Endogenous Selenoprotein P, a Liver-Derived Secretory Protein, Mediates Myocardial Ischemia/Reperfusion Injury in Mice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. SeP Gene Deletion Reduces I/R Injury in Mice
2.2. SeP Gene Deletion Reduces I/R-Induced Apoptosis in the Heart
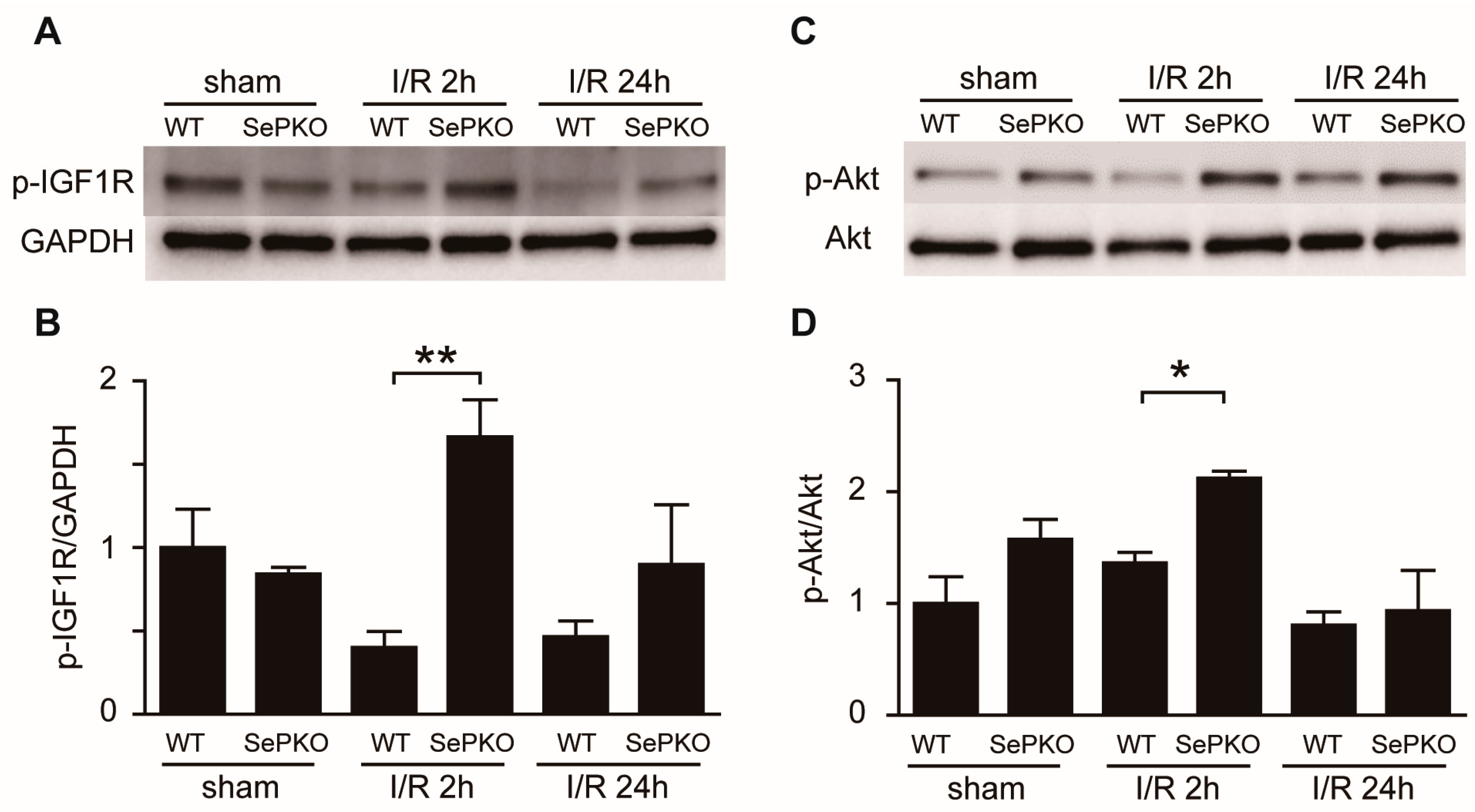
2.3. SeP Gene Deletion Increases the Phosphorylation of IGF-1 Receptor, Akt, Erk, and S6K
2.4. SeP Induces Myocardial I/R Injury in Mice
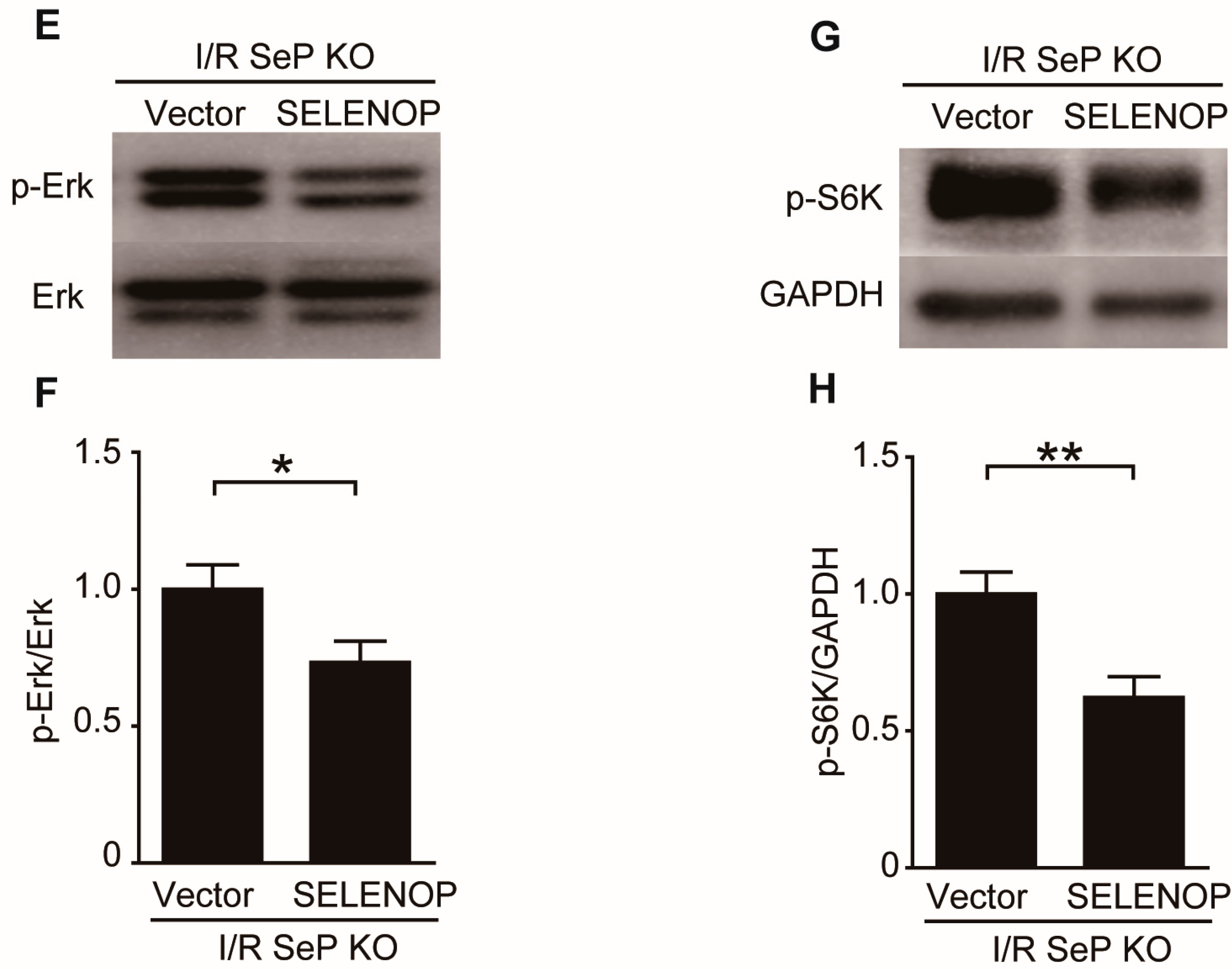
2.5. SeP Impaired the RISK Pathway during I/R Injury in Mice
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animal Models and Experimental Procedures
4.3. Evaluation of Apoptosis
4.4. Western Blotting
4.5. Preparation of Human SELENOP Plasmids and Overexpression of SeP in Mice
4.6. Total RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (QRT-PCR)
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| SeP | selenoprotein P |
| I/R | ischemia/reperfusion |
| KO | knockout |
| WT | wild-type |
| IA/AAR | myocardial infarction/area at risk |
| EB | Evans blue |
| TTC | 2,3,5-triphenyltetrazolium chloride |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick-end labeling |
| RISK | reperfusion injury salvage kinase |
| ACS | acute coronary syndrome |
| Erk1/2 | extracellular signal-regulated kinase 1/2 |
References
- Weaver, W.D.; Simes, R.J.; Betriu, A.; Grines, C.L.; Zijlstra, F.; Garcia, E.; Grinfeld, L.; Gibbons, R.J.; Ribeiro, E.E.; DeWood, M.A.; et al. Comparison of primary coronary angioplasty and intravenous thrombolytic therapy for acute myocardial infarction: A quantitative review. JAMA 1997, 278, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E.; Kloner, R.A. Myocardial reperfusion: A double-edged sword? J. Clin. Investig. 1985, 76, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Reperfusion injury salvage kinase signalling: Taking a risk for cardioprotection. Heart Fail. Rev. 2007, 12, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Rennert, G.; Saltz-Rennert, H.; Wanderman, K.; Weitzman, S. Size of acute myocardial infarcts in patients with diabetes mellitus. Am. J. Cardiol. 1985, 55, 1629–1630. [Google Scholar] [CrossRef]
