Signaling Crosstalk of TGF-β/ALK5 and PAR2/PAR1: A Complex Regulatory Network Controlling Fibrosis and Cancer

 ,
,  ,
, 

Abstract
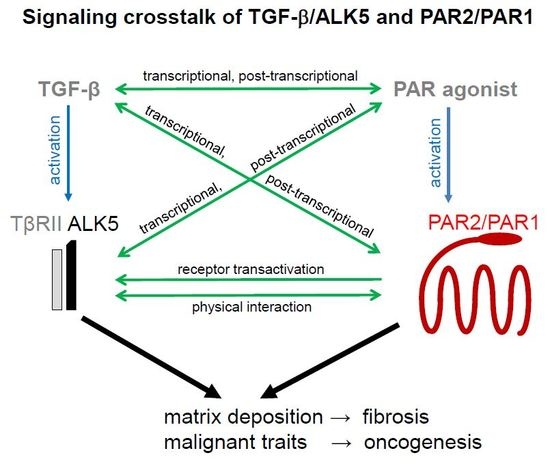
{kind=link}
1. Introduction
2. TGF-β, PARs, and Sphingosine-1-Phosphate as a Mediator between TGF-β and PAR Signaling
2.1. TGF-β
2.2. PARs
2.3. Sphingosine-1-Phosphate (S1P) as a Mediator between TGF-β and PAR Signaling
3. Inflammatory/Fibrotic Disorders
3.1. Lung Fibrosis
3.2. Kidney Fibrosis
3.3. Liver Fibrosis
3.4. Cardiac Fibrosis
3.5. Wound Healing
4. Cancer
5. Types of TGF-β–PAR2 and TGF-β–PAR1 Interactions
5.1. Regulation of TGF-β, TGF-β Receptors, and SMAD Proteins by PAR1 and PAR2 and their Ligands
5.2. Regulation of PAR1 and PAR2 Expression and Activation by TGF-β
6. Conclusions and Potential Therapeutical Implications
Acknowledgments
Conflicts of Interest
References
- Kitisin, K.; Saha, T.; Blake, T.; Golestaneh, N.; Deng, M.; Kim, C.; Tang, Y.; Shetty, K.; Mishra, B.; Mishra, L. Tgf-Beta signaling in development. Sci. STKE. 2007, 399, cm1. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Unchaining the beast; insights from structural and evolutionary studies on tgfb secretion, sequestration, and activation. Cytokine Growth Factor Rev. 2013, 24, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Sheppard, D. Cross talk among tgf-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harbor Perspect. Biol. 2011, 3, a005017. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. Tgf-beta activates erk map kinase signalling through direct phosphorylation of shca. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Jakowlew:, S.B. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006, 25, 435–457. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Yang, X. Smad4-mediated TGF-beta signaling in tumorigenesis. Int. J. Biol. Sci. 2010, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Ehata, S.; Koinuma, D. Tumour-promoting functions of transforming growth factor-β in progression of cancer. Ups. J. Med. Sci. 2012, 117, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Calone, I.; Souchelnytskyi, S. Inhibition of TGFβ signaling and its implication in anticancer treatments. Exp. Oncol. 2012, 34, 9–16. [Google Scholar] [PubMed]
- Fabregat, I.; Fernando, J.; Mainez, J.; Sancho, P. TGF-beta signaling in cancer treatment. Curr. Pharm. Des. 2014, 20, 2934–2947. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Derynck, R. Essential role of TGF-β signaling in glucose-induced cell hypertrophy. Dev. Cell. 2009, 17, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xu, P.; Lamouille, S.; Xu, J.; Derynck, R. TACE-mediated ectodomain shedding of the type I TGF-β receptor downregulates TGF-β signaling. Mol. Cell. 2009, 35, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Mestdagh, P.; Bostrom, A.K.; Impens, F.; Fredlund, E.; Van Peer, G.; De Antonellis, P.; von Stedingk, K.; Ghesquiere, B.; Schulte, S.; Dews, M.; et al. The miR-17–92 microRNA cluster regulates multiple components of the TGF-β pathway in neuroblastoma. Mol. Cell 2010, 40, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Ramachandran, R.; Hollenberg, M.D.; Muruve, D.A. Proteinase-activated receptor-2 transactivation of epidermal growth factor receptor and transforming growth factor-β receptor signaling pathways contributes to renal fibrosis. J. Biol. Chem. 2013, 288, 37319–37331. [Google Scholar] [CrossRef] [PubMed]
- Dayati, P.; Rezaei, H.B.; Sharifat, N.; Kamato, D.; Little, P.J. G protein coupled receptors can transduce signals through carboxy terminal and linker region phosphorylation of Smad transcription factors. Life Sci. 2018, 199, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Talati, N.; Kamato, D.; Piva, T.J.; Little, P.J.; Osman, N. Thrombin promotes PAI-1 expression and migration in keratinocytes via ERK dependent Smad linker region phosphorylation. Cell Signal. 2018, 47, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Takekawa, M.; Tatebayashi, K.; Itoh, F.; Adachi, M.; Imai, K.; Saito, H. Smad-dependent GADD45beta expression mediates delayed activation of p38 MAP kinase by TGF-beta. EMBO J. 2002, 21, 6473–6482. