Nanotechnology and Glycosaminoglycans: Paving the Way Forward for Ovarian Cancer Intervention

 ,
,  and
and
Abstract
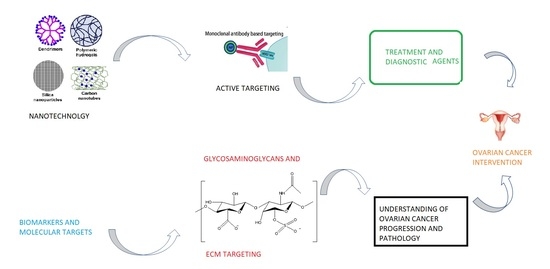
:
1. Introduction
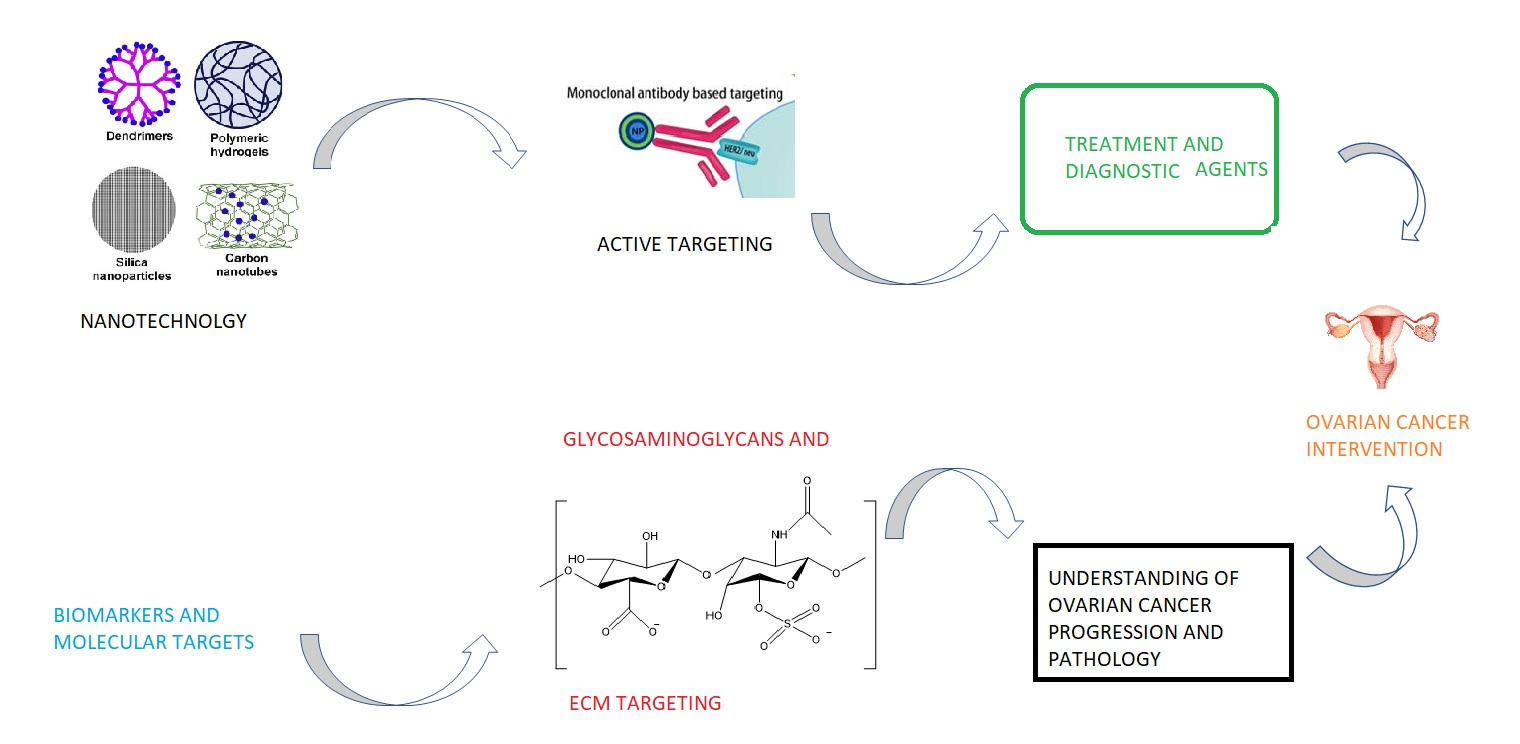
2. Active Targeting in Ovarian Cancer
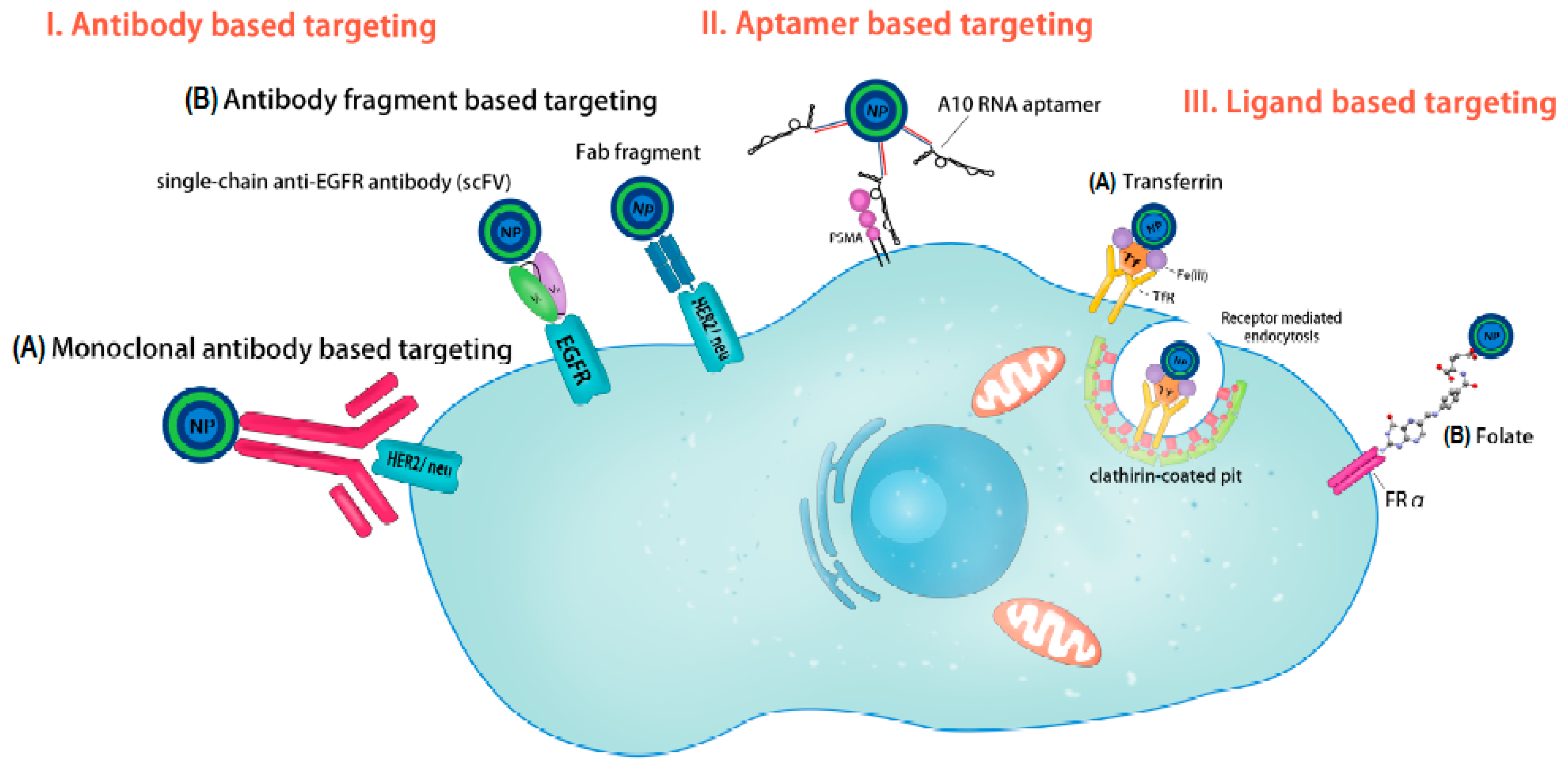
2.1. Antibody Based Targeting in OC
2.2. Aptamer Based Targeting and Detection in OC
2.2.1. Aptamers in OC Diagnostics
2.2.2. Aptamers in OC Treatment
2.3. Folate Receptor Targeting in OC
2.4. Is the Active Targeting Avenue Alone a Way Forward for Cancer Nanomedicines?
3. Nanocarrier Based Delivery in OC
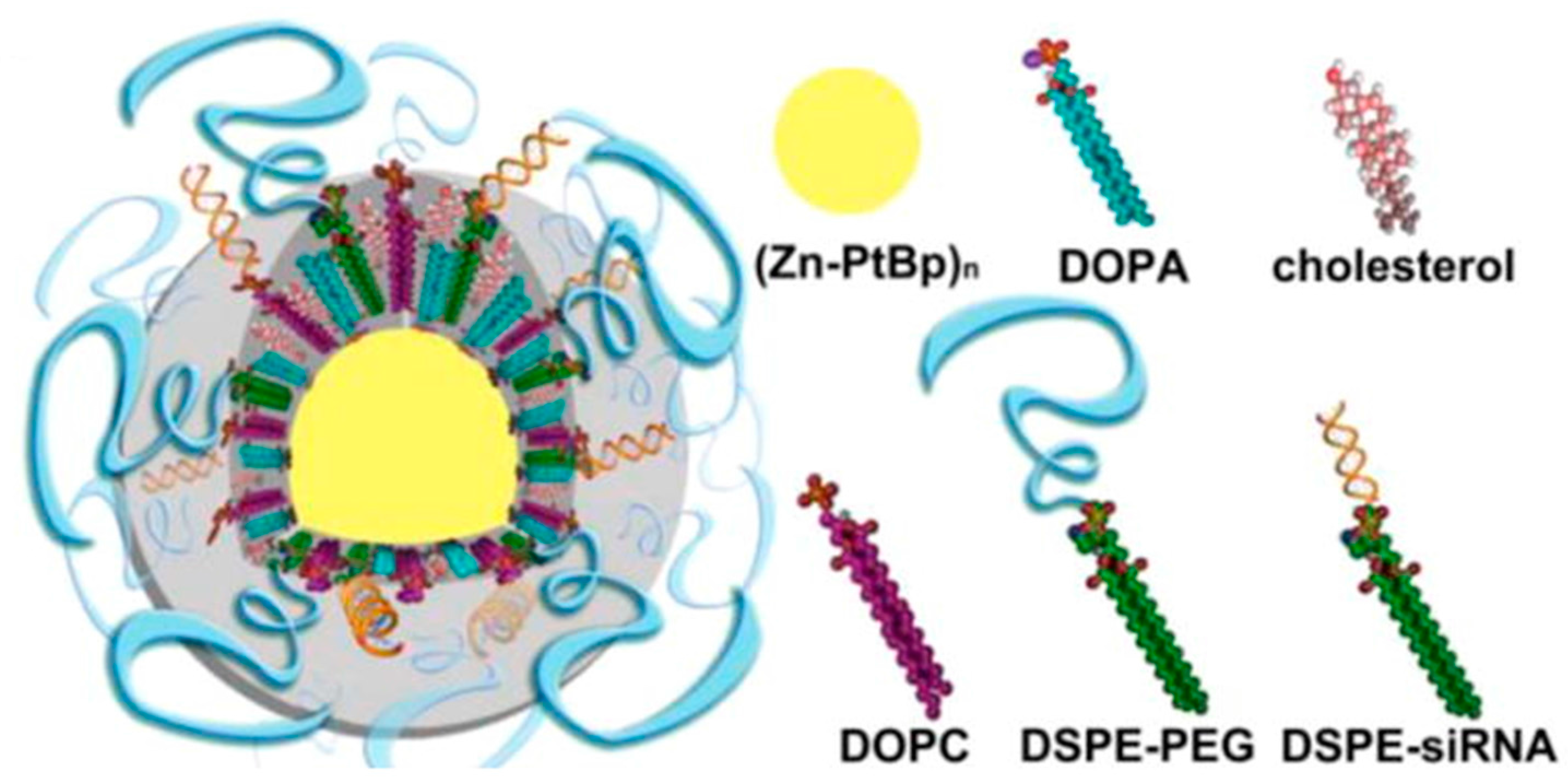
3.1. Nanocarriers for the Delivery of siRNAs to OC
3.2. Nanocarrier for the Delivery of OC Drugs
3.3. What Happens to These Nanosystems Beyond the Realms of Research?
4. Glycosaminoglycans as a Potential Molecular Target to Stop the Progression of OC
4.1. Chondroitin Sulphate as a Molecular Target
4.1.1. The Role of CS in OC
4.1.2. Targeting Chondroitin Sulphate-E
4.2. Hyaluronan (HA) As a Potential Molecular Target
4.2.1. The Role of Hyaluronan, CD44, Hyaluronidase and Hyaluronan Synthase in OC
4.2.2. Targeting Hyaluronan
5. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Iyer, A.K.; Singh, A.; Choy, E.; Hornicek, F.J.; Amiji, M.M.; Duan, Z. MDR1 siRNA loaded hyaluronic acid-based CD44 targeted nanoparticle systems circumvent paclitaxel resistance in ovarian cancer. Sci. Rep. 2015, 5, 8509. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.B.; Fu, L.W. Application of single-cell technology in cancer research. Biotechnol. Adv. 2017, 35, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Menderes, G.; Bonazzoli, E.; Bellone, S.; Black, J.; Altwerger, G.; Masserdotti, A.; Pettinella, F.; Zammataro, L.; Buza, N.; Hui, P.; et al. SYD985, a novel duocarmycin-based HER2-targeting antibody-drug conjugate, shows promising antitumor activity in epithelial ovarian carcinoma with HER2/Neu expression. Gynecol. Oncol. 2017, 146, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Alivisatos, A.P. Perspectives on the Physical Chemistry of Semiconductor Nanocrystals. J. Phys. Chem. 1996, 100, 13226–13239. [Google Scholar] [CrossRef]
- Sutherland, A.J. Quantum dots as luminescent probes in biological systems. Curr. Opin. Solid State Mater. Sci. 2002, 6, 365–370. [Google Scholar] [CrossRef]
- Coccia, M.; Wang, L. Path-breaking directions of nanotechnology-based chemotherapy and molecular cancer therapy. Technol. Forecast. Soc. Chang. 2015, 94, 155–169. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Devalapally, H.; Shahiwala, A.; Amiji, M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Control. Release 2008, 126, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Nobile, S.; Nobile, L. Nanotechnology for biomedical applications: Recent advances in neurosciences and bone tissue engineering. Polym. Eng. Sci. 2017, 57, 644–650. [Google Scholar] [CrossRef]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Singh, A.; Kulkarni, P.; Keeler, A.W.; Piroyan, A.; Sawant, R.R.; Patel, N.R.; Davis, B.; Ferris, C.; O’Neal, S.; et al. EGFR targeted theranostic Nanoemulsion for image-guided ovarian cancer therapy. Pharm. Res. 2015, 32, 2753–2763. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Patel, B.B.; Tiwari, S. Colloidal nanocarriers: A review on formulation technology, types and applications toward targeted drug delivery. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wang, J.; Wang, Y.; Li, L.; Guo, X.; Zhou, S. An implantable active-targeting micelle-in-nanofiber device for efficient and safe cancer therapy. ACS Nano 2015, 9, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Rhoda, K.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; Du Toit, L.C.; Pillay, V. Potential nanotechnologies and molecular targets in the quest for efficient chemotherapy in ovarian cancer. Expert Opin. Drug Deliv. 2015, 12, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Wadee, A.; Pillay, V.; Choonara, Y.E.; du Toit, L.C.; Penny, C.; Ndesendo, V.M.K.; Kumar, P.; Murphy, C.S. Recent advances in the design of drug-loaded polymeric implants for the treatment of solid tumors. Expert Opin. Drug Deliv. 2011, 8, 1323–1340. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.K.; Lee, S.C.; Han, B.; Park, K. Analysis on the current status of targeted drug delivery to tumors. J. Control. Release 2012, 164, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Raavé, R.; de Vries, R.B.M.; Massuger, L.F.; van Kuppevelt, T.H.; Daamen, W.F. Drug delivery systems for ovarian cancer treatment: A systematic review and meta-analysis of animal studies. PeerJ 2015, 3, e1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunliffe, D.; Kirby, A.; Alexander, C. Molecularly imprinted drug delivery systems. Adv. Drug Deliv. Rev. 2005, 57, 1836–1853. [Google Scholar] [CrossRef] [PubMed]
- Sudimack, J.; Lee, R.J. Targeted drug delivery via the folate receptor. Adv. Drug Deliv. Rev. 2000, 41, 147–162. [Google Scholar] [CrossRef]
