Connexins and Pannexins: Important Players in Tumorigenesis, Metastasis and Potential Therapeutics



Abstract
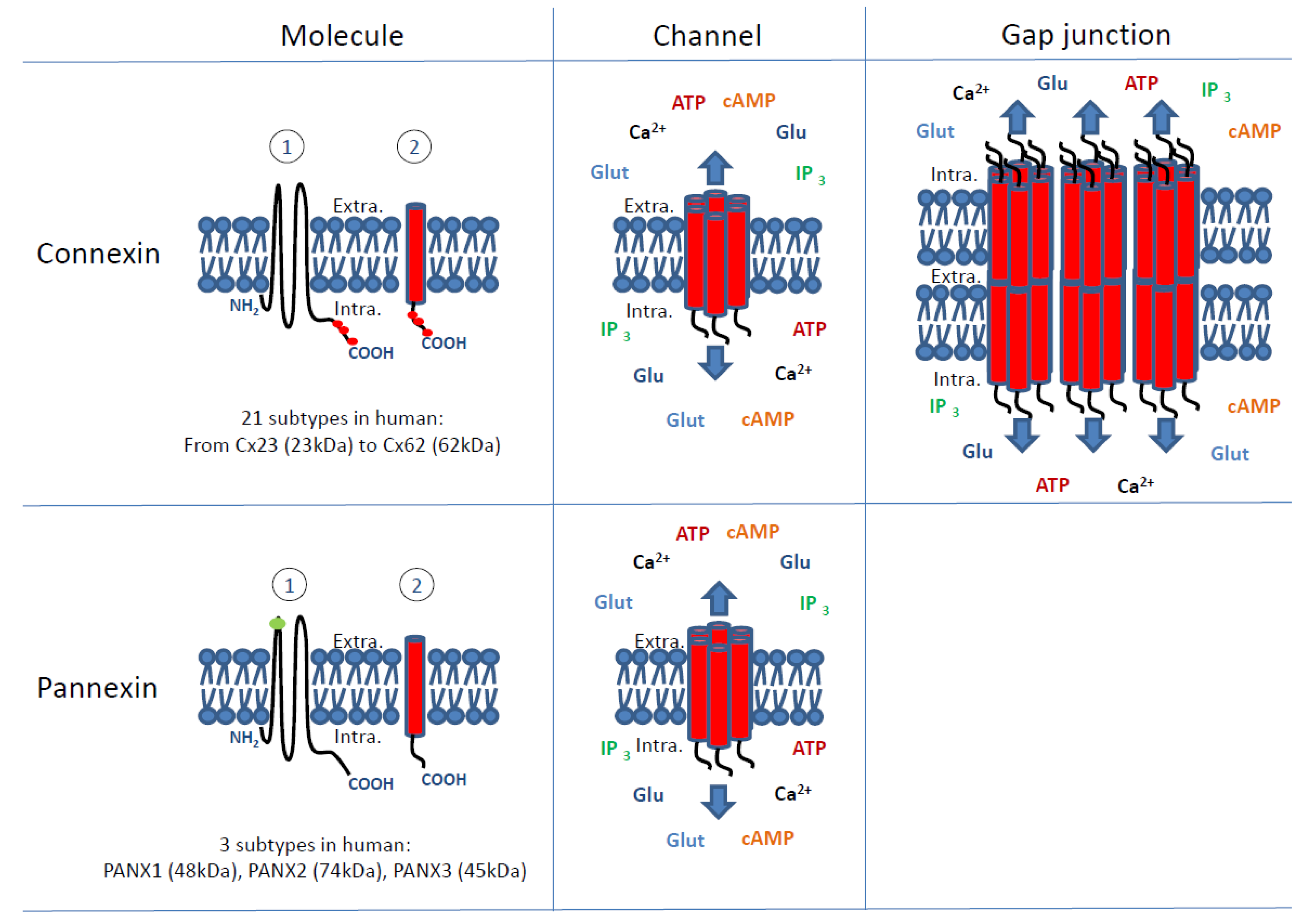
:1. Introduction
2. Connexins and Pannexins Involvement in Tumour Cell Growth
2.1. Connexins Involvement in Tumour Cell Growth
2.1.1. How Can the Presence of Connexins Regulate Cell Growth?
Gap-Junctional Intercellular Communication and Cell Growth Control
Cell Growth Control Independent from Gap-Junctional Intercellular Communication
2.1.2. What Does Prevent Connexin Expression or Function during Tumour Progression?
2.2. Pannexins Involvement in Tumour Cell Growth
3. Connexins and Pannexins: Involvement in Tumour Metastasis and Microenvironment
3.1. The Process of Metastasis
3.2. Connexin Involvement in Tumour Metastasis and Microenvironment
3.2.1. The Role of Connexins in Cancer Progression
3.2.2. Invasion and the Local Microenvironment
3.2.3. Promoting Metastasis: Connexins and Cell Motility
3.2.4. Involvement of Gap Junctions and Hemichannels in Metastasis
3.2.5. The Tumour Microenvironment
3.3. Pannexins and Metastasis
4. Connexin and Pannexin Channels in Potential Cancer Therapeutics
4.1. Connexin Channels in Potential Cancer Therapeutics
4.1.1. Chemical Compounds in Modulating Connexins and Potential Cancer Therapy
4.1.2. Connexin-Targeting Strategies in Potential Cancer Therapy
4.2. Pannexin Channels in Potential Cancer Therapeutics
4.2.1. Pannexin Channel Activation and Potential Cancer Therapy
4.2.2. Pannexin in Potential Cancer Diagnosis
4.2.3. Pannexin Channels in Pain Management Related to Cancer Treatment
5. Discussion and Conclusions
Acknowledgments
Conflicts of Interest
References
- Forman, D.; Ferlay, J. The global and regional burden of cancer. In World Cancer Report; Stewart, B.W., Wild, C.P., Eds.; International Agency for Research on Cancer: Lyon, France, 2014; pp. 16–53. ISBN 978-92-832-0429-9. [Google Scholar]
- Cronier, L.; Crespin, S.; Strale, P.O.; Defamie, N.; Mesnil, M. Gap junctions and cancer: New functions for an old story. Antioxid. Redox Signal. 2009, 11, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.; Laird, D.W. Implications and challenges of connexin connections to cancer. Nat. Rev. Cancer 2010, 10, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Hervé, J.C.; Bourmeyster, N.; Sarrouilhe, D.; Duffy, H.S. Gap junctional complexes: From partners to functions. Prog. Biophys. Mol. Biol. 2007, 94, 29–65. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T. Connexins: Junctional and non-junctional modulators of proliferation. Cell Tissue Res. 2015, 360, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Carvajal-Hausdorf, D.; Oyarzo, M.P. Possible role of hemichannels in cancer. Front. Physiol. 2014, 5, 237. [Google Scholar] [CrossRef] [PubMed]
- Willecke, K.; Eiberger, J.; Degen, J.; Eckardt, D.; Romualdi, A.; Güldenagel, M.; Deutsch, U.; Söhl, G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 2002, 383, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Panchin, Y.; Kelmanson, I.; Matz, M.; Lukyanov, K.; Usman, N.; Lukyanov, S. A ubiquitous family of putative gap junction molecules. Curr. Biol. 2000, 10, R473–474. [Google Scholar] [CrossRef]