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Groth, S.; Ruhnke, M.; Kalthoff, H.; Fändrich, F. Transforming growth factor-beta (TGF-beta) type I receptor/ALK5-dependent activation of the GADD45beta gene mediates the induction of biglycan expression by TGF-beta. J. Biol. Chem. 2005, 280, 2644–2652. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Lenschow, W.; Chen, W.B.; Faendrich, F.; Kalthoff, H. Regulation of biglycan gene expression by transforming growth factor-beta requires MKK6-p38 mitogen-activated protein Kinase signaling downstream of Smad signaling. J. Biol. Chem. 2003, 278, 11041–11049. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Gieseler, F.; Ungefroren, H.; Settmacher, U.; Hollenberg, M.D.; Kaufmann, R. Proteinase-activated receptors (PARs)—Focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun. Signal. 2013, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, M.D.; Mihara, K.; Polley, D.; Suen, J.Y.; Han, A.; Fairlie, D.P.; Ramachandran, R. Biased signalling and proteinase-activated receptors (PARs): Targeting inflammatory disease. Br. J. Pharmacol. 2014, 171, 1180–1194. [Google Scholar] [CrossRef] [PubMed]
- Mihara, K.; Ramachandran, R.; Saifeddine, M.; Hansen, K.K.; Renaux, B.; Polley, D.; Gibson, S.; Vanderboor, C.; Hollenberg, M.D. Thrombin-Mediated Direct Activation of Proteinase-Activated Receptor-2: Another Target for Thrombin Signaling. Mol. Pharmacol. 2016, 89, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Burch, M.L.; Ballinger, M.L.; Yang, S.N.; Getachew, R.; Itman, C.; Loveland, K.; Osman, N.; Little, P.J. Thrombin stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by protease-activated receptor-1 transactivation of the transforming growth factor beta type I receptor. J. Biol. Chem. 2010, 285, 26798–26805. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; von der Thüsen, J.; Daalhuisen, J.; ten Brink, M.; Crestani, B.; van der Poll, T.; Borensztajn, K.; Spek, C.A. Protease-activated receptor (PAR)-2 is required for PAR-1 signalling in pulmonary fibrosis. J. Cell. Mol. Med. 2015, 19, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Jaber, M.; Maoz, M.; Kancharla, A.; Agranovich, D.; Peretz, T.; Grisaru-Granovsky, S.; Uziely, B.; Bar-Shavit, R. Protease-activated-receptor-2 affects protease-activated-receptor-1-driven breast cancer. Cell. Mol. Life Sci. 2014, 71, 2517–2533. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.V.; Alvarez, S.E.; Spiegel, S.; Lebman, D.A. Sphingosine kinases and sphingosine-1-phosphate are critical for transforming growth factor beta-induced extracellular signal-regulated kinase 1 and 2 activation and promotion of migration and invasion of esophageal cancer cells. Mol. Cell. Biol. 2008, 28, 4142–4151. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Babelova, A.; Moreth, K.; Xin, C.; Eberhardt, W.; Doller, A.; Pavenstädt, H.; Schaefer, L.; Pfeilschifter, J.; Huwiler, A. Transforming growth factor-beta2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-beta2 by impeding CTGF expression. Kidney Int. 2009, 76, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Gellings Lowe, N.; Swaney, J.S.; Moreno, K.M.; Sabbadini, R.A. Sphingosine-1-phosphate and sphingosine kinase are critical for transforming growth factor-beta-stimulated collagen production by cardiac fibroblasts. Cardiovasc. Res. 2009, 82, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Böhm, A.; Flößer, A.; Ermler, S.; Fender, A.C.; Lüth, A.; Kleuser, B.; Schrör, K.; Rauch, B.H. Factor-Xa-induced mitogenesis and migration require sphingosine kinase activity and S1P formation in human vascular smooth muscle cells. Cardiovasc. Res. 2013, 99, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Billich, A.; Urtz, N.; Reuschel, R.; Baumruker, T. Sphingosine kinase 1 is essential for proteinase-activated receptor-1 signalling in epithelial and endothelial cells. Int. J. Biochem. Cell Biol. 2009, 41, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [PubMed]
