et al. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm 2013, 10, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- London, B.; Michalec, M.; Mehdi, H.; Zhu, X.; Kerchner, L.; Sanyal, S.; Viswanathan, P.C.; Pfahnl, A.E.; Shang, L.L.; Madhusudanan, M.; et al. Mutation in glycerol-3-phosphate dehydrogenase 1-like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 2007, 116, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, Y.-Q.; Fan, L.-L.; Guo, S.; Li, J.-J.; Jin, J.-Y.; Xiang, R. Whole-exome sequencing identifies a novel mutation of GPD1L (R189X) associated with familial conduction disease and sudden death. J. Cell. Mol. Med. 2018, 22, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Sato, A.; Marcou, C.A.; Tester, D.J.; Ackerman, M.J.; Crotti, L.; Schwartz, P.J.; On, Y.K.; Park, J.E.; Nakamura, K.; et al. A novel disease gene for Brugada syndrome: Sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ. Arrhythm. Electrophysiol. 2012, 5, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Kattygnarath, D.; Maugenre, S.; Neyroud, N.; Balse, E.; Ichai, C.; Denjoy, I.; Dilanian, G.; Martins, R.P.; Fressart, V.; Berthet, M.; et al. MOG1: A new susceptibility gene for Brugada syndrome. Circ. Cardiovasc. Genet. 2011, 4, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Olesen, M.S.; Holst, A.G.; Schmitt, N. Letter by Olesen et al. regarding article “MOG1: A New susceptibility gene for brugada syndrome”. Circ. Cardiovasc. Genet. 2011, 4, 2010–2011. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Berne, P.; Selga, E.; Allegue, C.; Iglesias, A.; Brugada, J.; Brugada, R. Brugada syndrome and p.E61X_RANGRF. Cardiol. J. 2014, 21, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Boczek, N.J.; Ye, D.; Johnson, E.K.; Wang, W.; Crotti, L.; Tester, D.J.; Dagradi, F.; Mizusawa, Y.; Torchio, M.; Alders, M.; et al. Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome. Circ. Res. 2014, 115, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, L.; Crotti, P.J.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Veerman, C.C.; Podliesna, S.; Tadros, R.; Lodder, E.M.; Mengarelli, I.; De Jonge, B.; Beekman, L.; Barc, J.; Wilders, R.; Wilde, A.A.M.; et al. The brugada syndrome susceptibility gene HEY2 modulates cardiac transmural ion channel patterning and electrical heterogeneity. Circ. Res. 2017, 121, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.S.; Tynan, M.; Braidwood, L.; Fitzgerald, G.R. Bidirectional tachycardia in a child. A study using His bundle electrography. Br. Heart J. 1975, 37, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Leenhardt, A.; Lucet, V.; Denjoy, I.; Grau, F.; do Ngoc, D.; Coumel, P. Catecholaminergic polymorphic ventricular tachycardia in children. Circulation 1995, 95, 1512–1519. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Ruan, Y.; Priori, S.G. Catecholaminergic Polymorphic Ventricular Tachycardia. Prog. Cardiovasc. Dis. 2008, 51, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, N. Current topics in catecholaminergic polymorphic ventricular tachycardia. J. Arrhythm. 2016, 32, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Bloise, R.; Memmi, M.; Priori, S.G. Clinical utility gene card for: Catecholaminergic polymorphic ventricular tachycardia (CPVT). Eur. J. Hum. Genet. 2014, 22, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Bloise, R.; Monteforte, N.; Priori, S.G. Sudden cardiac death and genetic ion channelopathies: Long QT, Brugada, short QT, catecholaminergic polymorphic ventricular tachycardia, and idiopathic ventricular fibrillation. Circulation 2012, 125, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Kannankeril, P.J.; Moore, J.P.; Cerrone, M.; Priori, S.G.; Kertesz, N.J.; Ro, P.S.; Batra, A.S.; Kaufman, E.S.; Fairbrother, D.L.; Saarel, E.V.; et al. Efficacy of flecainide in the treatment of catecholaminergic polymorphic ventricular tachycardia a randomized clinical trial. JAMA Cardiol. 2017, 2, 759–766. [Google Scholar] [PubMed]
- Roston, T.M.; Vinocur, J.M.; Maginot, K.R.; Mohammed, S.; Salerno, J.C.; Etheridge, S.P.; Cohen, M.; Hamilton, R.M.; Pflaumer, A.; Kanter, R.J.; et al. Catecholaminergic Polymorphic Ventricular Tachycardia in Children: Analysis of Therapeutic Strategies and Outcomes from an International Multicenter Registry. Circ. Arrhythm. Electrophysiol. 2015, 8, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Lieve, K.V.; Wilde, A.A.; van der Werf, C. The Role of Flecainide in the Management of Catecholaminergic Polymorphic Ventricular Tachycardia. Arrhythm. Electrophysiol. Rev. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Chopra, N.; Laver, D.; Hwang, H.S.; Davies, S.S.; Roach, D.E.; Duff, H.J.; Roden, D.M.; Wilde, A.A.M.; Knollmann, B.C. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat. Med. 2009, 15, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Denegri, M.; Ruan, Y.; Avelino-Cruz, J.E.; Perissi, A.; Negri, S.; Napolitano, C.; Coetzee, W.A.; Boyden, P.A.; Priori, S.G. Short communication: Flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity. Circ. Res. 2011, 109, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the Cardiac Ryanodine Receptor Gene (hRyR2) Underlie Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Tiso, N.; Salamon, M.; Bagattin, A.; Danieli, G.A.; Argenton, F.; Bortolussi, M. The binding of the RyR2 calcium channel to its gating protein FKBP12.6 is oppositely affected by ARVD2 and VTSIP mutations. Biochem. Biophys. Res. Commun. 2002, 299, 594–598. [Google Scholar] [CrossRef]