- Bond, S.R.; Naus, C.C. The pannexins: Past and present. Front. Physiol. 2014, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim. Biophys. Acta 2018, 1860, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Hervé, J.C.; Derangeon, M.; Sarrouilhe, D.; Giepmans, B.N.; Bourmeyster, N. Gap junctional channels are parts of multiprotein complexes. Biochim. Biophys. Acta 2012, 1818, 1844–1865. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.; Giaume, C. Bridging the gap to therapeutic strategies based on connexin/pannexin biology. J. Transl. Med. 2016, 14, 330. [Google Scholar] [CrossRef] [PubMed]
- Kandouz, M.; Batist, G. Gap junctions and connexins as therapeutic targets in cancer. Expert Opin. Ther. Targets 2010, 14, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R. Junctional intercellular communication and the control of growth. Biochim. Biophys. Acta 1979, 560, 1–65. [Google Scholar] [CrossRef]
- Loewenstein, W.R.; Socolar, S.J.; Higashino, S.; Kanno, Y.; Davidson, N. Intercellular Communication: Renal, Urinary Bladder, Sensory, and Salivary Gland Cells. Science 1965, 149, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Loewenstein, W.R. Cell-to-cell passage of large molecules. Nature 1966, 212, 629–630. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and the control of tissue growth: Lack of communication between cancer cells. Nature 1966, 209, 1248–1249. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and tissue growth. I. Cancerous growth. J. Cell Biol. 1967, 33, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Yotti, L.P.; Chang, C.C.; Trosko, J.E. Elimination of metabolic cooperation in Chinese hamster cells by a tumor promoter. Science 1979, 206, 1089–1091. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.W.; Fitzgerald, D.J. Tumor promoters inhibit metabolic cooperation in cocultures of epidermal and 3T3 cells. Biochem. Biophys. Res. Commun. 1979, 91, 395–401. [Google Scholar] [CrossRef]
- Yamasaki, H. Cell-cell interaction and carcinogenesis. Toxicol. Pathol. 1986, 14, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E.; Jone, C.; Chang, C.C. Oncogenes, inhibited intercellular communication and tumor promotion. Princess Takamatsu Symp. 1983, 14, 101–113. [Google Scholar] [PubMed]
- Atkinson, M.M.; Anderson, S.K.; Sheridan, J.D. Modification of gap junctions in cells transformed by a temperature-sensitive mutant of Rous sarcoma virus. J. Membr. Biol. 1986, 91, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Kakunaga, T.; Kuroki, T.; Neubert, D.; Trosko, J.E.; Vasiliev, J.M.; Williams, G.M.; Yamasaki, H. Short-term assays to predict carcinogenicity. In-vitro assays that may be predictive of tumour-promoting agents. IARC Sci. Publ. 1986, 287–302. [Google Scholar]
- Temme, A.; Buchmann, A.; Gabriel, H.D.; Nelles, E.; Schwarz, M.; Willecke, K. High incidence of spontaneous and chemically induced liver tumors in mice deficient for connexin32. Curr. Biol. 1997, 7, 713–716. [Google Scholar] [CrossRef]
- Avanzo, J.L.; Mesnil, M.; Hernandez-Blazquez, F.J.; Mackowiak, I.I.; Mori, C.M.; da Silva, T.C.; Oloris, S.C.; Gárate, A.P.; Massironi, S.M.; Yamasaki, H.; et al. Increased susceptibility to urethane-induced lung tumors in mice with decreased expression of connexin43. Carcinogenesis 2004, 25, 1973–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, D.J.; Mesnil, M.; Oyamada, M.; Tsuda, H.; Ito, N.; Yamasaki, H. Changes in gap junction protein (connexin 32) gene expression during rat liver carcinogenesis. J. Cell Biochem. 