- Rauch, B.H. Sphingosine 1-phosphate as a link between blood coagulation and inflammation. Cell. Physiol. Biochem. 2014, 34, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Moritz, E.; Wegner, D.; Gross, S.; Bahls, M.; Dörr, M.; Felix, S.B.; Ittermann, T.; Oswald, S.; Nauck, M.; Friedrich, N.; et al. Reference intervals for serum sphingosine-1-phosphate in the population-based Study of Health in Pomerania. Clin. Chim. Acta 2017, 468, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Mahajan-Thakur, S.; Sostmann, B.D.; Fender, A.C.; Behrendt, D.; Felix, S.B.; Schrör, K.; Rauch, B.H. Sphingosine-1-phosphate induces thrombin receptor PAR-4 expression to enhance cell migration and COX-2 formation in human monocytes. J. Leuk. Biol. 2014, 96, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Mahajan-Thakur, S.; Böhm, A.; Jedlitschky, G.; Schrör, K.; Rauch, B.H. Sphingosine-1-Phosphate and Its Receptors: A Mutual Link between Blood Coagulation and Inflammation. Mediators Inflamm. 2015, 831059. [Google Scholar] [CrossRef] [PubMed]
- Ulrych, T.; Böhm, A.; Polzin, A.; Daum, G.; Nusing, R.M.; Geisslinger, G.; Hohlfeld, T.; Schrör, K.; Rauch, B.H. Release of sphingosine-1-phosphate from human platelets is dependent on thromboxane formation. J. Thromb. Haemost. 2011, 9, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Polzin, A.; Rassaf, T.; Bohm, A.; Luth, A.; Kleuser, B.; Zeus, T.; Kelm, M.; Kroemer, H.K.; Schrör, K.; Rauch, B.H. Aspirin inhibits release of platelet-derived sphingosine-1-phosphate in acute myocardial infarction. Int. J. Cardiol. 2013, 170, e23–e24. [Google Scholar] [CrossRef] [PubMed]
- Vogt, K.; Mahajan-Thakur, S.; Wolf, R.; Bröderdorf, S.; Vogel, C.; Böhm, A.; Ritter, C.A.; Gräler, M.; Oswald, S.; Greinacher, A.; et al. Release of platelet-derived sphingosine-1-phosphate involves multidrug resistance protein 4 (MRP4/ABCC4) and is inhibited by statins. Thromb. Haemost. 2018, 118, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Weske, S.; Vaidya, M.; Reese, A.; von Wnuck Lipinski, K.; Keul, P.; Bayer, J.K.; Fischer, J.W.; Flögel, U.; Nelsen, J.; et al. Targeting sphingosine-1-phosphate lyase as an anabolic therapy for bone loss. Nat. Med. 2018, 24, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Coward, W.R.; Saini, G.; Jenkins, G. The pathogenesis of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2010, 4, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; von der Thüsen, J.; Daalhuisen, J.; ten Brink, M.; Crestani, B.; van der Poll, T.; Borensztajn, K.; Spek, C.A. Pharmacological Targeting of Protease-Activated Receptor 2 Affords Protection from Bleomycin-Induced Pulmonary Fibrosis. Mol. Med. 2015, 21, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Bardou, O.; Menou, A.; François, C.; Duitman, J.W.; von der Thüsen, J.H.; Borie, R.; Sales, K.U.; Mutze, K.; Castier, Y.; Sage, E.; et al. Membrane-anchored Serine Protease Matriptase Is a Trigger of Pulmonary Fibrogenesis. Am. J. Respir. Crit. Care Med. 2016, 193, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Borensztajn, K.; Spek, C.A. Targeting coagulation factor receptors—Protease-activated receptors in idiopathic pulmonary fibrosis. J. Thromb. Haemost. 2017, 15, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C. Procoagulant signalling mechanisms in lung inflammation andfibrosis: Novel opportunities for pharmacological intervention? Br. J. Pharmacol. 2008, 153, S367–378. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C.; Scotton, C.J. Coagulation cascade proteinases in lung injury and fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Mercer, P.F.; Williams, A.E.; Scotton, C.J.; José, R.J.; Sulikowski, M.; Moffatt, J.D.; Murray, L.A.; Chambers, R.C. Proteinase-activated receptor-1, CCL2, and CCL7 regulate acute neutrophilic lung inflammation. Am. J. Respir. Cell Mol. Biol. 2014, 50, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Wygrecka, M.; Kwapiszewska, G.; Jablonska, E.; von Gerlach, S.; Henneke, I.; Zakrzewicz, D.; Guenther, A.; Preissner, K.T.; Markart, P. Role of protease-activated receptor-2 in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Soumyakrishnan, S.; Divya, T.; Kalayarasan, S.; Sriram, N.; Sudhandiran, G. Daidzein exhibits anti-fibrotic effect by reducing the expressions of Proteinase activated receptor 2 and TGFβ1/smad mediated inflammation and apoptosis in Bleomycin-induced experimental pulmonary fibrosis. Biochimie 2014, 103, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Rezaee, F.; Waasdorp, M.; Shi, K.; van der Poll, T.; Borensztajn, K.; Spek, C.A. Protease activated receptor-1 regulates macrophage-mediated cellular senescence: a risk for idiopathic pulmonary fibrosis. Oncotarget 2015, 6, 35304–35314. [Google Scholar] [CrossRef] [PubMed]