- Xu, X.; Ho, W.; Zhang, X.; Bertrand, N.; Farokhzad, O. Cancer nanomedicine: From targeted delivery to combination therapy. Trends Mol. Med. 2015, 21, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Bazak, R.; Houri, M.; El Achy, S.; Kamel, S.; Refaat, T. Cancer active targeting by nanoparticles: A comprehensive review of literature. J. Cancer Res. Clin. Oncol. 2015, 141, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Kuan, C.H.; Wang, L.W.; Wu, H.C.; Chen, Y.; Chang, C.W.; Huang, R.Y.; Wang, T.W. Integrated self-assembling drug delivery system possessing dual responsive and active targeting for orthotopic ovarian cancer theranostics. Biomaterials 2016, 90, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Kulhari, H.; Pooja, D.; Kota, R.; Reddy, T.S.; Tabor, R.F.; Shukla, R.; Adams, D.J.; Sistla, R.; Bansal, V. Cyclic RGDfK Peptide Functionalized Polymeric Nanocarriers for Targeting Gemcitabine to Ovarian Cancer Cells. Mol. Pharm. 2016, 13, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.Y.; Guo, H.; Barengo, N.; Naora, H. Inhibition of ovarian cancer growth by a tumor-targeting peptide that binds eukaryotic translation initiation factor 4E. Clin. Cancer Res. 2009, 15, 4336–4347. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Li, Y.; Lee, J.S.; Gonik, A.M.; Dong, T.; Fung, G.; Sanchez, E.; Xing, L.; Cheng, H.R.; Luo, J.; et al. “OA02” peptide facilitates the precise targeting of paclitaxel-loaded micellar nanoparticles to ovarian cancer In Vivo. Cancer Res. 2012, 72, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.S.; Puranik, A.S.; Peppas, N.A. Intelligent nanoparticles for advanced drug delivery in cancer treatment. Curr. Opin. Chem. Eng. 2015, 7, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Dharap, S.S.; Wang, Y.; Chandna, P.; Khandare, J.J.; Qiu, B.; Gunaseelan, S.; Sinko, P.J.; Stein, S.; Farmanfarmaian, A.; Minko, T. Tumor-specific targeting of an anticancer drug delivery system by LHRH peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 12962–12967. [Google Scholar] [CrossRef] [PubMed]
- Apte, A.; Koren, E.; Koshkaryev, A.; Torchilin, V.P. Doxorubicin in TAT peptide-modified multifunctional immunoliposomes demonstrates increased activity against both drug-sensitive and drug-resistant ovarian cancer models. Cancer Biol. Ther. 2014, 15, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Tuefferd, M.; Couturier, J.; Penault-Llorca, F.; Vincent-Salomon, A.; Broët, P.; Guastalla, J.P.; Allouache, D.; Combe, M.; Weber, B.; Pujade-Lauraine, E.; et al. HER2 status in ovarian carcinomas: A multicenter GINECO study of 320 patients. PLoS ONE 2007, 2, e1138. [Google Scholar] [CrossRef] [PubMed]
- Peoples, G.; Goedegebuure, P.; Smith, R.; Linehan, D.; Yoshino, I.; Eberlein, T. Breast and ovarian cancer-specific cytotoxic T lymphocytes recognize the same HER2/neu-derived peptide. Proc. Natl. Acad. Sci. USA 1995, 92, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.P.; Sims, A.H. HER2-Targeted Antibody Treatment for Ovarian Cancer—Future Opportunities. J. Mol. Pharm. Org. Process. Res. 2016, 4, e125. [Google Scholar] [CrossRef]
- Palanca-Wessels, M.C.; Booth, G.C.; Convertine, A.J.; Lundy, B.B.; Berguig, G.Y.; Press, M.F.; Stayton, P.S.; Press, O.W. Antibody targeting facilitates effective intratumoral siRNA nanoparticle delivery to HER2-overexpressing cancer cells. Oncotarget 2016, 7, 9561–9575. [Google Scholar] [CrossRef] [PubMed]
- Kasten, B.B.; Arend, R.C.; Katre, A.A.; Kim, H.; Fan, J.; Ferrone, S.; Zinn, K.R.; Buchsbaum, D.J. B7-H3-targeted 212Pb radioimmunotherapy of ovarian cancer in preclinical models. Nucl. Med. Biol. 2017, 47, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Sullivan, P.S.; Soslow, R.A.; Waitz, R.; Reuter, V.E.; Wilton, A.; Thaler, H.T.; Arul, M.; Slovin, S.F.; Wei, J.; et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. Mod. Pathol. 2010, 23, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Fauci, J.M.; Straughn, J.M.; Ferrone, S.; Buchsbaum, D.J. A review of B7-H3 and B7-H4 immune molecules and their role in ovarian cancer. Gynecol. Oncol. 2012, 127, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.; Ling, V.; Carreno, B.M. The B7 family of immune-regulatory ligands. Genome Biol. 2005, 6, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-W.; Tekle, C.; Fodstad, O. The immunoregulatory protein human B7H3 is a tumor-associated antigen that regulates tumor cell migration and invasion. Cancer Drug Targets 2008, 8, 404–413. [Google Scholar] [CrossRef]
- Lamberti, I.; Scarano, S.; Esposito, C.L.; Antoccia, A.; Antonini, G.; Tanzarella, C.; De Franciscis, V.; Minunni, M. In vitro selection of RNA aptamers against CA125 tumor marker in ovarian cancer and its study by optical biosensing. Methods 2016, 97, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Shigdar, S.; Qiao, G.; Wang, T.; Kouzani, A.Z.; Zhou, S.F.; Kong, L.; Li, Y.; Pu, C.; Duan, W. Nucleic acid aptamer-guided cancer therapeutics and diagnostics: The next generation of cancer medicine. Theranostics 2015, 5, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Qian, C.; Lv, L.; Pu, C.; Li, Y.; Li, L.; Marappan, M.; Lin, J.; Wang, L.; Duan, W. The Use of Sensitive Chemical Antibodies for Diagnosis: Detection of Low Levels of Epcam in Breast Cancer. PLoS ONE 2013, 8, e57613. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xie, J.; Chen, H.; Gu, S.; Zhao, R.; Shao, J.; Jia, L. Nanotechnology-based intelligent drug design for cancer metastasis treatment. Biotechnol. Adv. 2014, 32, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, J.; Wu, M.; Zhao, J.X. Aptamers: Active targeting ligands for cancer diagnosis and therapy. Theranostics 2015, 5, 322–344. [Google Scholar] [CrossRef] [PubMed]
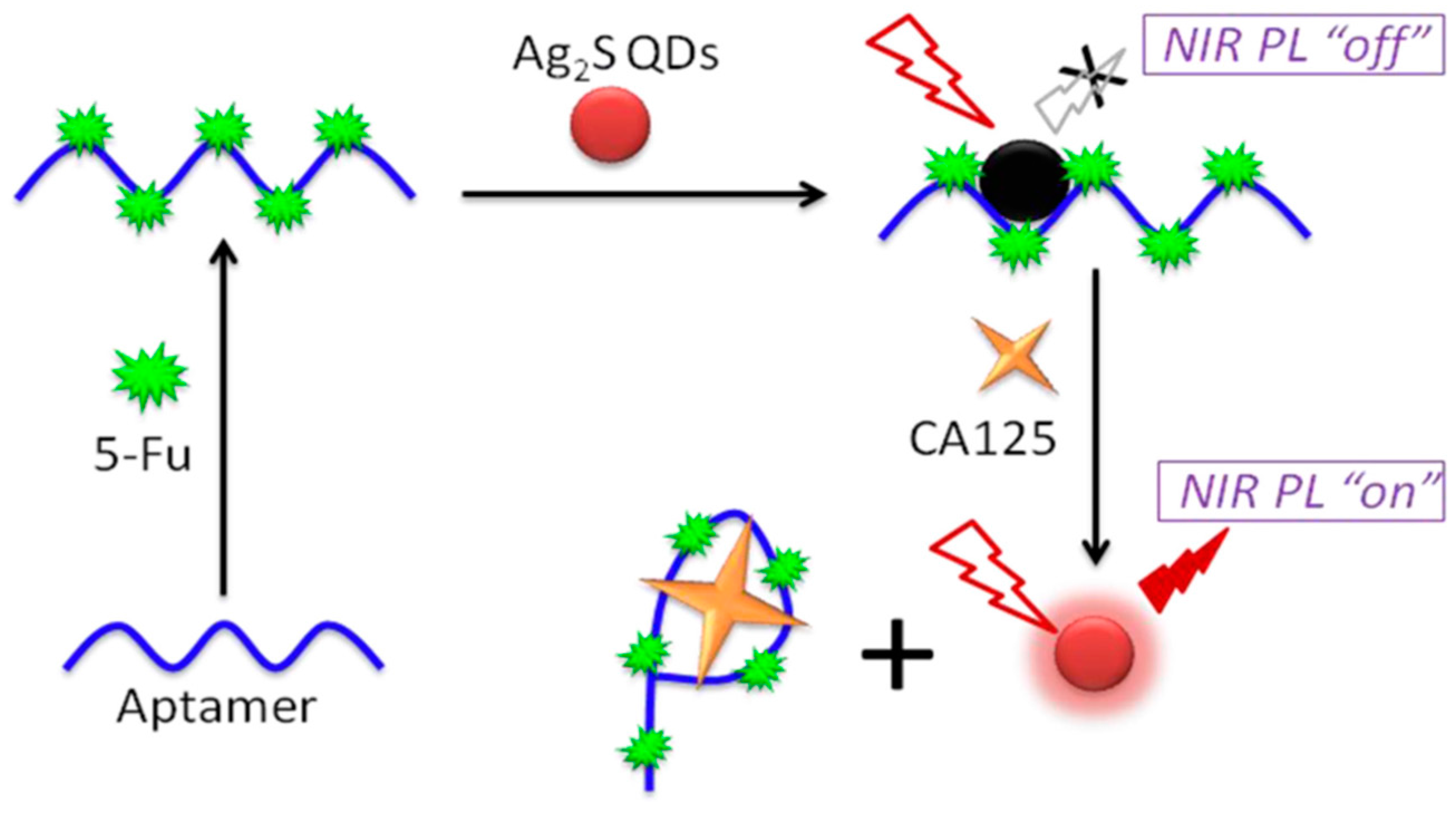
- Jin, H.; Gui, R.; Gong, J.; Huang, W. Aptamer and 5-fluorouracil dual-loading Ag2S quantum dots used as a sensitive label-free probe for near-infrared photoluminescence turn-on detection of CA125 antigen. Biosens. Bioelectron. 2017, 92, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Brody, E.N.; Gold, L. Aptamers as therapeutic and diagnostic agents. J. Biotechnol. 2000, 74, 5–13. [Google Scholar] [CrossRef]
- Zhang, Y.; Hong, H.; Cai, W. Tumor-targeted drug delivery with aptamers. Curr. Med. Chem. 2011, 18, 4185–4194. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Huang, R.; Deng, Y.; He, N. Progress in selection and biomedical applications of aptamers. J. Biomed. Nanotechnol. 2014, 10, 3043–3062. [Google Scholar] [CrossRef] [PubMed]