- Postma, A.V. Absence of Calsequestrin 2 Causes Severe Forms of Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2002, 91, 21e–26e. [Google Scholar] [CrossRef]
- Song, L.; Alcalai, R.; Arad, M.; Wolf, C.M.; Toka, O.; Conner, D.A.; Berul, C.I.; Eldar, M.; Seidman, C.E.; Seidman, J.G. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2007, 117, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.; Bagnall, R.D.; Lam, L.; Ingles, J.; Turner, C.; Haan, E.; Davis, A.; Yang, P.C.; Clancy, C.E.; Sy, R.W.; et al. A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2016, 13, 1652–1660. [Google Scholar] [CrossRef] [PubMed]
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in bedouin families from Israel. Am. J. Hum. Genet. 2002, 67, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Di Barletta, M.R.; Viatchenko-Karpinski, S.; Nori, A.; Memmi, M.; Terentyev, D.; Turcato, F.; Valle, G.; Rizzi, N.; Napolitano, C.; Gyorke, S.; et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation 2006, 114, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Györke, I.; Hester, N.; Jones, L.R.; Györke, S. The Role of Calsequestrin, Triadin, and Junctin in Conferring Cardiac Ryanodine Receptor Responsiveness to Luminal Calcium. Biophys. J. 2004, 86, 2121–2128. [Google Scholar] [CrossRef]
- Roux-buisson, N.; Cacheux, M.; Fourest-lieuvin, A.; Fauconnier, J.; Brocard, J.; Denjoy, I.; Durand, P.; Guicheney, P.; Kyndt, F.; Leenhardt, A.; et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 2012, 21, 2759–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyegaard, M.; Overgaard, M.T.; Sondergaard, M.T.; Vranas, M.; Behr, E.R.; Hildebrandt, L.L.; Lund, J.; Hedley, P.L.; Camm, A.J.; Wettrell, G.; et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 2012, 91, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Hurtado, N.; Boczek, N.J.; Kryshtal, D.O.; Johnson, C.N.; Sun, J.; Nitu, F.R.; Cornea, R.L.; Chazin, W.J.; Calvert, M.L.; Tester, D.J.; et al. Novel CPVT-Associated Calmodulin Mutation in CALM3 (CALM3-A103V) Activates Arrhythmogenic Ca Waves and Sparks. Circ. Arrhythm. Electrophysiol. 2016, 9, e004161. [Google Scholar] [CrossRef] [PubMed]
- Devalla, H.D.; Gélinas, R.; Aburawi, E.H.; Beqqali, A.; Goyette, P.; Freund, C.; Chaix, M.; Tadros, R.; Jiang, H.; Le Béchec, A.; et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol. Med. 2016, 8, 1390–1408. [Google Scholar] [CrossRef] [PubMed]
- Mohler, P.J.; Splawski, I.; Napolitano, C.; Bottelli, G.; Sharpe, L.; Timothy, K.; Priori, S.G.; Keating, M.T.; Bennett, V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc. Natl. Acad. Sci. USA 2004, 101, 9137–9142. [Google Scholar] [CrossRef] [PubMed]
- El Refaey, M.M.; Mohler, P.J. Ankyrins and spectrins in cardiovascular biology and disease. Front. Physiol. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Van der Werf, C.; Wilde, A.A.M. Catecholaminergic polymorphic ventricular tachycardia: From bench to bedside. Heart 2013, 99, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Abriel, H.; Rougier, J.S.; Jalife, J. Ion Channel Macromolecular Complexes in Cardiomyocytes: Roles in Sudden Cardiac Death. Circ. Res. 2015, 344, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(A) Cardiac action potential and transmembrane ionic currents that participate in each phase. Phase 0: rapid depolarization due to entrance of Na+ currents into the cell. Phase 1: early repolarization initiated by outward K+ Ito currents. Phase 2: a plateau phase marked by the Ca2+ entry into the cell against K+ outward repolarizing currents. Phase 3: end of repolarization produced by K+ currents upon Ca2+ channel-inactivation. Phase 4: resting membrane potential (≈−90 mV) determined by inward-rectifier K+ currents. OW: outward currents. IW: inward currents. (B) Excitation-contraction coupling: during action potential, Ca2+ entry in phase 2 induces a large release of Ca2+ from the sarcoplasmic reticulum through the RyR2 receptor that allows cell contraction. After repolarization, Ca2+ is extruded from the cell through the Na+/Ca2+ exchanger or taken back into the sarcoplasmic reticulum through SERCA2a to allow cell relaxation.
Figure 1.
(A) Cardiac action potential and transmembrane ionic currents that participate in each phase. Phase 0: rapid depolarization due to entrance of Na+ currents into the cell. Phase 1: early repolarization initiated by outward K+ Ito currents. Phase 2: a plateau phase marked by the Ca2+ entry into the cell against K+ outward repolarizing currents. Phase 3: end of repolarization produced by K+ currents upon Ca2+ channel-inactivation. Phase 4: resting membrane potential (≈−90 mV) determined by inward-rectifier K+ currents. OW: outward currents. IW: inward currents. (B) Excitation-contraction coupling: during action potential, Ca2+ entry in phase 2 induces a large release of Ca2+ from the sarcoplasmic reticulum through the RyR2 receptor that allows cell contraction. After repolarization, Ca2+ is extruded from the cell through the Na+/Ca2+ exchanger or taken back into the sarcoplasmic reticulum through SERCA2a to allow cell relaxation.