1989, 41, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Krutovskikh, V.; Mazzoleni, G.; Mironov, N.; Omori, Y.; Aguelon, A.M.; Mesnil, M.; Berger, F.; Partensky, C.; Yamasaki, H. Altered homologous and heterologous gap-junctional intercellular communication in primary human liver tumors associated with aberrant protein localization but not gene mutation of connexin 32. Int. J. Cancer 1994, 56, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.Z.; Wilkens, L.R.; Mehta, P.P.; Loewenstein, W.; Bertram, J.S. Enhancement of gap junctional communication by retinoids correlates with their ability to inhibit neoplastic transformation. Carcinogenesis 1989, 10, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Vine, A.L.; Bertram, J.S. Cancer chemoprevention by connexins. Cancer Metastasis Rev. 2002, 21, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Mesnil, M.; Crespin, S.; Avanzo, J.L.; Zaidan-Dagli, M.L. Defective gap junctional intercellular communication in the carcinogenic process. Biochim. Biophys. Acta 2005, 1719, 125–145. [Google Scholar] [CrossRef] [PubMed]
- Mesnil, M.; Krutovskikh, V.; Piccoli, C.; Elfgang, C.; Traub, O.; Willecke, K.; Yamasaki, H. Negative growth control of HeLa cells by connexin genes: Connexin species specificity. Cancer Res. 1995, 55, 629–639. [Google Scholar] [PubMed]
- Mesnil, M. Connexins and cancer. Biol. Cell 2002, 94, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.C.; Pelletier, D.B.; Ao, P.; Boynton, A.L. Connexin43 reverses the phenotype of transformed cells and alters their expression of cyclin/cyclin-dependent kinases. Cell Growth Differ. 1995, 6, 681–690. [Google Scholar] [PubMed]
- Zhang, Y.W.; Morita, I.; Ikeda, M.; Ma, K.W.; Murota, S. Connexin43 suppresses proliferation of osteosarcoma U2OS cells through post-transcriptional regulation of p27. Oncogene 2001, 20, 4138–4149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koffler, L.; Roshong, S.; Kyu Park, I.; Cesen-Cummings, K.; Thompson, D.C.; Dwyer-Nield, L.D.; Rice, P.; Mamay, C.; Malkinson, A.M.; Ruch, R.J. Growth inhibition in G(1) and altered expression of cyclin D1 and p27(kip-1) after forced connexin expression in lung and liver carcinoma cells. J. Cell Biochem. 2000, 79, 347–354. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Kaneda, M.; Morita, I. The gap junction-independent tumor-suppressing effect of connexin 43. J. Biol. Chem. 2003, 278, 44852–44856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Nakayama, K.; Nakayama, K.; Morita, I. A novel route for connexin 43 to inhibit cell proliferation: Negative regulation of S-phase kinase-associated protein (Skp2). Cancer Res. 2003, 63, 1623–1630. [Google Scholar] [PubMed]
- Goldberg, G.S.; Bechberger, J.F.; Tajima, Y.; Merritt, M.; Omori, Y.; Gawinowicz, M.A.; Narayanan, R.; Tan, Y.; Sanai, Y.; Yamasaki, H.; et al. Connexin43 suppresses MFG-E8 while inducing contact growth inhibition of glioma cells. Cancer Res. 2000, 60, 6018–6026. [Google Scholar] [PubMed]
- Goldberg, G.S.; Moreno, A.P.; Lampe, P.D. Gap junctions between cells expressing connexin 43 or 32 show inverse permselectivity to adenosine and ATP. J. Biol. Chem. 2002, 277, 36725–36730. [Google Scholar] [CrossRef] [PubMed]