- Hamd-Ghadareh, S.; Salimi, A.; Fathi, F.; Bahrami, S. An amplified comparative fluorescence resonance energy transfer immunosensing of CA125 tumor marker and ovarian cancer cells using green and economic carbon dots for bio-applications in labeling, imaging and sensing. Biosens. Bioelectron. 2017, 96, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Akyol, S.; Gercel-Taylor, C.; Reynolds, L.C.; Taylor, D.D. HSP-10 in ovarian cancer: Expression and suppression of T-cell signaling. Gynecol. Oncol. 2006, 101, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.B.; Neves, M.A.D.; Thompson, M. Biosensor surface attachment of the ovarian cancer biomarker HSP10 via His-tag modification. Sens. Biosens. Res. 2016, 11, 107–112. [Google Scholar] [CrossRef]
- Pi, F.; Zhang, H.; Li, H.; Thiviyanathan, V.; Gorenstein, D.G.; Sood, A.K.; Guo, P. RNA nanoparticles harboring Annexin A2 aptamer can target ovarian cancer for tumor-specific doxorubicin delivery. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Kanlikilicer, P.; Ozpolat, B.; Aslan, B.; Bayraktar, R.; Gurbuz, N.; Rodriguez-Aguayo, C.; Lokesh, G.L. Therapeutic Targeting of AXL Receptor Tyrosine Kinase Inhibits Tumor Growth and Intraperitoneal Metastasis in Ovarian Cancer Models. Mol. Ther. Nucleic Acids 2017, 9, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Binzel, D.W.; Khisamutdinov, E.F.; Guo, P. Entropy-driven one-step formation of phi29 pRNA 3WJ from three RNA fragments. Biochemistry 2014, 53, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.; Shu, Y.; Haque, F.; Abdelmawla, S.; Guo, P. Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol. 2011, 6, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Somasunderam, A.; Thiviyanathan, V.; Tanaka, T.; Li, X.; Neerathilingam, M.; Lokesh, G.L.R.; Mann, A.; Peng, Y.; Ferrari, M.; Klostergaard, J.; et al. Combinatorial selection of DNA thioaptamers targeted to the HA binding domain of human CD44. Biochemistry 2010, 49, 9106–9112. [Google Scholar] [CrossRef] [PubMed]
- Lokman, N.A.; Elder, A.S.F.; Ween, M.P.; Pyragius, C.E.; Hoffmann, P.; Oehler, M.K.; Ricciardelli, C. Annexin A2 is regulated by ovarian cancer-peritoneal cell interactions and promotes metastasis. Oncotarget 2013, 4, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Huang, K.W.; Reebye, V.; Spalding, D.; Przytycka, T.M.; Wang, Y.; Swiderski, P.; Li, L.; Armstrong, B.; Reccia, I.; et al. Aptamer-Drug Conjugates of Active Metabolites of Nucleoside Analogs and Cytotoxic Agents Inhibit Pancreatic Tumor Cell Growth. Mol. Ther. Nucleic Acids 2017, 6, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Huang, K.; Reebye, V.; Mintz, P.; Tien, Y.; Lai, H.S.; Sætrom, P.; Reccia, I.; Swiderski, P.; Armstrong, B.; et al. Targeted Delivery of C/EBPα-saRNA by Pancreatic Ductal Adenocarcinoma-specific RNA Aptamers Inhibits Tumor Growth in Vivo. Mol. Ther. 2016, 24, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Rossi, J. Treatment of Pancreatic Cancer by Aptamer Conjugated C/EBPα-saRNA. RNA Act. 2017, 983, 173–188. [Google Scholar] [CrossRef]
- Sharma, T.K.; Bruno, J.G.; Dhiman, A. ABCs of DNA aptamer and related assay development. Biotechnol. Adv. 2017, 35, 275–301. [Google Scholar] [CrossRef] [PubMed]
- Low, P.S.; Henne, W.A.; Doorneweerd, D.D. Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc. Chem. Res. 2008, 41, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Song, Y.; Shi, W.; Li, X.; Ma, H. Distinguishing folate-receptor-positive cells from folate-receptor-negative cells using a fluorescence off-on nanoprobe. Anal. Chem. 2013, 85, 6530–6535. [Google Scholar] [CrossRef] [PubMed]
- Garin-Chesa, P.; Campbell, I.; Saigo, P.E.; Lewis, J.L.; Old, L.J.; Rettig, W.J. Trophoblast and ovarian cancer antigen LK26. Sensitivity and specificity in immunopathology and molecular identification as a folate-binding protein. Am. J. Pathol. 1993, 142, 557–567. [Google Scholar] [PubMed]
- Lv, L.; Zhuang, Y.X.; Zhang, H.W.; Tian, N.N.; Dang, W.Z.; Wu, S.Y. Capsaicin-loaded folic acid-conjugated lipid nanoparticles for enhanced therapeutic efficacy in ovarian cancers. Biomed. Pharmacother. 2017, 91, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.; Raczka, E.; Piehler, L.; Lee, I.; Myc, A.; Majoros, I.; Patri, A.K.; Thomas, T.; Mulé, J.; Baker, J.R. Design and function of a dendrimer-based therapeutic nanodevice targeted to tumor cells through the folate receptor. Pharm. Res. 2002, 19, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Hami, Z.; Rezayat, SM.; Gilani, K.; Amini, M.; Ghazi-Khansari, M. In Vitro cytotoxicity and combination effects of the docetaxel-conjugated and doxorubicin-conjugated poly(lactic acid)-poly(ethylene glycol)-folate-based polymeric micelles in human ovarian cancer cells. J. Pharm. Pharmacol. 2017, 69, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Alberti, D.; Protti, N.; Franck, M.; Stefania, R.; Bortolussi, S.; Altieri, S.; Deagostino, A.; Aime, S.; Geninatti Crich, S. Theranostic Nanoparticles Loaded with Imaging Probes and Rubrocurcumin for Combined Cancer Therapy by Folate Receptor Targeting. ChemMedChem 2017, 12, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Jain, R.K. Strategies for advancing cancer nanomedicine. Nat. Mater. 2013, 12, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Multifunctional nanocarriers. Adv. Drug Deliv. Rev. 2012, 64, 302–315. [Google Scholar] [CrossRef]
- Sivakumar, S.; Bansal, V.; Cortez, C.; Chong, S.F.; Zelikin, A.N.; Caruso, F. Degradable, surfactant-free, monodisperse polymer-encapsulated emulsions as anticancer drug carriers. Adv. Mater. 2009, 21, 1820–1824. [Google Scholar] [CrossRef]