Figure 2.
Electrocardiographic findings in the four primary electrical disorders or channelopathies. (A) LQTS, with prolonged QT interval; (B) SQTS, with shortened QT interval; (C) BrS. In this case, ECG at baseline was normal (C1), but the typical pattern, with ST-segment elevation in right precordial leads, was unmasked after a provocative test with sodium-blockers (D2); (D) CPVT. ECG is normal at baseline (D1), but premature ventricular complexes and occurrence of bidirectional tachycardia appear with exercise (D2). Characteristic ECG features of each disorder are circled in red.
Figure 2.
Electrocardiographic findings in the four primary electrical disorders or channelopathies. (A) LQTS, with prolonged QT interval; (B) SQTS, with shortened QT interval; (C) BrS. In this case, ECG at baseline was normal (C1), but the typical pattern, with ST-segment elevation in right precordial leads, was unmasked after a provocative test with sodium-blockers (D2); (D) CPVT. ECG is normal at baseline (D1), but premature ventricular complexes and occurrence of bidirectional tachycardia appear with exercise (D2). Characteristic ECG features of each disorder are circled in red.

Figure 3.
Schematic representation of the pathophysiological mechanisms involved in the four main primary electrical disorders. (A) in LQTS, either by a decrease in K+ currents (A1) or an increase in Na+ currents (A2), AP duration is prolonged, and so is the QTc interval on the ECG (depicted by the bottom horizontal lines: in black normal QT, in red long-QT interval). This situation favors the development of early afterdepolarizations, the trigger of ventricular arrhythmias in LQTS patients (A3). (B) in SQTS, an increase in K+ currents accelerates repolarization (B1), and manifests as short QTc interval in the ECG (depicted by the bottom horizontal lines: in black normal QT, in red short-QT interval); in those cases of SQTS caused by loss-of-function mutations in the calcium channel (B2), besides shortening of the AP duration there is transmural gradient in early phases of repolarization, leading to ST-segment elevation like the one seen in BrS (combined phenotype, QT interval depicted by horizontal lines and ST segment elevation by the red arrow). In SQTS, an increased dispersion of repolarization favors the appearance of atrial and ventricular arrhythmias (B3). (C) In BrS, a decrease in Na+ currents (C1) or, less commonly, an increase in Ito currents (C2); produces a ionic imbalance in early repolarization, giving rise to the characteristic ST-segment elevation seen in the ECG (depicted by the red arrows). The consequent epicardial and transmural dispersion of repolarization favors ventricular arrhythmias by a mechanism of phase-2 reentry (C3). (D) CPVT is produced by abnormal Ca2+ leak from the sarcoplasmic reticulum (D1), which favors the occurrence of delayed afterdepolarizations, which in turn can trigger ventricular arrhythmias (D2). Modified from [6].
Figure 3.
Schematic representation of the pathophysiological mechanisms involved in the four main primary electrical disorders. (A) in LQTS, either by a decrease in K+ currents (A1) or an increase in Na+ currents (A2), AP duration is prolonged, and so is the QTc interval on the ECG (depicted by the bottom horizontal lines: in black normal QT, in red long-QT interval). This situation favors the development of early afterdepolarizations, the trigger of ventricular arrhythmias in LQTS patients (A3). (B) in SQTS, an increase in K+ currents accelerates repolarization (B1), and manifests as short QTc interval in the ECG (depicted by the bottom horizontal lines: in black normal QT, in red short-QT interval); in those cases of SQTS caused by loss-of-function mutations in the calcium channel (B2), besides shortening of the AP duration there is transmural gradient in early phases of repolarization, leading to ST-segment elevation like the one seen in BrS (combined phenotype, QT interval depicted by horizontal lines and ST segment elevation by the red arrow). In SQTS, an increased dispersion of repolarization favors the appearance of atrial and ventricular arrhythmias (B3). (C) In BrS, a decrease in Na+ currents (C1) or, less commonly, an increase in Ito currents (C2); produces a ionic imbalance in early repolarization, giving rise to the characteristic ST-segment elevation seen in the ECG (depicted by the red arrows). The consequent epicardial and transmural dispersion of repolarization favors ventricular arrhythmias by a mechanism of phase-2 reentry (C3). (D) CPVT is produced by abnormal Ca2+ leak from the sarcoplasmic reticulum (D1), which favors the occurrence of delayed afterdepolarizations, which in turn can trigger ventricular arrhythmias (D2). Modified from [6].