- Howell, D.C.; Johns, R.H.; Lasky, J.A.; Shan, B.; Scotton, C.J.; Laurent, G.J.; Chambers, R.C. Absence of proteinase-activated receptor-1 signaling affords protection from bleomycin-induced lung inflammation and fibrosis. Am. J. Pathol. 2005, 166, 1353–1365. [Google Scholar] [CrossRef]
- Huang, L.S.; Berdyshev, E.; Mathew, B.; Fu, P.; Gorshkova, I.A.; He, D.; Ma, W.; Noth, I.; Ma, S.F.; Pendyala, S.; et al. Targeting sphingosine kinase 1 attenuates bleomycin-induced pulmonary fibrosis. FASEB J. 2013, 27, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.G.; Su, X.; Su, G.; Scotton, C.J.; Camerer, E.; Laurent, G.J.; Davis, G.E.; Chambers, R.C.; Matthay, M.A.; Sheppard, D. Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J. Clin. Investig. 2006, 116, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; He, Y.; Wang, M.; Lv, P.; Liu, J.; Wang, J. Role of Protease-Activated Receptor 2 in Regulating Focal Segmental Glomerulosclerosis. Cell. Physiol. Biochem. 2017, 41, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Grandaliano, G.; Pontrelli, P.; Cerullo, G.; Monno, R.; Ranieri, E.; Ursi, M.; Loverre, A.; Gesualdo, L.; Schena, F.P. Protease-activated receptor-2 expression in IgA nephropathy: a potential role in the pathogenesis of interstitial fibrosis. J. Am. Soc. Nephrol. 2003, 14, 2072–2083. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, S.; Beyer, S.; Frey, H.; Haceni, R.; Grammatikos, G.; Thomas, D.; Geisslinger, G.; Schaefer, L.; Huwiler, A.; Pfeilschifter, J. Sphingosine kinase-2 deficiency ameliorates kidney fibrosis by up-regulating Smad7 in a mouse model of unilateral ureteral obstruction. Am. J. Pathol. 2017, 187, 2413–2429. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Kim, I.S.; Rezaie, A.R. Thrombin down-regulates the TGF-beta-mediated synthesis of collagen and fibronectin by human proximal tubule epithelial cells through the EPCR-dependent activation of PAR-1. J. Cell. Physiol. 2010, 225, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.M.; Rana, R.; Austin, K.; Baleja, J.D.; Nguyen, N.; Bohm, A.; Covic, L.; Kuliopulos, A. Targeting liver fibrosis with a cell-penetrating protease-activated receptor-2 (PAR2) pepducin. J. Biol. Chem. 2016, 291, 23188–23198. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, K.; Saile, B.; Ramadori, G. Liver fibrosis and altered matrix synthesis. Can. J. Gastroenterol. 2001, 15, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Sakai, T. Biological significance of Local TGF-beta activation in liver diseases. Front. Physiol. 2012, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Xiu, L.; Chang, N.; Yang, L.; Liu, X.; Yang, L.; Ge, J.; Li, L. Intracellular sphingosine 1-phosphate contributes to collagen expression of hepatic myofibroblasts in human liver fibrosis independent of its receptors. Am. J. Pathol. 2015, 185, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Knight, V.; Tchongue, J.; Lourensz, D.; Tipping, P.; Sievert, W. Protease-activated receptor 2 promotes experimental liver fibrosis in mice and activates human hepatic stellate cells. Hepatology 2012, 55, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Mußbach, F.; Ungefroren, H.; Günther, B.; Katenkamp, K.; Henklein, P.; Westermann, M.; Settmacher, U.; Lenk, L.; Sebens, S.; Müller, J.P.; et al. Proteinase-activated receptor 2 (PAR2) in hepatic stellate cells - evidence for a role in hepatocellular carcinoma growth in vivo. Mol. Cancer. 2016, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Kitasato, L.; Yamaoka-Tojo, M.; Hashikata, T.; Ishii, S.; Kameda, R.; Shimohama, T.; Tojo, T.; Ako, J. Factor Xa in mouse fibroblasts may induce fibrosis more than thrombin. Int. Heart J. 2014, 55, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Sonin, D.L.; Wakatsuki, T.; Routhu, K.V.; Harmann, L.M.; Petersen, M.; Meyer, J.; Strande, J.L. Protease-activated receptor 1 inhibition by SCH79797 attenuates left ventricular remodeling and profibrotic activities of cardiac fibroblasts. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Rattenholl, A.; Seeliger, S.; Buddenkotte, J.; Schön, M.; Schön, M.P.; Ständer, S.; Vergnolle, N.; Steinhoff, M. Proteinase-activated receptor-2 (PAR2): a tumor suppressor in skin carcinogenesis. J. Investig. Dermatol. 2007, 127, 2245–2252. [Google Scholar] [CrossRef] [PubMed]