- Swenson, K.I.; Piwnica-Worms, H.; McNamee, H.; Paul, D.L. Tyrosine phosphorylation of the gap junction protein connexin43 is required for the pp60v-src-induced inhibition of communication. Cell Regul. 1990, 1, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Stains, J.P.; Civitelli, R. Cell-to-cell interactions in bone. Biochem. Biophys. Res. Commun. 2005, 328, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, A.; Kalmykov, E.A.; Polusani, S.R.; Mathis, S.A.; Zucker, S.N.; Nicholson, B.J. Intercellular redistribution of cAMP underlies selective suppression of cancer cell growth by connexin26. PLoS ONE 2013, 8, e82335. [Google Scholar] [CrossRef] [PubMed]
- Suzhi, Z.; Liang, T.; Yuexia, P.; Lucy, L.; Xiaoting, H.; Yuan, Z.; Qin, W. Gap Junctions Enhance the Antiproliferative Effect of MicroRNA-124–3p in Glioblastoma Cells. J. Cell Physiol. 2015, 230, 2476–2488. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Jego, G.; Berthenet, K.; Hammann, A.; Solary, E.; Garrido, C. Gap junction-mediated transfer of miR-145–5p from microvascular endothelial cells to colon cancer cells inhibits angiogenesis. Oncotarget 2016, 7, 28160–28168. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.R.; Martins-Marques, T.; Ribeiro-Rodrigues, T.; Ferreira, J.V.; Catarino, S.; Pinho, M.J.; Zuzarte, M.; Isabel Anjo, S.; Manadas, B.P.G.; Sluijter, J.; et al. Gap junctional protein Cx43 is involved in the communication between extracellular vesicles and mammalian cells. Sci. Rep. 2015, 5, 13243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laird, D.W. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2010, 20, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Duffy, H.S.; Iacobas, I.; Hotchkiss, K.; Hirst-Jensen, B.J.; Bosco, A.; Dandachi, N.; Dermietzel, R.; Sorgen, P.L.; Spray, D.C. The gap junction protein connexin32 interacts with the Src homology3/hook domain of discs large homolog 1. J. Biol. Chem. 2007, 282, 9789–9796. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.T.; Bechberger, J.F.; Ozog, M.A.; Perbal, B.; Naus, C.C. CCN3 (NOV) interacts with connexin43 in C6 glioma cells: Possible mechanism of connexin-mediated growth suppression. J. Biol. Chem. 2004, 279, 36943–36950. [Google Scholar] [CrossRef] [PubMed]
- Gellhaus, A.; Dong, X.; Propson, S.; Maass, K.; Klein-Hitpass, L.; Kibschull, M.; Traub, O.; Willecke, K.; Perbal, B.; Lye, S.J.; et al. Connexin43 interacts with NOV: A possible mechanism for negative regulation of cell growth in choriocarcinoma cells. J. Biol. Chem. 2004, 279, 36931–36942. [Google Scholar] [CrossRef] [PubMed]
- Penes, M.C.; Li, X.; Nagy, J.I. Expression of zonula occludens-1 (ZO-1) and the transcription factor ZO-1-associated nucleic acid-binding protein (ZONAB)-MsY3 in glial cells and colocalization at oligodendrocyte and astrocyte gap junctions in mouse brain. Eur. J. Neurosci. 2005, 22, 404–418. [Google Scholar] [CrossRef] [PubMed]
- González-Sánchez, A.; Jaraíz-Rodríguez, M.; Domínguez-Prieto, M.; Herrero-González, S.; Medina, J.M.; Tabernero, A. Connexin43 recruits PTEN and Csk to inhibit c-Src activity in glioma cells and astrocytes. Oncotarget 2016, 7, 49819–49833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.P.; Fan, Y.; Hossain, M.Z.; Peng, A.; Zeng, Z.L.; Boynton, A.L. Reversion of the neoplastic phenotype of human glioblastoma cells by connexin 43 (cx43). Cancer Res. 1998, 58, 5089–5096. [Google Scholar] [PubMed]