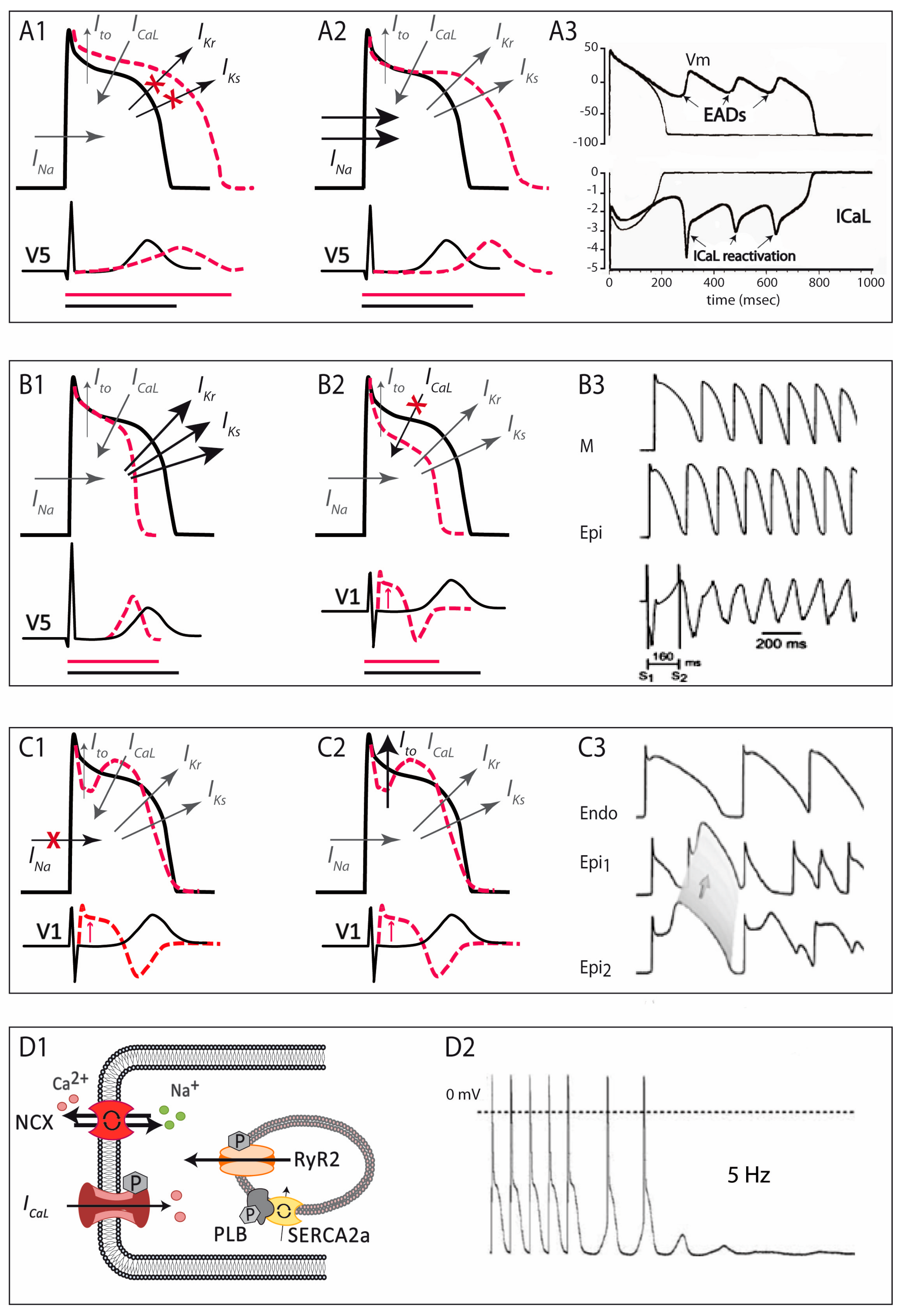

{kind=link}
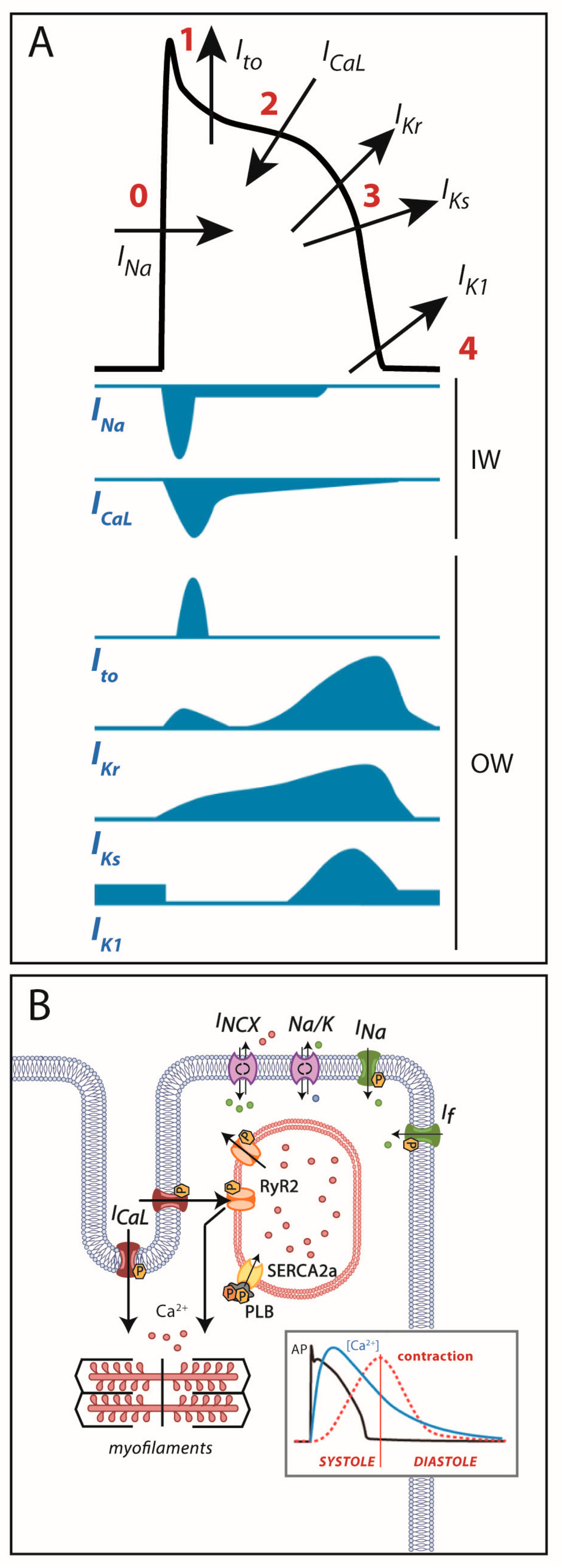{kind=link}
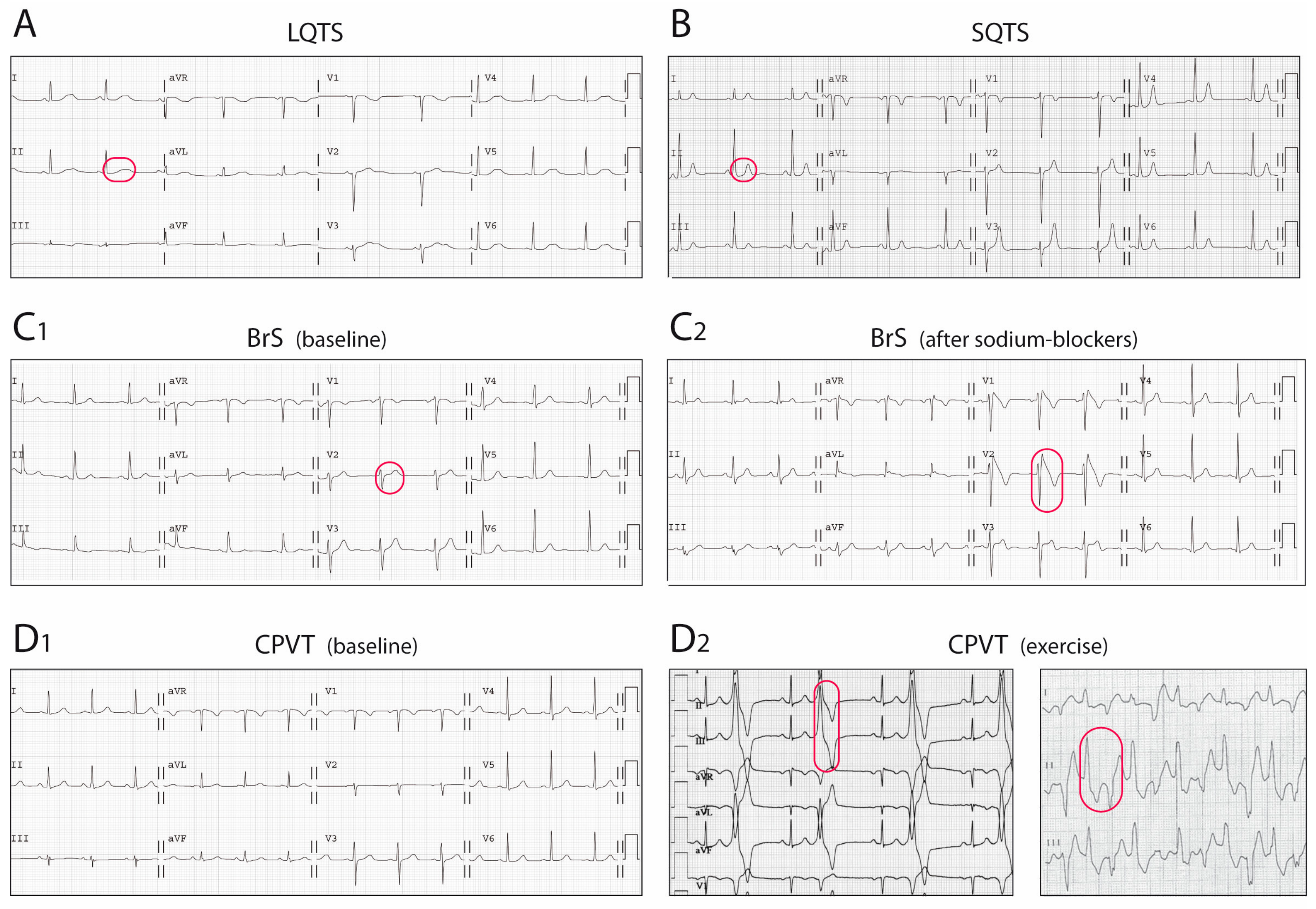{kind=link}
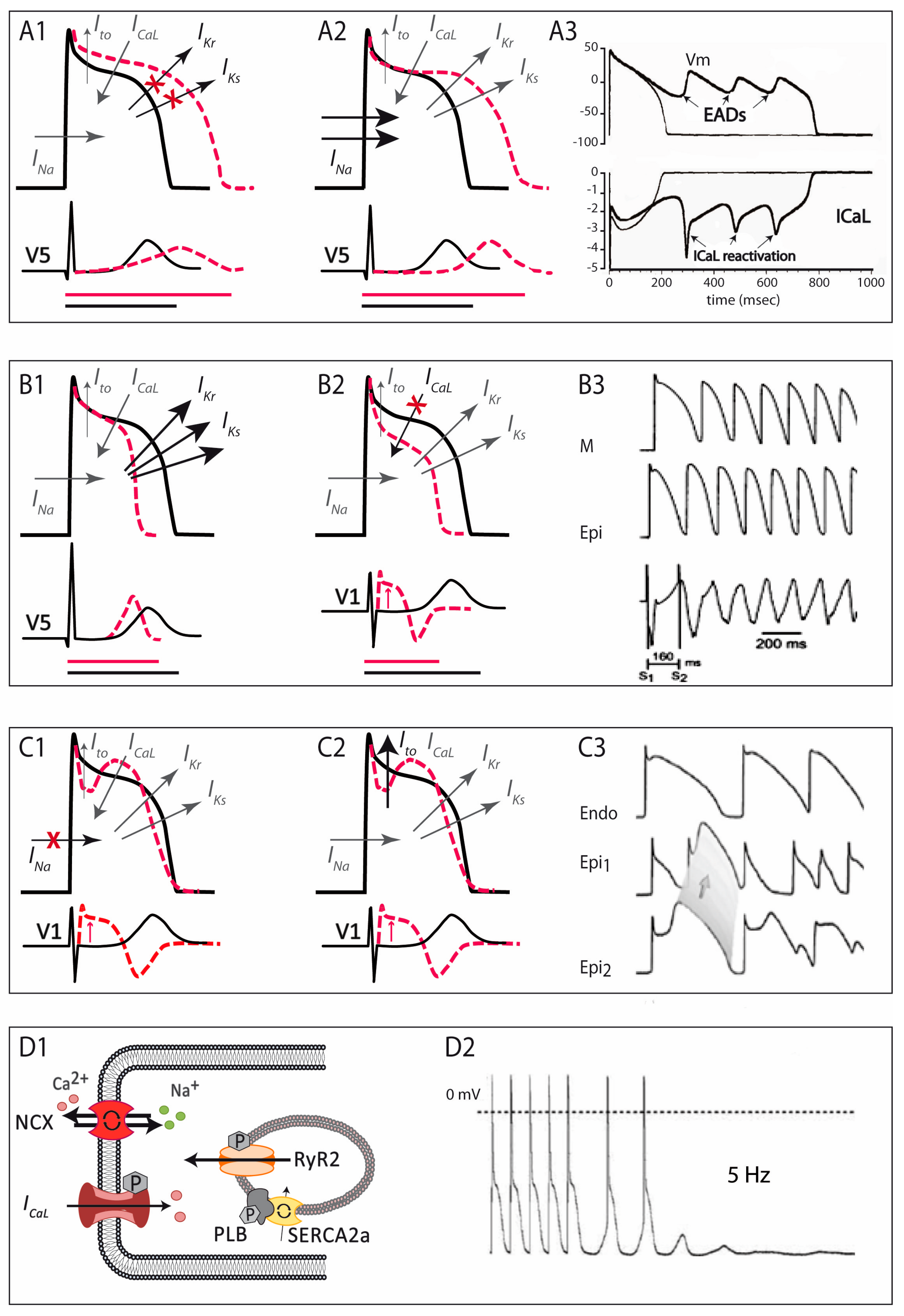{kind=link}
Table 1.
Mutations associated with the LQTS.
| Gene | Protein | Current | Effect | Function | Prevalence |
|---|---|---|---|---|---|
| GENES ENCODING ION CHANNEL SUBUNITS | |||||
| 1. Major LQTS-susceptibility genes | |||||
| KCNQ1 | KV7.1 (α-subunit of the voltage-dependent K+ channel) | ↓ IKs | loss-of-function | mediator of the slow component of the delayed rectifying potassium IKs current | ⋍40% (LQT1) |
| KCNH2 | KV11.1/hERG (α-subunit of the voltage-dependent K+ channel) | ↓ IKr | loss-of-function | mediator of the rapid component of the delayed rectifying potassium IKr current | ⋍30% (LQT2) |
| SCN5A | NaV1.5 (α-subunit of the voltage-dependent Na+ channel) | ↑ INa | gain-of-function | mediator of the depolarizing inward sodium INa current | ⋍10% (LQT3) |
| 2. Rare LQTS-susceptibility genes | |||||
| By reducing outward currents | |||||
| KCNE1 | minK (β1-subunit of the voltage-dependent K+ channel) | ↓ IKs | loss-of-function | auxiliary protein modulator of KV7.1 and the IKs current | <1% |
| KCNE2 | MiRP1 (β2-subunit of the voltage-dependent K+ channel) | ↓ IKr | loss-of-function | auxiliary protein modulator of KV11.1 and the IKr current | <1% |