- Borensztajn, K.; Stiekema, J.; Nijmeijer, S.; Reitsma, P.H.; Peppelenbosch, M.P.; Spek, C.A. Factor Xa stimulates proinflammatory and profibrotic responses in fibroblasts via protease-activated receptor-2 activation. Am. J. Pathol. 2008, 172, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Materazzi, S.; Pellerito, S.; Di Serio, C.; Paglierani, M.; Naldini, A.; Ardinghi, C.; Carraro, F.; Geppetti, P.; Cirino, G.; Santucci, M.; et al. Analysis of protease-activated receptor-1 and -2 in human scar formation. J. Pathol. 2007, 212, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Duitman, J.; Ruela-de-Sousa, R.R.; Shi, K.; de Boer, O.J.; Borensztajn, K.S.; Florquin, S.; Peppelenbosch, M.P.; Spek, C.A. Protease activated receptor-1 deficiency diminishes bleomycin-induced skin fibrosis. Mol. Med. 2014, 20, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Schniewind, B.; Groth, S.; Sebens Muerkoster, S.; Sipos, B.; Schafer, H.; Kalthoff, H.; Ungefroren, H. Dissecting the role of TGF-beta type I receptor/ALK5 in pancreatic ductal adenocarcinoma: Smad activation is crucial for both the tumor suppressive and prometastatic function. Oncogene 2007, 26, 4850–4862. [Google Scholar] [CrossRef] [PubMed]
- Jikuhara, A.; Yoshii, M.; Iwagaki, H.; Mori, S.; Nishibori, M.; Tanaka, N. MAP kinase-mediated proliferation of DLD-1 carcinoma by the stimulation of protease-activated receptor 2. Life Sci. 2003, 73, 2817–2829. [Google Scholar] [CrossRef]
- Ikeda, O.; Egami, H.; Ishiko, T.; Ishikawa, S.; Kamohara, H.; Hidaka, H.; Mita, S.; Ogawa, M. Expression of proteinase-activated receptor-2 in human pancreatic cancer: A possible relation to cancer invasion and induction of fibrosis. Int. J. Oncol. 2003, 22, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Darmoul, D.; Gratio, V.; Devaud, H.; Laburthe, M. Protease-activated receptor 2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J. Biol. Chem. 2004, 279, 20927–20934. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Shenoy, S.; Lefkowitz, R.; DeFea, K. Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both beta-arrestin-1 and -2. J. Biol. Chem. 2004, 279, 55419–55424. [Google Scholar] [CrossRef] [PubMed]
- Hjortoe, G.; Petersen, L.; Albrektsen, T.; Sorensen, B.; Norby, P.; Mandal, S.; Pendurthi, U.; Rao, L. Tissue factor-factor VIIa-specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated by PAR-2 and results in increased cell migration. Blood 2004, 103, 3029–3037. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gangadharan, B.; Brass, L.F.; Ruf, W.; Mueller, B.M. Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol. Cancer Res. 2004, 2, 395–402. [Google Scholar] [PubMed]
- Shimamoto, R.; Sawada, T.; Uchima, Y.; Inoue, M.; Kimura, K.; Yamashita, Y.; Yamada, N.; Nishihara, T.; Ohira, M.; Hirakawa, K. A role for protease-activated receptor-2 in pancreatic cancer cell proliferation. Int. J. Oncol. 2004, 24, 1401–1406. [Google Scholar] [PubMed]
- Morris, D.; Ding, Y.; Ricks, T.; Gullapalli, A.; Wolfe, B.; Trejo, J. Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 2006, 66, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Schaffner, F.; Kerver, M.; Ellies, L.G.; Andrade-Gordon, P.; Mueller, B.M.; Ruf, W. Protease-activated receptor (PAR) 2, but not PAR1, signaling promotes the development of mammary adenocarcinoma in polyoma middle T mice. Cancer Res. 2008, 68, 7219–7227. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, K.; Shibata, K.; Ohta, M.; Endo, Y.; Uchida, H.; Tominaga, M.; Okunaga, R.; Kai, S.; Kitano, S. A small interfering RNA targeting proteinase-activated receptor-2 is effective in suppression of tumor growth in a Panc1 xenograft model. Int. J. Cancer 2008, 122, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Bocheva, G.; Rattenholl, A.; Kempkes, C.; Goerge, T.; Lin, C.; D'Andrea, M.; Ständer, S.; Steinhoff, M. Role of matriptase and proteinase-activated receptor-2 in nonmelanoma skin cancer. J. Investig. Dermatol. 