- Wang, Y.; Bansal, V.; Zelikin, A.N.; Caruso, F. Templated synthesis of single-component polymer capsules and their application in drug delivery. Nano Lett. 2008, 8, 1741–1745. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.K.; Ramanathan, R.; Rakwal, R.; Agrawal, G.K.; Bansal, V. Moving forward in plant food safety and security through NanoBioSensors: Adopt or adapt biomedical technologies? Proteomics 2015, 15, 1680–1692. [Google Scholar] [CrossRef] [PubMed]
- Goethals, E.C.; Shukla, R.; Mistry, V.; Bhargava, S.K.; Bansal, V. Role of the templating approach in influencing the suitability of polymeric nanocapsules for drug delivery: LbL vs. SC/MS. Langmuir 2013, 29, 12212–12219. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.L.; Arora, J.; Cowell, S.F.; Garg, A.; Eu, P.; Bhargava, S.K.; Bansal, V. Quasi-cubic magnetite/silica core-shell nanoparticles as enhanced mri contrast agents for cancer imaging. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Shukla, R.; Chanda, N.; Zambre, A.; Upendran, A.; Katti, K.V.; Kulkarni, R.R.; Nune, S.K.; Casteel, S.W.; Smith, C.J.; Vimal, J.; et al. Laminin receptor specific therapeutic gold nanoparticles (198AuNP-EGCg) show efficacy in treating prostate cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 12426–12431. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.; Garbuzenko, O.B.; Ber, E.; Chandna, P.; Khandare, J.J.; Pozharov, V.P.; Minko, T. Receptor targeted polymers, dendrimers, liposomes: Which nanocarrier is the most efficient for tumor-specific treatment and imaging? J. Control. Release 2008, 130, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Hajba, L.; Guttman, A. The use of magnetic nanoparticles in cancer theranostics: Toward handheld diagnostic devices. Biotechnol. Adv. 2016, 34, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Smetsers, T.F.C.M.; Van De Westerlo, E.M.A.; Ten Dam, G.B.; Overes, I.M.; Schalkwijk, J.; Van Muijen, G.N.P.; Van Kuppevelt, T.H. Human single-chain antibodies reactive with native chondroitin sulfate detect chondroitin sulfate alterations in melanoma and psoriasis. J. Investig. Dermatol. 2004, 122, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Liu, X.; Li, L.; Mallapragada, S.; Narasimhan, B.; Wang, Q. Multifunctional nanoparticles for targeted delivery of immune activating and cancer therapeutic agents. J. Control. Release 2013, 172, 1020–1034. [Google Scholar] [CrossRef] [PubMed]
- Saraswathy, M.; Gong, S. Different strategies to overcome multidrug resistance in cancer. Biotechnol. Adv. 2013, 31, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Safari, J.; Zarnegar, Z. Advanced drug delivery systems: Nanotechnology of health design: A review. J. Saudi Chem. Soc. 2014, 18, 85–99. [Google Scholar] [CrossRef]
- Ramasamy, T.; Kim, J.; Choi, J.; Tran, T.; Choi, H.; Yong, C.; Kim, J. pH sensitive polyelectrolyte complex micelles for highly effective combination chemotherapy. J. Mater. Chem. B 2014, 2, 6324–6333. [Google Scholar] [CrossRef]
- Nicolas, J.; Mura, S.; Brammbilla, D.; Mackiewicz, N.; Couvreur, P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem. Soc. Rev. 2013, 42, 1147–1235. [Google Scholar] [CrossRef] [PubMed]
- Arora, D.; Jaglan, S. Nanocarriers based delivery of nutraceuticals for cancer prevention and treatment: A review of recent research developments. Trends Food Sci. Technol. 2016, 54, 114–126. [Google Scholar] [CrossRef]
- He, C.; Poon, C.; Chan, C.; Yamada, S.D.; Lin, W. Nanoscale coordination polymers codeliver chemotherapeutics and sirnas to eradicate tumors of cisplatin-resistant ovarian cancer. J. Am. Chem. Soc. 2016, 138, 6010–6019. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Bourre, L.; Soden, D.M.; O’Sullivan, G.C.; O’Driscoll, C. Can non-viral technologies knockdown the barriers to siRNA delivery and achieve the next generation of cancer therapeutics? Biotechnol. Adv. 2011, 29, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Ozpolat, B.; Sood, A.K.; Lopez-Berestein, G. Nanomedicine based approaches for the delivery of siRNA in cancer. J. Intern. Med. 2010, 267, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Peng, W.; Furuuchi, N.; Gerhart, J.; Rhodes, K.; Mukherjee, N.; Sawicki, J.A. Delivery of therapeutics targeting the mRNA-binding protein HuR using 3DNA nanocarriers suppresses ovarian tumor growth. Cancer Res. 2016, 76, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Lu, K.; Liu, D.; Lin, W. Nanoscale metal-organic frameworks for the co-delivery of cisplatin and pooled siRNAs to enhance therapeutic efficacy in drug-resistant ovarian cancer cells. J. Am. Chem. Soc. 2014, 136, 5181–5184. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Taratula, O.; Garbuzenko, O.B.; Taratula, O.R.; Rodriguez-Rodriguez, L.; Minko, T. Targeted nanomedicine for suppression of CD44 and simultaneous cell death induction in ovarian cancer: An optimal delivery of siRNA and anticancer drug. Clin. Cancer Res. 2013, 19, 6193–6204. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Liong, M.; Xia, T.; Li, Z.; Ji, Z.; Zink, J.I.; Nel, A.E. Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and p-glycoprotein siRNA to overcome drug resistance in a cancer cell line. ACS Nano 2010, 4, 4539–4550. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Cao, N.; Cheng, D.; Zheng, R.; Wang, J.; Zhu, K.; Shuai, X. Enhanced apoptosis of ovarian cancer cells via nanocarrier-mediated codelivery of siRNA and doxorubicin. Int. J. Nanomed. 2012, 7, 3823–3835. [Google Scholar] [CrossRef]
- Ren, Y.; Cheung, H.W.; von Maltzhan, G.; Agrawal, A.; Cowley, G.S.; Weir, B.A.; Boehm, J.S.; Tamayo, P.; Karst, A.M.; Liu, J.F.; et al. Targeted Tumor-Penetrating siRNA Nanocomplexes for Credentialing the Ovarian Cancer Oncogene ID4. Sci. Transl. Med. 2012, 4, 147ra112. [Google Scholar] [CrossRef] [PubMed]