| KCNJ2 | Kir2.1 (inward rectifying K+ channel) | ↓ IK1 | loss-of-function, extra-cardiac manifestations | mediator of the inward rectifying potassium IK1 current | <1% (Andersen-Tawil syndrome, LQT7) |
| KCNJ5 | Kir3.4 (G protein-activated inward rectifying K+ channel 4) | ↓ IK,Ach | loss-of-function | mediator of the acetylcholine/adenosine-induced potassium IK,Ach current | <1% |
| By increasing inward currents | |||||
| SCN1B | β1-subunit of the voltage-dependent Na+ channel | ↑ INa | gain-of-function | auxiliary protein modulator of NaV1.5 and the INa current | <1% |
| SCN4B | β4-subunit of the voltage-dependent Na+ channel | ↑ INa | gain-of-function | auxiliary protein modulator of NaV1.5 and the INa current | <1% |
| CACNA1C | CaV1.2 (α1C-subunit of the voltage-dependent L-type Ca2+ channel) | ↑ ICaL | gain-of-function, extra-cardiac manifestations | mediator of the inward calcium ICaL current | <1% (Timothy syndrome, LQT8) |
| GENES ENCODING AUXILIARY PROTEINS | |||||
| By reducing outward currents | |||||
| AKAP9 | A-kinase anchor protein-9 | ↓ IKs | disruption of KV7.1/PKA interaction | scaffolding protein assembling PKA and KV7.1 | <1% |
| By increasing inward currents | |||||
| ANK2 | ankyrin B | ↑ ICaL | disruption of Na+/K+ exchanger, Na+/Ca2+ exchanger/IP3 interaction | scaffolding protein assembling Na+/K+ exchanger, Na+/Ca2+ exchanger and IP3 receptor | <1% |