2009, 129, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, R.; Oettel, C.; Horn, A.; Halbhuber, K.J.; Eitner, A.; Krieg, R.; Katenkamp, K.; Henklein, P.; Westermann, M.; Böhmer, F.D.; et al. Met receptor tyrosine kinase transactivation is involved in proteinase-activated receptor-2-mediated hepatocellular carcinoma cell invasion. Carcinogenesis 2009, 30, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Queiroz, K.C.; Roelofs, J.J.; van Noesel, C.J.; Richel, D.J.; Spek, C.A. Protease-activated receptor 2 suppresses lymphangiogenesis and subsequent lymph node metastasis in a murine pancreatic cancer model. J. Pathol. 2014, 234, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Segal, L.; Katz, L.S.; Lupu-Meiri, M.; Shapira, H.; Sandbank, J.; Gershengorn, M.C.; Oron, Y. Proteinase-activated receptors differentially modulate in vitro invasion of human pancreatic adenocarcinoma PANC1 cells in correlation with changes in the expression of CDC42 protein. Pancreas 2014, 43, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Protease-activated receptors (PARs)--biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 2015, 34, 775–796. [Google Scholar] [CrossRef] [PubMed]
- Gamperl, H.; Plattfaut, C.; Freund, A.; Quecke, T.; Theophil, F.; Gieseler, F. Extracellular vesicles from malignant effusions induce tumor cell migration: inhibitory effect of LMWH tinzaparin. Cell Biol. Int. 2016, 40, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, P.B.; Yao, Y.F.; Xiu, A.Y.; Peng, Z.; Bai, Y.H.; Gao, Y.J. Proteinase-activated receptor 2 promotes tumor cell proliferation and metastasis by inducing epithelial-mesenchymal transition and predicts poor prognosis in hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shavit, R.; Turm, H.; Salah, Z.; Maoz, M.; Cohen, I.; Weiss, E.; Uziely, B.; Grisaru-Granovsky, S. PAR1 plays a role in epithelial malignancies: transcriptional regulation and novel signaling pathway. IUBMB Life 2011, 63, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, J.; Song, S.; Yue, X.; Li, Q. Protease-activated receptor-1 (PAR-1): a promising molecular target for cancer. Oncotarget 2017, 8, 107334–107345. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Witte, D.; Fiedler, C.; Gädeken, T.; Kaufmann, R.; Lehnert, H.; Gieseler, F.; Rauch, B.H. The role of ERK activation in PAR2 agonist and TGF-beta1-induced cell migration. Int. J. Mol. Sci. 2017, 18, E2776. [Google Scholar] [CrossRef] [PubMed]
- Stayrook, K.R.; Mack, J.K.; Cerabona, D.; Edwards, D.F.; Bui, H.H.; Niewolna, M.; Fournier, P.G.; Mohammad, K.S.; Waning, D.L.; Guise, T.A. TGFβ-Mediated induction of SphK1 as a potential determinant in human MDA-MB-231 breast cancer cell bone metastasis. Bonekey Rep. 2015, 4, 719. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, O.; Egami, H.; Ishiko, T.; Ishikawa, S.; Kamohara, H.; Hidaka, H.; Takahashi, M.; Ogawa, M. Signal of proteinase-activated receptor-2 contributes to highly malignant potential of human pancreatic cancer by up-regulation of interleukin-8 release. Int. J. Oncol. 2006, 28, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Zarogoulidis, P.; Katsikogianni, F.; Tsiouda, T.; Sakkas, A.; Katsikogiannis, N.; Zarogoulidis, K. Interleukin-8 and interleukin-17 for cancer. Cancer Investig. 2014, 32, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jiao, J.; Zhang, H.; Zhou, W.; Li, Z.; Han, S.; Wang, J.; Yang, X.; Huang, Q.; Wu, Z.; et al. TGF-β induced PAR-1 expression promotes tumor progression and osteoclast differentiation in giant cell tumor of bone. Int. J. Cancer. 2017, 141, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Smoktunowicz, N.; Platé, M.; Stern, A.O.; D'Antongiovanni, V.; Robinson, E.; Chudasama, V.; Caddick, S.; Scotton, C.J.; Jarai, G.; Chambers, R.C. TGFβ upregulates PAR-1 expression and signalling responses in A549 lung adenocarcinoma cells. Oncotarget 2016, 7, 65471–65484. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zhang, S.; Miao, L.; Wang, J.; Jin, Z.; Gu, B.; Duan, Z.; Zhao, Z.; Ma, S.; Zhang, W.; et al. Activation of platelet protease-activated receptor-1 induces epithelial-mesenchymal transition and chemotaxis of colon cancer cell line SW620. Oncol. Rep. 2015, 33, 2681–2688. [Google Scholar] [CrossRef] [PubMed]