- Ishihara, M.; Inoue, I.; Kawagoe, T.; Shimatani, Y.; Kurisu, S.; Nishioka, K.; Kouno, Y.; Umemura, T.; Nakamura, S.; Sato, H. Diabetes mellitus prevents ischemic preconditioning in patients with a first acute anterior wall myocardial infarction. J. Am. Coll. Cardiol. 2001, 38, 1007–1011. [Google Scholar] [CrossRef]
- Kristiansen, S.B.; Lofgren, B.; Stottrup, N.B.; Khatir, D.; Nielsen-Kudsk, J.E.; Nielsen, T.T.; Botker, H.E.; Flyvbjerg, A. Ischaemic preconditioning does not protect the heart in obese and lean animal models of type 2 diabetes. Diabetologia 2004, 47, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Novoselov, S.V.; Kumaraswamy, E.; Lee, B.J.; Anver, M.R.; Gladyshev, V.N.; Hatfield, D.L. Specific excision of the selenocysteine tRNA[Ser]Sec (Trsp) gene in mouse liver demonstrates an essential role of selenoproteins in liver function. J. Biol. Chem. 2004, 279, 8011–8017. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Takahashi, K. Characterization of selenoprotein P as a selenium supply protein. Eur. J. Biochem. 2002, 269, 5746–5751. [Google Scholar] [CrossRef] [PubMed]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein p, causes insulin resistance. Cell Metab. 2010, 12, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Mita, Y.; Nakayama, K.; Inari, S.; Nishito, Y.; Yoshioka, Y.; Sakai, N.; Sotani, K.; Nagamura, T.; Kuzuhara, Y.; Inagaki, K.; et al. Selenoprotein p-neutralizing antibodies improve insulin secretion and glucose sensitivity in type 2 diabetes mouse models. Nat. Commun. 2017, 8, 1658. [Google Scholar] [CrossRef] [PubMed]
- Misu, H.; Takayama, H.; Saito, Y.; Mita, Y.; Kikuchi, A.; Ishii, K.A.; Chikamoto, K.; Kanamori, T.; Tajima, N.; Lan, F.; et al. Deficiency of the hepatokine selenoprotein p increases responsiveness to exercise in mice through upregulation of reactive oxygen species and amp-activated protein kinase in muscle. Nat. Med. 2017, 23, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Ishikura, K.; Misu, H.; Kumazaki, M.; Takayama, H.; Matsuzawa-Nagata, N.; Tajima, N.; Chikamoto, K.; Lan, F.; Ando, H.; Ota, T.; et al. Selenoprotein P as a diabetes-associated hepatokine that impairs angiogenesis by inducing VEGF resistance in vascular endothelial cells. Diabetologia 2014, 57, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E. Selenoprotein P: An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr. 2005, 25, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Stone, G.W.; Holmes, D.R.; Gersh, B. Reperfusion injury, microvascular dysfunction, and cardioprotection: The “dark side” of reperfusion. Circulation 2009, 120, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Baxter, G.F. Reperfusion injury revisited: Is there a role for growth factor signaling in limiting lethal reperfusion injury? Trends Cardiovasc. Med. 1999, 9, 245–249. [Google Scholar] [CrossRef]
- Whittington, H.J.; Harding, I.; Stephenson, C.I.; Bell, R.; Hausenloy, D.J.; Mocanu, M.M.; Yellon, D.M. Cardioprotection in the aging, diabetic heart: The loss of protective akt signalling. Cardiovasc. Res. 2013, 99, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. Diabetes abolishes morphine-induced cardioprotection via multiple pathways upstream of glycogen synthase kinase-3β. Diabetes 2007, 56, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Kloner, R.A. Cardioprotection of insulin-like growth factor-1 during reperfusion therapy: What is the underlying mechanism or mechanisms? Circ. Cardiovasc. Interv. 2011, 4, 311–313. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.F.; Leblond, A.L.; Kelly, G.; Kumar, A.H.; Metharom, P.; Buneker, C.K.; Alizadeh-Vikali, N.; Hristova, I.; Hynes, B.G.; O’Connor, R.; et al. Potent long-term cardioprotective effects of single low-dose insulin-like growth factor-1 treatment postmyocardial infarction. Circ. Cardiovasc. Interv. 