- Fleige, E.; Quadir, M.A.; Haag, R. Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: Concepts and applications. Adv. Drug Deliv. Rev. 2012, 64, 866–884. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur. J. Pharm. Biopharm. 2009, 71, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Lee, S.; Chen, X. Nanoparticle-based theranostic agents. Adv. Drug Deliv. Rev. 2010, 62, 1064–1079. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Emons, G.; Pinski, J.; Schally, A.V. AEZS-108: A targeted cytotoxic analog of LHRH for the treatment of cancers positive for LHRH receptors. Expert Opin. Investig. Drugs 2012, 21, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Saravanakumar, G.; Park, J.H.; Park, K. Hyaluronic acid-based nanocarriers for intracellular targeting: Interfacial interactions with proteins in cancer. Colloids Surfaces B Biointerfaces 2012, 99, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, Y.; Kumar, A.; Tan, A.; Jin, S.; Mozhi, A.; Liang, X.J. PH-Sensitive nano-systems for drug delivery in cancer therapy. Biotechnol. Adv. 2014, 32, 693–710. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Le Breton, A.; Préat, V. RGD-based strategies to target α(v) β(3) integrin in cancer therapy and diagnosis. Mol. Pharm. 2012, 9, 2961–2973. [Google Scholar] [CrossRef] [PubMed]
- Landen, C.N.; Kim, T.-J.; Lin, Y.G.; Merritt, W.M.; Kamat, A.A.; Han, L.Y.; Spannuth, W.A.; Nick, A.M.; Jennnings, N.B.; Kinch, M.S.; et al. Tumor-selective response to antibody-mediated targeting of αvβ3 integrin in ovarian cancer. Neoplasia 2008, 10, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Lössner, D.; Abou-Ajram, C.; Benge, A.; Reuning, U. Integrin αvβ3 mediates upregulation of epidermal growth-factor receptor expression and activity in human ovarian cancer cells. Int. J. Biochem. Cell Biol. 2008, 40, 2746–2761. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Zhong, G.G.; Eun, S.L.; You, H.B. In vivo evaluation of doxorubicin-loaded polymeric micelles targeting folate receptors and early endosomal pH in drug-resistant ovarian cancer. Mol. Pharm. 2009, 6, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Desale, S.S.; Cohen, S.M.; Zhao, Y.; Kabanov, A.V.; Bronich, T.K. Biodegradable hybrid polymer micelles for combination drug therapy in ovarian cancer. J. Control. Release 2013, 171, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Sarisozen, C.; Abouzeid, A.; Torchilin, V. The effect of co-delivery of paclitaxel and curcumin by transferrin-targeted PEG-PE-based mixed micelles on resistant ovarian cancer in 3-D spheroids and in vivo tumors. Eur. J. Pharm. Biopharm. 2014, 88, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Bregoli, L.; Movia, D.; Gavigan-Imedio, J.D.; Lysaght, J.; Reynolds, J.; Prina-Mello, A. Nanomedicine applied to translational oncology: A future perspective on cancer treatment. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.N.; Doemeny, L.J.; Geraci, C.L.; Schmidt, D.G. Nanotechnology Overview: Opportunities and Challenges. ACS Symp. Ser. 2016, 1220, 1–12. [Google Scholar] [CrossRef]
- Pothacharoen, P.; Siriaunkgul, S.; Ong-Chai, S.; Supabandhu, J.; Kumja, P.; Wanaphirak, C.; Sugahara, K.; Hardingham, T.; Kongtawelert, P. Raised serum chondroitin sulfate epitope level in ovarian epithelial cancer. J. Biochem. 2006, 140, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavão, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key players in cancer cell biology and treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.E.; Senger, D.R. Endothelial extracellular matrix: Biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef] [PubMed]
- Engels, B.; Rowley, D.A.; Schreiber, H. Targeting stroma to treat cancers. Semin. Cancer Biol. 2012, 22, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Asimakopoulou, A.P.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N.K. The biological role of chondroitin sulfate in cancer and chondroitin-based anticancer agents. In Vivo (Brooklyn) 2008, 22, 385–390. [Google Scholar]
- Valcarcel, J.; Novoa-Carballal, R.; Pérez-Martín, R.I.; Reis, R.L.; Vázquez, J.A. Glycosaminoglycans from marine sources as therapeutic agents. Biotechnol. Adv. 2017, 35, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, P.; Choonara, Y.E.; Kumar, P.; Pillay, V. “On-the-spot” arresting of chondroitin sulphate proteoglycans: Implications for ovarian adenocarcinoma recognition and intervention. Int. J. Mol. Sci. 2016, 17, 1136. [Google Scholar] [CrossRef] [PubMed]
- Vallen, M.J.E.; Schmidt, S.; Oosterhof, A.; Bulten, J.; Massuger, L.F.A.G.; Van Kuppevelt, T.H. Primary ovarian carcinomas and abdominal metastasis contain 4,6-disulfated chondroitin sulfate rich regions, which provide adhesive properties to tumour cells. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, R.; Willis, C.; van Kuppevelt, T.; Kluppel, M. Biphasic role of chondroitin sulphate in cardiac differentiation of embryonic stem cells through inhibition of wnt/β-catenin signalling. PLoS ONE 2014, 9, e92381. [Google Scholar] [CrossRef] [PubMed]
- Adany, R.; Heimer, R.; Caterson, B.; Sorrell, J.M.; Iozzo, R.V. Altered expression of chondroitin sulfate proteoglycan in the stroma of human colon carcinoma. Hypomethylation of PG-40 gene correlates with increased PG-40 content and mRNA levels. J. Biol. Chem. 1990, 265, 11389–11396. [Google Scholar] [PubMed]
- Isogai, Z.; Shinomura, T.; Yamakawa, N.; Takeuchi, J.; Tsuji, T.; Heinegård, D.; Kimata, K. 2B1 antigen characteristically expressed on extracellular matrices of human malignant tumors is a large chondroitin sulfate proteoglycan, PG- M/versican. Cancer Res. 1996, 56, 3902–3908. [Google Scholar] [PubMed]