- He, R.Q.; Tang, X.F.; Zhang, B.L.; Li, X.D.; Hong, M.N.; Chen, Q.Z.; Han, W.Q.; Gao, P.J. Protease-activated receptor 1 and 2 contribute to angiotensin II-induced activation of adventitial fibroblasts from rat aorta. Biochem. Biophys. Res. Commun. 2016, 473, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.E.; Levis, J.E.; Schneider, D.J.; Bambace, N.M.; Sharma, D.; Lal, I.; Wood, M.E.; Muss, H.B. Platelet phenotype changes associated with breast cancer and its treatment. Platelets 2016, 27, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Little, P.J.; Burch, M.L.; Al-aryahi, S.; Zheng, W. The paradigm of G protein receptor transactivation: a mechanistic definition and novel example. Sci. World J. 2011, 11, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Bourd-Boittin, K.; Bonnier, D.; Leyme, A.; Mari, B.; Tuffery, P.; Samson, M.; Ezan, F.; Baffet, G.; Theret, N. Protease profiling of liver fibrosis reveals the ADAM metallopeptidase with thrombospondin type 1 motif, 1 as a central activator of transforming growth factor beta. Hepatology 2011, 54, 2173–2184. [Google Scholar] [CrossRef] [PubMed]
- Pagel, C.N.; Song, S.J.; Loh, L.H.; Tudor, E.M.; Murray-Rust, T.A.; Pike, R.N.; Mackie, E.J. Thrombin-stimulated growth factor and cytokine expression in osteoblasts is mediated by protease-activated receptor-1 and prostanoids. Bone. 2009, 44, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Bi, M.; Liu, Y.; Wang, Y.; Pan, F.; Qiu, L.; Guo, A.; Lv, H.; Yao, P.; Zhang, N.; et al. Thrombin promotes airway remodeling via protease-activated receptor-1 and transforming growth factor-β1 in ovalbumin-allergic rats. Inhal. Toxicol. 2013, 25, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Nakajima, K.; Tohyama, Y.; Kohsaka, S. Characteristic response of astrocytes to plasminogen/plasmin to upregulate transforming growth factor beta 3 (TGFbeta3) production/secretion through proteinase-activated receptor-1 (PAR-1) and the downstream phosphatidylinositol 3-kinase (PI3K)-Akt/PKB signaling cascade. Brain Res. 2009, 1305, 1–13. [Google Scholar] [PubMed]
- Ma, Y.; Bao-Han, W.; Lv, X.; Su, Y.; Zhao, X.; Yin, Y.; Zhang, X.; Zhou, Z.; MacNaughton, W.K.; Wang, H. MicroRNA-34a mediates the autocrine signaling of PAR2-activating proteinase and its role in colonic cancer cell proliferation. PLoS ONE 2013, 8, e72383. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Low, B.; Rutherford, S.A.; Hao, Q. Thrombin induces endocytosis of endoglin and type-II TGF-beta receptor and down-regulation of TGF-beta signaling in endothelial cells. Blood 2005, 105, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; An, S.; Sassmann, A.; Liu, M.; Mastej, V.; Mittal, M.; Zhang, W.; Hong, Z.; Offermanns, S.; Rehman, J.; et al. PAR1 Scaffolds TGFβRII to downregulate TGF-β signaling and activate ESC differentiation to endothelial cells. Stem Cell Rep. 2016, 7, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Zeeh, F.; Witte, D.; Gädeken, T.; Rauch, B.H.; Grage-Griebenow, E.; Leinung, N.; Fromm, S.J.; Stölting, S.; Mihara, K.; Kaufmann, R.; et al. Proteinase-activated receptor 2 promotes TGF-β-dependent cell motility in pancreatic cancer cells by sustaining expression of the TGF-β type I receptor ALK5. Oncotarget 2016, 7, 41095–41109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, X.; Xie, F.; Zhang, Z.; van Dam, H.; Zhang, L.; Zhou, F. The regulation of TGF-β/SMAD signaling by protein deubiquitination. Protein Cell 2014, 5, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.R.; McIntosh, K.A.; Pediani, J.D.; Robben, J.; Cooke, A.E.; Nilsson, M.; Gould, G.W.; Mundell, S.; Milligan, G.; Plevin, R. Novel role for proteinase-activated receptor 2 (PAR2) in membrane trafficking of proteinase-activated receptor 4 (PAR4). J. Biol. Chem. 2012, 287, 16656–16669. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Witte, D.; Mihara, K.; Rauch, B.H.; Henklein, P.; Johren, O.; Bonni, S.; Settmacher, U.; Lehnert, H.; Hollenberg, M.D.; et al. TGF-β1/ALK5-mediated cell migration is dependent on the protein PAR2 but not on PAR2-stimulated Gq-calcium signaling. Mol. Pharmacol. 2017, 92, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Osuga, Y.; Yoshino, O.; Takamura, M.; Hirata, T.; Hirota, Y.; Koga, K.; Harada, M.; Takemura, Y.; Yano, T.; et al. TGF-β1 induces proteinase-activated receptor 2 (PAR2) expression in endometriotic stromal cells and stimulates PAR2 activation-induced secretion of IL-6. Hum. Reprod. 2011, 26, 1892–1898. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H. University Hospital Schleswig-Holstein, Lübeck, Germany. Unpublished work. 2017. [Google Scholar]