2011, 4, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Hsieh, P.C.; Takahashi, T.; Song, Q.; Zhang, S.; Kamm, R.D.; Grodzinsky, A.J.; Anversa, P.; Lee, R.T. Local myocardial insulin-like growth factor 1 (IGF-1) delivery with biotinylated peptide nanofibers improves cell therapy for myocardial infarction. Proc. Natl. Acad. Sci. USA 2006, 103, 8155–8160. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Shan, Y.X.; Yang, T.L.; Lin, H.D.; Chen, J.W.; Lin, S.J.; Wang, P.H. Insulin deficiency downregulated heat shock protein 60 and IGF-1 receptor signaling in diabetic myocardium. Diabetes 2005, 54, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Engler, R.L. Apoptosis in myocardial ischemia-reperfusion. Ann. N. Y. Acad. Sci. 1999, 874, 412–426. [Google Scholar] [CrossRef] [PubMed]
- MacLellan, W.R.; Schneider, M.D. Death by design. Programmed cell death in cardiovascular biology and disease. Circ. Res. 1997, 81, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Konhilas, J.P.; Kelly, D.P.; Leinwand, L.A. Molecular mechanisms underlying cardiac adaptation to exercise. Cell Metab. 2017, 25, 1012–1026. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.M.; Mekary, R.A.; da Cruz Rodrigues, K.C.; Anaruma, C.P.; Ropelle, E.R.; da Silva, A.S.R.; Cintra, D.E.; Pauli, J.R.; de Moura, L.P. Protective molecular mechanisms of clusterin against apoptosis in cardiomyocytes. Heart Fail. Rev. 2017, 23, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.E.; Zhou, J.; McMahan, W.J.; Motley, A.K.; Atkins, J.F.; Gesteland, R.F.; Burk, R.F. Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 2003, 278, 13640–13646. [Google Scholar] [CrossRef] [PubMed]
- Takatori, O.; Usui, S.; Okajima, M.; Kaneko, S.; Ootsuji, H.; Takashima, S.I.; Kobayashi, D.; Murai, H.; Furusho, H.; Takamura, M. Sodium 4-phenylbutyrate attenuates myocardial reperfusion injury by reducing the unfolded protein response. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Kitano, K.; Usui, S.; Ootsuji, H.; Takashima, S.; Kobayashi, D.; Murai, H.; Furusho, H.; Nomura, A.; Kaneko, S.; Takamura, M. Rho-kinase activation in leukocytes plays a pivotal role in myocardial ischemia/reperfusion injury. PLoS ONE 2014, 9, e92242. [Google Scholar] [CrossRef] [PubMed]