- Van Der Steen, S.C.H.A.; Van Tilborg, A.A.G.; Vallen, M.J.E.; Bulten, J.; Van Kuppevelt, T.H.; Massuger, L.F.A.G. Prognostic significance of highly sulfated chondroitin sulfates in ovarian cancer defined by the single chain antibody GD3A11. Gynecol. Oncol. 2016, 140, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Vallen, M.J.E.; Massuger, L.F.A.G.; Ten Dam, G.B.; Bulten, J.; Van Kuppevelt, T.H. Highly sulfated chondroitin sulfates, a novel class of prognostic biomarkers in ovarian cancer tissue. Gynecol. Oncol. 2012, 127, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.A.; Learmonth, D.A.; Sousa, R.A.; Salgado, A.J. αvβ3 and α5β1 integrin-specific ligands: From tumor angiogenesis inhibitors to vascularization promoters in regenerative medicine? Biotechnol. Adv. 2017. [Google Scholar] [CrossRef] [PubMed]
- Vallen, M.J.E.; Van Der Steen, S.C.H.A.; Van Tilborg, A.A.G.; Massuger, L.F.A.G.; Van Kuppevelt, T.H. Sulfated sugars in the extracellular matrix orchestrate ovarian cancer development: “When sweet turns sour”. Gynecol. Oncol. 2014, 135, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Ten Dam, G.B.; van de Westerlo, E.M.A.; Purushothaman, A.; Stan, R.V.; Bulten, J.; Sweep, F.C.G.J.; Massuger, L.F.; Sugahara, K.; van Kuppevelt, T.H. Antibody GD3G7 selected against embryonic glycosaminoglycans defines chondroitin sulfate-E domains highly up-regulated in ovarian cancer and involved in vascular endothelial growth factor binding. Am. J. Pathol. 2007, 171, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef] [PubMed]
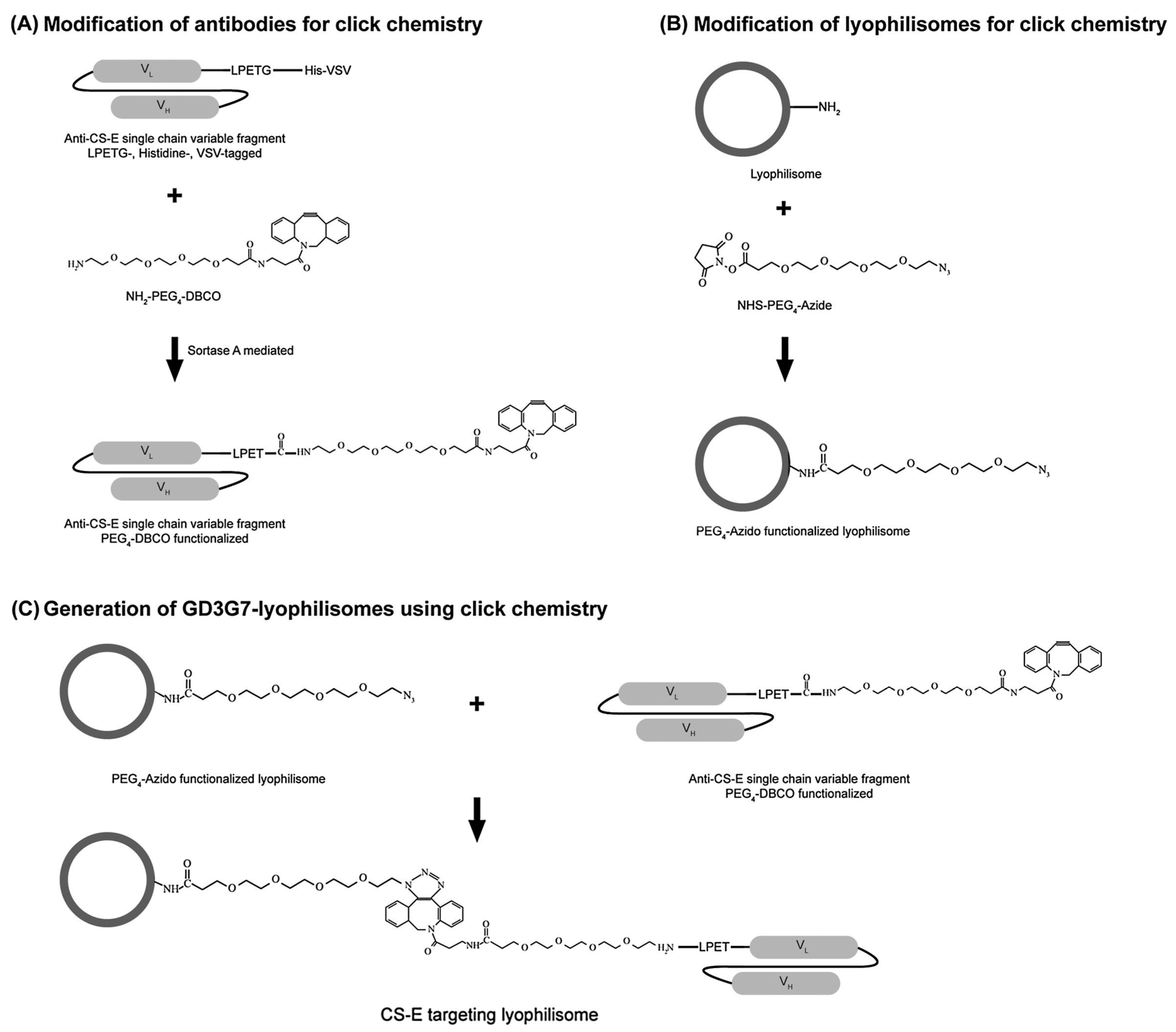
- Van der Steen, S.C.H.A.; Raave, R.; Langerak, S.; van Houdt, L.; van Duijnhoven, S.M.J.; van Lith, S.A.M.; Massuger, L.F.A.G.; Daamen, W.F.; Leenders, W.P.; van Kuppevelt, T.H. Targeting the extracellular matrix of ovarian cancer using functionalized, drug loaded lyophilisomes. Eur. J. Pharm. Biopharm. 2017, 113, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Li, X.; Chen, B.; Wang, B.; Zhao, Y.; Zhuang, Y.; Shen, H.; Zhang, Z.; Dai, J. A collagen-binding EGFR single-chain Fv antibody fragment for the targeted cancer therapy. J. Control. Release 2015, 209, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, M.; Manabe, S.; Tarin, D.; Matsumura, Y. Cancer-stroma targeting therapy by cytotoxic immunoconjugate bound to the collagen 4 network in the tumor tissue. Bioconjug. Chem. 2011, 22, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, I.; Hikita, T.; Mizuno, H.; Sekita, R.; Minami, A.; Ishii, A.; Minamisawa, Y.; Suzuki, K.; Maeda, H.; Hidari, K.I.P.J.; et al. Isolation and characterization of monoclonal antibodies specific for chondroitin sulfate E. Glycobiology 2015, 25, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, M.; Kimata, K.; Oike, Y.; Tani, K.; Maeda, N.; Yoshida, K.; Shimomura, Y.; Yoneda, M.; Suzuki, S. A monoclonal antibody that specifically recognizes a glucuronic acid 2-sulfate-containing determinant in intact chondroitin sulfate chain. J. Biol. Chem. 1987, 262, 4146–4152. [Google Scholar] [PubMed]
- Weigel, P.H.; DeAngelis, P.L. Hyaluronan synthases: A decade-plus of novel glycosyltransferases. J. Biol. Chem. 2007, 282, 36777–36781. [Google Scholar] [CrossRef] [PubMed]
- Delpech, B.; Girard, N.; Bertrand, P.; Courel, M.N.; Chauzy, C.; Delpech, A. Hyaluronan: Fundamental principles and applications in cancer. J. Intern. Med. 1997, 242, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Anttila, M.A.; Tammi, R.H.; Tammi, M.I.; Syrjänen, K.J.; Saarikoski, S.V.; Kosma, V.M. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res. 2000, 60, 150–155. [Google Scholar] [PubMed]
- Yeo, T.K.; Nagy, J.A.; Yeo, K.T.; Dvorak, H.F.; Toole, B.P. Increased hyaluronan at sites of attachment to mesentery by CD44-positive mouse ovarian and breast tumor cells. Am. J. Pathol. 1996, 148, 1733–1740. [Google Scholar] [PubMed]
- Csoka, A.B.; Frost, G.I.; Stern, R. The six hyaluronidase-like genes in the human and mouse genomes. Matrix Biol. 2001, 20, 499–508. [Google Scholar] [CrossRef]
- Montagner, I.M.; Merlo, A.; Carpanese, D.; Zuccolotto, G.; Renier, D.; Campisi, M.; Pasut, G.; Zanovello, P.; Rosato, A. Drug conjugation to hyaluronan widens therapeutic indications for ovarian cancer. Oncoscience 2015, 2, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.W.; Peyrollier, K.; Xia, W.; Gilad, E. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J. Biol. Chem. 2008, 283, 17635–17651. [Google Scholar] [CrossRef] [PubMed]
- Ween, M.P.; Oehler, M.K.; Ricciardelli, C. Role of versican, hyaluronan and CD44 in ovarian cancer metastasis. Int. J. Mol. Sci. 2011, 12, 1009–1029. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Gayol, A.; Jothy, S. Effects of hyaluronan on the invasive properties of human breast cancer cells in vitro. Int. J. Exp. Pathol. 2001, 82, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.W.; Zhu, H.; Chu, A.; Iida, N.; Zhang, L.; Hung, M.C. Interaction between the adhesion receptor, CD44, and the oncogene product, p185(HER2), promotes human ovarian tumor cell activation. J. Biol. Chem. 1997, 272, 27913–27918. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.W.; Gilad, E.; Peyrollier, K. Heregulin-mediated ErbB2-ERK signaling activates hyaluronan synthases leading to CD44-dependent ovarian tumor cell growth and migration. J. Biol. Chem. 2007, 282, 19426–19441. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, E.L.J.; Anttila, M.; Kultti, A.; Ropponen, K.; Penttinen, J.; Yliskoski, M.; Kuronen, A.T.; Juhola, M.; Tammi, R.; Tammi, M.; et al. Elevated hyaluronan concentration without hyaluronidase activation in malignant epithelial ovarian tumors. Cancer Res. 2002, 62, 6410–6413. [Google Scholar] [PubMed]
- Tamakoshi, K.; Kikkawa, F.; Maeda, O.; Suganuma, N.; Yamagata, S.; Yamagata, T.; Tomoda, Y. Hyaluronidase activity in gynaecological cancer tissues with different metastatic forms. Br. J. Cancer 1997, 75, 1807–1811. [Google Scholar] [CrossRef] [PubMed]