| CALM1 | calmodulin (CaM) | ↑ ICaL | disorder in CaV1.2 functioning | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 (and others) | <1% |
| CALM2 | calmodulin (CaM) | ↑ ICaL | disorder in CaV1.2 functioning | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 (and others) | <1% |
| CALM3 | calmodulin (CaM) | ↑ ICaL | disorder in CaV1.2 functioning | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 (and others) | <1% |
| SNTA1 | α1-syntrophin | ↑ INa | disruption of NaV1.5/NOS-PMCA4b complex interaction | scaffolding protein that associates NaV1.5 channels with the NOS-PMCA4b complex | <1% |
| TRDN | triadin | ↑ ICaL | reduction of ICaL inactivation | regulator of ryanodine receptors and CaV1.2 | <1% |
| Less established mechanisms | |||||
| CAV3 | caveolin-3 | ↑ INa?/↓ IK1? | changes in membrane expression of NaV1.5/Kir2.1 | scaffolding protein regulating ion channels in caveolae | <1% |
| TRPM4 | Transient receptor potential melastatin 4 | loss-of-function | regulator of conduction and cellular electrical activity which impact heart development | <1% | |
| RYR2 | ryanodine receptor 2 (RyR2) | not described | mediator of Ca2+ release from the SR | <1% | |
↑: increased current; ↓: decreased current; ?: suspected but not confirmed mechanism.