- Yu, N.; Kozlowski, J.M.; Park, II.; Chen, L.; Zhang, Q.; Xu, D.; Doll, J.A.; Crawford, S.E.; Brendler, C.B.; Lee, C. Over-expression of transforming growth factor β1 in malignant prostate cells is partly caused by a runaway of TGF-β1 auto-induction mediated through a defective recruitment of protein phosphatase 2A by TGF-β type I receptor. Urology. 2010, 76, 1519e8–e13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, N.; Lee, C. Vicious cycle of TGF-β signaling in tumor progression and metastasis. Am. J. Clin. Exp. Urol. 2014, 2, 149–155. [Google Scholar] [PubMed]
- Liu, X.; Hu, H.; Yin, J.Q. Therapeutic strategies against TGF-beta signaling pathway in hepatic fibrosis. Liver Int. 2006, 26, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; De Ceunynck, K. Targeting PAR1: Now What? Trends Pharmacol. Sci. 2017, 38, 701–716. [Google Scholar] [CrossRef] [PubMed]
- Serebruany, V.L.; Kogushi, M.; Dastros-Pitei, D.; Flather, M.; Bhatt, D.L. The in-vitro effects of E5555, a protease-activated receptor (PAR)-1 antagonist, on platelet biomarkers in healthy volunteers and patients with coronary artery disease. Thromb. Haemost. 2009, 102, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Goh, F.G.; Ng, P.Y.; Nilsson, M.; Kanke, T.; Plevin, R. Dual effect of the novel peptide antagonist K-14585 on proteinase-activated receptor-2-mediated signalling. Br. J. Pharmacol. 2009, 158, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Lohman, R.J.; Cotterell, A.J.; Suen, J.; Liu, L.; Do, A.T.; Vesey, D.A.; Fairlie, D.P. Antagonism of protease activated receptor 2 protects against experimental colitis. J. Pharmacol. Exp. Ther. 2012, 340, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Barry, G.D.; Suen, J.Y.; Le, G.T.; Cotterell, A.; Reid, R.C.; Fairlie, D.P. Novel agonists and antagonists for human protease activated receptor 2. J. Med. Chem. 2010, 53, 7428–7440. [Google Scholar] [CrossRef] [PubMed]
- Suen, J.Y.; Gardiner, B.; Grimmond, S.; Fairlie, D.P. Profiling gene expression induced by protease-activated receptor 2 (PAR2) activation in human kidney cells. PLoS ONE 2010, 5, e13809. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Noorbakhsh, F.; Defea, K.; Hollenberg, M.D. Targeting proteinase-activated receptors: Therapeutic potential and challenges. Nat. Rev. Drug Discov. 2012, 11, 69–86. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungefroren, H.; Gieseler, F.; Kaufmann, R.; Settmacher, U.; Lehnert, H.; Rauch, B.H. Signaling Crosstalk of TGF-β/ALK5 and PAR2/PAR1: A Complex Regulatory Network Controlling Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1568. https://doi.org/10.3390/ijms19061568
Ungefroren H, Gieseler F, Kaufmann R, Settmacher U, Lehnert H, Rauch BH. Signaling Crosstalk of TGF-β/ALK5 and PAR2/PAR1: A Complex Regulatory Network Controlling Fibrosis and Cancer. International Journal of Molecular Sciences. 2018; 19(6):1568. https://doi.org/10.3390/ijms19061568
Chicago/Turabian StyleUngefroren, Hendrik, Frank Gieseler, Roland Kaufmann, Utz Settmacher, Hendrik Lehnert, and Bernhard H. Rauch. 2018. "Signaling Crosstalk of TGF-β/ALK5 and PAR2/PAR1: A Complex Regulatory Network Controlling Fibrosis and Cancer" International Journal of Molecular Sciences 19, no. 6: 1568. https://doi.org/10.3390/ijms19061568
APA StyleUngefroren, H., Gieseler, F., Kaufmann, R., Settmacher, U., Lehnert, H., & Rauch, B. H. (2018). Signaling Crosstalk of TGF-β/ALK5 and PAR2/PAR1: A Complex Regulatory Network Controlling Fibrosis and Cancer. International Journal of Molecular Sciences, 19(6), 1568. https://doi.org/10.3390/ijms19061568