- Weiss, I.; Trope, C.G.; Reich, R.; Davidson, B. Hyaluronan synthase and hyaluronidase expression in serous ovarian carcinoma is related to anatomic site and chemotherapy exposure. Int. J. Mol. Sci. 2012, 13, 12925–12938. [Google Scholar] [CrossRef] [PubMed]
- Nykopp, T.K.; Rilla, K.; Sironen, R.; Tammi, M.I.; Tammi, R.H.; Hämäläinen, K.; Heikkinen, A.-M.; Komulainen, M.; Kosma, V.-M.; Anttila, M. Expression of hyaluronan synthases (HAS1-3) and hyaluronidases (HYAL1-2) in serous ovarian carcinomas: Inverse correlation between HYAL1 and hyaluronan content. BMC Cancer 2009, 9, 143. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Min, K.H.; Na, J.H.; Choi, K.; Kim, K.; Park, J.H.; Kwon, I.C.; Jeong, S.Y. Self-assembled hyaluronic acid nanoparticles as a potential drug carrier for cancer therapy: Synthesis, characterization, and in vivo biodistribution. J. Mater. Chem. 2009, 19, 4102. [Google Scholar] [CrossRef]
- Auzenne, E.; Ghosh, S.C.; Khodadadian, M.; Rivera, B.; Farquhar, D.; Price, R.E.; Ravoori, M.; Kundra, V.; Freedman, R.S.; Klostergaard, J. Hyaluronic acid-paclitaxel: Antitumor efficacy against CD44(+) human ovarian carcinoma xenografts. Neoplasia 2007, 9, 479–486. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Targeting Moiety | Nanocarrier | Response | Reference |
|---|---|---|---|---|
| Cisplatin, GEM and siRNA | - | Lipid based nanocarrier, with a self -assembled core-shell NCP | - NCP-siRNAs NPs efficiently downregulated the Bcl-2 gene expression in SKOV-3 and A2780/CDDP cells by >70%. - NCP-siRNAs NPs successfully eradicated tumours causing 100% survival in mouse models for > 90 days. - 92 days after tumour inoculation, NCP-1/siRNAs treated mice were sacrificed with no evidence tumours. | [86] |
| siRNA | FA to target HuR Overexpression | Derivatized DNA dendrimer | - When Mice were injected twice weekly with FA-3DNA-siHuR for 4 weeks, the median survival of FA-3DNA-siHuR-treated mice were approximately 1.5 times longer than the controls. | [89] |
| Co-delivery of cisplatin and siRNAs | - | NMOFs | - The cisplatin IC50 values of free cisplatin, UiO-Cis, pooled siRNAs/UiO-Cis, free cisplatin plus free pooled siRNAs, and free cisplatin plus pooled siRNAs/UiO were 53.9 ± 4.7, 53.2 ± 4.4, 4.7 ± 1.8, 45.1 ± 7.0, and 6.6 ± 0.3 μM, respectively. - Co-delivering of pooled siRNAs and cisplatin employing NMOFs decreased the IC50 value >11-fold, in contrast to free cisplatin and UiO-Cis. | [90] |
| PTX and siRNA | LHRH peptide | Nanoscale PPI dendrimer | - LHRH-PPI-siRNA and LHRH-PPI-PTX combination enhanced the cytotoxicity of the conjugate. - The viability of ascitic cells were decreased almost 10-fold in comparison to the control cells, more than 5-fold when compared with free PTX and more than 2-fold when compared with non-targeted PPI-PTX-siRNA complex. - The combination caused almost complete tumour shrinkage within 28-days. | [91] |
| siRNA | HA-NP system targeting CD44 receptors | HA based self-assembling NPs | - Tumour volume of mice treated with HA-PEG/MDR1 siRNAs targeted NPs was approximately 3-fold lower than in mice treated with native PTX. | [3] |
| DOX and siRNA | NA | MSNP | - MSNP with siRNAs caused increased cellular accumulation of DOX. - The IC50 value of the siRNA delivering MSNP was approximately 2.5 times lower than the IC50 of free Dox or other Dox loaded particles. | [92] |
| Blc2-siRNA and DOX | FA-targeting overexpressed FR | copolymer self-assembled cationic micelles | - The highest apoptosis of 77.5% were observed in cells incubated with FA-DOX-Bcl2 siRNA-NPs leading to potent synergistic effects inducing cell apoptosis. | [93] |
| siID4 | tandem tumour-penetrating and membrane-translocating peptide to target ID4 | TPN | - The tumour burden in mice that received TPN/siID4 remained low compared to controls, 80% of these recipients survived >60 days, despite treatments stopping at day 50. - No visible tumour lesions in 4 out of 5 TPN/siID4–treated mice occurred indicating tumour regression. - Histological analysis revealed significant reduction in ID4 levels and increased apoptosis in the tumour parenchyma of mice treated with TPN/siID4. | [94] |
| Drug | Targeting Moiety | Nanocarrier | Response | Reference |
|---|---|---|---|---|
| DOX | FA to target FR | pH-sensitive micelles | - The micelle formulation effectively decreased the growth of existing MDR tumours in mice for at least 50 days by three i.v. injections at a 3-day interval at a dose of 10 mg DOX/kg. - Tumour growth rate of the micelle group was delayed when compared with free DOX group. | [104] |
| PTX | OA02 peptide | Micellar NPs formed using PEG-block-dendritic CA copolymers (PTX-OA02-NPs) | - PTX-OA02-NPs displayed superior tumour growth inhibition than Taxol®, at equivalent PTX dose of 10 mg/kg. - The median survival time of mice in the group of PBS control, Taxol®, PTX-NPs (10, 30 mg/kg), and PTX-OA02-NPs (10, 30 mg/kg) were 20, 27, 29, 69, 32, and 95 days, respectively. | [27] |
| PTX and CDDP | NA | Biodegradable, biocompatible polypeptide-based polymeric micelles | - MTT assays on A2780 cells revealed IC50 values Free CDDP is 1.5 in contrast to (CDDP + PTX)/cl-micelles of 0.14 exhibiting superior killing properties. | [105] |
| DOX | TATp | PEG-Hz-PE conjugated immunoliposomes | - In vitro cytotoxicity revealed the lowest cell viability was obtained Lipodox-TATp in both SKOV-3 sensitive and SKOV-3 resistant cells yielding IC50 values of 0.36 and 3.12 respectively in contrast to lipodox yielding IC50 values of 6.25 and 100.00 respectively. | [30] |
| Co-delivery of PTX and curcumin | TF to target resistant OC spheroids and in vivo tumours | PEG-PE based polymeric micelles | - The TF-targeted PTX system displayed and enhanced micelle penetration into spheroids reducing cell viability to 35.3 ± 2.7% at 6.9 µM of micellar PTX concentrations when compared to free PTX at 80 ± 22% at 6.9 µM dosage. | [106] |