Table 2.
Mutations associated with the SQTS.
| Gene | Protein | Current | Effect | Function | Prevalence |
|---|---|---|---|---|---|
| GENES ENCODING ION CHANNEL SUBUNITS | |||||
| By increasing outward currents | |||||
| KCNH2 | KV11.1/hERG (α-subunit of the voltage-dependent K+ channel) | ↑ IKr | gain-of-function | mediator of the rapid component of the delayed rectifying potassium IKr current | ⋍15% (SQT1) |
| KCNQ1 | KV7.1 (α-subunit of the voltage-dependent K+ channel) | ↑ IKs | gain-of-function | mediator of the slow component of the delayed rectifying potassium IKs current | <1% |
| KCNJ2 | Kir2.1 (inward rectifying K+ channel) | ↑ IK1 | gain-of-function | mediator of the inward rectifying potassium IK1 current | <1% |
| By decreasing inward currents | |||||
| CACNA1C | CaV1.2 (α1C-subunit of the voltage-dependent L-type Ca2+ channel) | ↓ ICaL | loss-of-function, combined phenotype of SQTS and BrS | mediator of the inward calcium ICaL current | <1% |
| CACNB2b | β2-subunit of the voltage-dependent L-type Ca2+ channel | ↓ ICaL | loss-of-function, combined phenotype of SQTS and BrS | auxiliary protein modulator of CaV1.2 and the ICaL current | <1% |
| Less established mechanisms | |||||
| CACNA2D1 | α2/δ-subunit of the voltage-dependent L-type Ca2+ channel | ↓ ICaL? | loss-of-function? | auxiliary protein modulator of CaV1.2 and the ICaL current | <1% |
↑: increased current; ↓: decreased current; ?: suspected but not confirmed mechanism.
Table 3.
Mutations associated with the BrS.
| Gene | Protein | Current | Effect | Function | Prevalence |
|---|---|---|---|---|---|
| GENES ENCODING ION CHANNEL SUBUNITS | |||||
| 1. Major BrS-susceptibility genes | |||||
| SCN5A | NaV1.5 (α-subunit of the voltage-dependent Na+ channel) | ↓ INa | loss-of-function | mediator of the depolarizing inward sodium INa current | ⋍25% (BrS1) |
| 2. Rare BrS-susceptibility genes | |||||
| By decreasing inward currents | |||||
| SCN1B | β1-subunit of the voltage-dependent Na+ channel | ↓ INa | loss-of-function | auxiliary protein modulator of NaV1.5 and the INa current | <1% |
| SCN2B | β2-subunit of the voltage-dependent Na+ channel | ↓ INa | loss-of-function | auxiliary protein modulator of NaV1.5 and the INa current | <1% |
| SCN3B | β3-subunit of the voltage-dependent Na+ channel | ↓ INa | loss-of-function | auxiliary protein modulator of NaV1.5 and the INa current | <1% |
| SCN10A | NaV1.8 (α-subunit of the neuronal voltage-dependent Na+ channel) | ↓ INa | loss-of-function | mediator of the depolarizing phase of the neural AP, associated with pain perception | ⋍10%? |
| CACNA1C | CaV1.2 (α1C-subunit of the volatge-dependent L-type Ca2+ channel) | ↓ ICaL | loss-of-function, combined phenotype of BrS and SQTS | mediator of the inward calcium ICaL current | <1% |
| CACNB2b | β2-subunit of the voltage-dependent L-type Ca2+ channel | ↓ ICaL | loss-of-function, combined phenotype of BrS and SQTS | auxiliary protein modulator of CaV1.2 and the ICaL current | <1% |
| By increasing outward currents | |||||
| KCND3 | KV4.3 (α-subunit of the voltage-dependent K+ channel) | ↑ Ito | gain-of-function | mediator of the transient outward K+ Ito current | <1% |
| KCNE3 | minK-related peptide 2 (β-subunit of the voltage-dependent K+ channel) | ↑ Ito | gain-of-function | regulator of KV4.3 | <1% |
| KCNAB2 | β2-subunit of the voltage-dependent K+ channel | ↑ Ito | gain-of-function | interaction with KV4.3 | <1% |
| KCND2 | KV4.2 (voltage-dependent K+ channel) | ↑ Ito | gain-of-function | contributor to the transient outward K+ Ito current | <1% |
| KCNE5 | minK-related peptide 4 (β-subunit of the voltage-dependent K+ channel) | ↑ Ito | gain-of-function | inhibitor of the delayed rectifying KV7.1 channel and modulator of KV4.3 | <1% |
| KCNJ8 | Kir6.1 (inward-rectifier K+ channel, subunit of the ATP-sensitive K+ channel) | ↑ IK-ATP | gain-of-function | mediator of the IK-ATP currents | <1% |
| ABCC9 | SUR2 (sulfonylurea receptor, subunit of the ATP-sensitive K+ channel) | ↑ IK-ATP | gain-of-function | modulator of IK-ATP currents | <1% |