- Usui, S.; Maejima, Y.; Pain, J.; Hong, C.; Cho, J.; Park, J.Y.; Zablocki, D.; Tian, B.; Glass, D.J.; Sadoshima, J. Endogenous muscle atrophy F-Box mediates pressure overload-induced cardiac hypertrophy through regulation of NF-κB. Circ. Res. 2011, 109, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Renko, K.; Werner, M.; Renner-Muller, I.; Cooper, T.G.; Yeung, C.H.; Hollenbach, B.; Scharpf, M.; Kohrle, J.; Schomburg, L.; Schweizer, U. Hepatic selenoprotein P (SePP) expression restores selenium transport and prevents infertility and motor-incoordination in Sepp-knockout mice. Biochem. J. 2008, 409, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Takamura, M.; Kurokawa, K.; Ootsuji, H.; Inoue, O.; Okada, H.; Nomura, A.; Kaneko, S.; Usui, S. Long-term administration of eicosapentaenoic acid improves post-myocardial infarction cardiac remodeling in mice by regulating macrophage polarization. J. Am. Heart Assoc. 2017, 6, e004560. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chadani, H.; Usui, S.; Inoue, O.; Kusayama, T.; Takashima, S.-i.; Kato, T.; Murai, H.; Furusho, H.; Nomura, A.; Misu, H.; et al. Endogenous Selenoprotein P, a Liver-Derived Secretory Protein, Mediates Myocardial Ischemia/Reperfusion Injury in Mice. Int. J. Mol. Sci. 2018, 19, 878. https://doi.org/10.3390/ijms19030878
Chadani H, Usui S, Inoue O, Kusayama T, Takashima S-i, Kato T, Murai H, Furusho H, Nomura A, Misu H, et al. Endogenous Selenoprotein P, a Liver-Derived Secretory Protein, Mediates Myocardial Ischemia/Reperfusion Injury in Mice. International Journal of Molecular Sciences. 2018; 19(3):878. https://doi.org/10.3390/ijms19030878
Chicago/Turabian StyleChadani, Hiroshi, Soichiro Usui, Oto Inoue, Takashi Kusayama, Shin-ichiro Takashima, Takeshi Kato, Hisayoshi Murai, Hiroshi Furusho, Ayano Nomura, Hirofumi Misu, and et al. 2018. "Endogenous Selenoprotein P, a Liver-Derived Secretory Protein, Mediates Myocardial Ischemia/Reperfusion Injury in Mice" International Journal of Molecular Sciences 19, no. 3: 878. https://doi.org/10.3390/ijms19030878