| GEM | cRGDfK peptide targeting αvβ3 integrin receptors | PLGA based NPs | - The IC50 values after 48 hours of incubation in SKOV3 cells for GEM, GEM-NPs and GEM-RGDfK-NPs were 0.572 ± 0.013 μg/ml, 0.148 ± 0.01 μg/mL and 0.034 ± 0.004 μg/mL respectively. - The improved anticancer efficacy may be attributed to the targeting properties of the peptide. | [25] |
| DOX coupled with a NIR dye for theranostic application | LLHRH | self-assembling HA NPs, | - Results demonstrate that LHRH-conjugated NPs possess a desirable tumour imaging capability and an excellent anticancer effect, such that almost 30% of the original tumour size was reduced in 20 days. | [24] |
| Implication in OC | References |
|---|---|
| CS-E displays strong up regulation in primary ovarian carcinomas which is responsible for the poor prognostic parameters, including high tumour grade and advanced FIGO stages. | [115,121] |
| CS-E can strongly bind to VEGF which is the most important pro- angiogenic stimulator. Furthermore, high CS-E levels correlate to high VEGF, causing further neo-vascularization development in the tumour stroma causing ovarian spheroid formation this spheroid formation is associated with the highly aggressive and invasive characteristics of OC. | [115,116,122] |
| The elevated CS-E aids the adhesion of tumours as it is responsible for increasing the adhesive properties of adhesion molecules N-cadherin and E- cadherin. In addition, integrins also play a role in adhesion as they can interact directly with CS chains, blocking of integrin receptors result in inhibition of OC cell adhesion. | [115,123] |
| The overexpression of CS-E improves the adhesive properties of tumour cells. | [115] |
| CS-E is responsible for the invasion and migration of tumours. MMPs is a group of enzymes responsible for OC progression. The activation and regulation are strongly influenced by CS, Furthermore, CS-E can interact with MPPs such as pro-MMP7, contributing to the activation and metastasis of tumour cells. | [123] |
| CS-E expression is predominantly seen in the stromal compartment of both primary ovarian carcinomas and metastasis. | [116] |
| The expression of mRNA for GalNAc4S-6ST, an enzyme which is responsible for the biosynthesis of CS-E, is up-regulated in OC. | [115,124] |
| CHST15, the only sulfotransferase responsible for biosynthesis of CS-E presents an altered transcription pattern in OC, furthermore increased CHST15 levels lead to increased CS-E levels. | [123,124] |
| CS has shown to be involved tumour cell proliferation, growth, angiogenesis, adhesion, migration, invasion, and survival of OC tumours. | [123] |
| Antibody | Specificity | Target Selectivity of CS Isomer | References |
|---|---|---|---|
| GD3G7 antibody |
| [124] | |
| The mAb 473HD | DSD |
| [124] |
| mAb CS-56 |
| [124] | |
| mAb MO-225 |
| [130] | |
| 473HD | CS-D |
| [130] |
| IO3D9 |
| [79] | |
| IO3H10 |
| [79] | |
| IO3H12 |
| [79] | |
| 12C and E-18H | CS-E specific |
| [129] |
| GD3A11 | CS-E specific |
| [120] |
| Implication in OC | References |
|---|---|
| CD44 is proposed to be involved in increased motility of cancer cells as well as differentiation of cancer stem cells | [91] |
| Co-overexpression of CD44 and multiple drug resistance proteins such as MDR1 and MDR2 correlate to OC progression. | [138] |
| RHAMM and CD44 are vital components for tumour progression. | [139] |
| When HA synthase and HA are down regulated directly using siRNAs it causes impaired cytoskeletal activation and decreased migration of tumours. | [141] |
| HA mediates the physical linkage between CD44s and Her2/ErbB2 tyrosine kinase, which results in the rapid stimulation of ovarian carcinoma cell growth. | [140] |
| Elevated CD44 expression is observed in OC in contrast to benign and borderline tumours. | [138] |
| Elevated HA concentration provides and additional growth advantage of primary ovarian tumours due to the cells ability to produce a HA rich stroma environment. | [142] |
| The high stromal HA levels are significantly associated with poor differentiation, serous histological type, advanced stage, and large primary residual tumour in epithelial OC. | [133] |
| CD44 expressions correlate with the expression of the drug efflux pump Pgp causing resistance and progression of metastases. | [91] |
| CD44 inhibition following treatment of the CD44 monoclonal antibody inhibits OC cell motility no significant impact on invasion | [3] |
| Anti-CD44 antibody has been shown to decrease the number of total peritoneal OC metastases in mice. | [3] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoosen, Y.; Pradeep, P.; Kumar, P.; Du Toit, L.C.; Choonara, Y.E.; Pillay, V. Nanotechnology and Glycosaminoglycans: Paving the Way Forward for Ovarian Cancer Intervention. Int. J. Mol. Sci. 2018, 19, 731. https://doi.org/10.3390/ijms19030731
Hoosen Y, Pradeep P, Kumar P, Du Toit LC, Choonara YE, Pillay V. Nanotechnology and Glycosaminoglycans: Paving the Way Forward for Ovarian Cancer Intervention. International Journal of Molecular Sciences. 2018; 19(3):731. https://doi.org/10.3390/ijms19030731
Chicago/Turabian StyleHoosen, Yasar, Priyamvada Pradeep, Pradeep Kumar, Lisa C. Du Toit, Yahya E. Choonara, and Viness Pillay. 2018. "Nanotechnology and Glycosaminoglycans: Paving the Way Forward for Ovarian Cancer Intervention" International Journal of Molecular Sciences 19, no. 3: 731. https://doi.org/10.3390/ijms19030731
APA StyleHoosen, Y., Pradeep, P., Kumar, P., Du Toit, L. C., Choonara, Y. E., & Pillay, V. (2018). Nanotechnology and Glycosaminoglycans: Paving the Way Forward for Ovarian Cancer Intervention. International Journal of Molecular Sciences, 19(3), 731. https://doi.org/10.3390/ijms19030731