| KCNH2 | KV11.1/hERG (α-subunit of the voltage-dependent K+ channel) | ↑ IKr | gain-of-function | mediator of the rapid component of the delayed rectifying potassium IKr current | <1% |
| Less established mechanisms | |||||
| CACNA2D1 | α2/δ subunit of the volatge-dependent L-type Ca2+ channel | ↓ ICaL? | loss-of-function?, combined phenotype of SQTS and BrS | auxiliary protein modulator of CaV1.2 and the ICaL current | <1% |
| HCN4 | hyperpolarization-activated, cyclic nucleotide-gated ion channel 4 | ↓ If? | loss-of-function? | mediator of the pacemaker current, If | <1% |
| TRPM4 | Transient receptor potential melastatin 4 | loss-of-function/gain-of-function | regulator of conduction and cellular electrical activity which impact heart development | <1% | |
| GENES ENCONDING AUXILIARY PROTEINS | |||||
| FGF12 | fibroblast growth factor 12 | ↓ INa | interaction with NaV1.5 trafficking | modulator of Nav1.5 and the INa current | <1% |
| GPD1L | glycerol-3-phosphate dehydrogenase 1-like | ↓ INa | interaction with NaV1.5 trafficking | modulator of Na1.5 and the INa current | <1% |
| SLMAP | sarcolemma associated protein (striatin-interacting phosphatase and kinase complex) | ↓ INa | interaction with NaV1.5 trafficking | present in the T-tubules, regulator of excitation-contraction coupling | <1% |
| PKP2 | plakophillin-2 | ↓ INa | changes in NaV1.5 expression in intercalated disc | binds to and modulates NaV1.5 and the INa current | <1% |
| SEMA3A | semaphorin-3A | ↑ Ito | loss-of-function | inhibitor of the KV4.3 channel | <1% |
| Less established mechanisms | |||||
| RANGRF | MOG1 (multicopy suppressor of Gsp1) | ↓ INa? | interaction with NaV1.5 trafficking | involved in nuclear protein import—regulates cell surface location of NaV1.5 | <1% |
| HEY2 | CHF1 (cardiovascular helix-loop-helix factor 1) | ↑ Ito? | interaction with KCNIP2 | transcriptional regulator of cardiac electrical function | <1% |
↑: increased current; ↓: decreased current; ?: suspected but not confirmed mechanism.
Table 4.
Mutations associated with the CPVT.
| Gene | Protein | Effect | Function | Prevalence |
|---|---|---|---|---|
| GENES ENCODING ION CHANNELS AND AUXILIARY PROTEINS | ||||
| 1. Major CPVT-susceptibility genes | ||||
| RYR2 | ryanodine receptor 2 (RyR2) | cytoplasmic Ca2+ overload, due to Ca2+ leak from the SR | mediator of the release of stored Ca2+ ions from the SR | ⋍50–60% (CPVT1) |
| CASQ2 | calsequestrin 2 | decreased Ca2+ content in the SR and abnormal Ca2+ regulation | Ca2+ storage protein, controls Ca2+ release from the SR | ⋍5% |
| 2. Rare CPVT-susceptibility genes | ||||
| TRDN | triadin | cytoplasmic Ca2+ overload, due to Ca2+ leak from the SR | regulator of ryanodine receptors, controls the Ca2+ release from the SR | <1% |
| CALM1 | calmodulin (CaM) | Ca2+ leak from the SR due to loss of interaction CaM-RyR2 | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 or RyR2 (and others) | <1% |
| CALM2 | calmodulin (CaM) | reduction in Ca2+-binding affinity in the CaM C-domain | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 or RyR2 (and others) | <1% |
| CALM3 | calmodulin (CaM) | reduction in Ca2+-binding affinity in the CaM C-domain and leak from the SR | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 or RyR2 (and others) | <1% |
| TECLR | trans-2,3-enoyl-CoA reductase- like | decreased Ca2+ content in the SR and abnormal Ca2+ regulation | participates in the synthesis of fatty acids | <1% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Garcia-Elias, A.; Benito, B. Ion Channel Disorders and Sudden Cardiac Death. Int. J. Mol. Sci. 2018, 19, 692. https://doi.org/10.3390/ijms19030692
AMA Style
Garcia-Elias A, Benito B. Ion Channel Disorders and Sudden Cardiac Death. International Journal of Molecular Sciences. 2018; 19(3):692. https://doi.org/10.3390/ijms19030692
Chicago/Turabian StyleGarcia-Elias, Anna, and Begoña Benito. 2018. "Ion Channel Disorders and Sudden Cardiac Death" International Journal of Molecular Sciences 19, no. 3: 692. https://doi.org/10.3390/ijms19030692
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.