- Dang, X.; Doble, B.W.; Kardami, E. The carboxy-tail of connexin-43 localizes to the nucleus and inhibits cell growth. Mol. Cell Biochem. 2003, 242, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Moorby, C.; Patel, M. Dual functions for connexins: Cx43 regulates growth independently of gap junction formation. Exp. Cell Res. 2001, 271, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Jeyaraman, M.; Kardami, E. Regulation of connexin-43-mediated growth inhibition by a phosphorylatable amino-acid is independent of gap junction-forming ability. Mol. Cell. Biochem. 2006, 289, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Ul-Hussain, M.; Olk, S.; Schoenebeck, B.; Wasielewski, B.; Meier, C.; Prochnow, N.; May, C.; Galozzi, S.; Marcus, K.; Zoidl, G.; et al. Internal ribosomal entry site (IRES) activity generates endogenous carboxyl-terminal domains of Cx43 and is responsive to hypoxic conditions. J. Biol. Chem. 2014, 289, 20979–20990. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.W.; Shaw, R.M. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 2013, 5, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, S.S.; Xiao, S.; Basheer, W.A.; Baum, R.; Epifantseva, I.; Hong, T.; Shaw, R.M. Cx43 Isoform GJA1–20k Promotes Microtubule Dependent Mitochondrial Transport. Front. Physiol. 2017, 8, 905. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, S.; Guida, L.; Zocchi, E.; Franco, L.; De Flora, A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001, 15, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Liu, X.; Liu, R.; Yang, L.; Zuo, J.; Liu, W. Connexin 43 hemichannel regulates H9c2 cell proliferation by modulating intracellular ATP and (Ca2+). Acta Biochim. Biophys. Sin. 2010, 42, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Z.; Riquelme, M.A.; Gu, S.; Kar, R.; Gao, X.; Sun, L.; Jiang, J.X. Osteocytic connexin hemichannels suppress breast cancer growth and bone metastasis. Oncogene 2016, 35, 5597–5607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, H.; Omori, Y.; Zaidan-Dagli, M.L.; Mironov, N.; Mesnil, M.; Krutovskikh, V. Genetic and epigenetic changes of intercellular communication genes during multistage carcinogenesis. Cancer Detect Prev. 1999, 23, 273–279. [Google Scholar] [CrossRef] [PubMed]
- King, T.J.; Fukushima, L.H.; Donlon, T.A.; Hieber, A.D.; Shimabukuro, K.A.; Bertram, J.S. Correlation between growth control, neoplastic potential and endogenous connexin43 expression in HeLa cell lines: Implications for tumor progression. Carcinogenesis 2000, 21, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Ito, F.; Yamasaki, H.; Hagiwara, K.; Ozasa, H.; Nakazawa, H.; Toma, H. Epigenetic inactivation of connexin 32 in renal cell carcinoma from hemodialytic patients. Kidney Int. 2004, 65, 1519. [Google Scholar] [CrossRef] [PubMed]
- Sumiko, S.; Hiromi, H.; Hiromi, S.; Keiko, F.; Shigeto, K.; Taiichiro, S.; Toyohiko, A.; Kiyokazu, H.; Hiroshi, Y.; Tomohiro, Y. Prevention of renal cell carcinoma from hemodialysis patients by regulating epigenetic factors. Kidney Int. 2005, 67, 2506–2507. [Google Scholar] [CrossRef] [PubMed]
- Jinn, Y.; Inase, N. Connexin 43, E-cadherin, beta-catenin and ZO-1 expression, and aberrant methylation of the connexin 43 gene in NSCLC. Anticancer Res. 2010, 30, 2271–2278. [Google Scholar] [PubMed]
- Sirnes, S.; Honne, H.; Ahmed, D.; Danielsen, S.A.; Rognum, T.O.; Meling, G.I.; Leithe, E.; Rivedal, E.; Lothe, R.A.; Lind, G.E. DNA methylation analyses of the connexin gene family reveal silencing of GJC1 (Connexin45) by promoter hypermethylation in colorectal cancer. Epigenetics 2011, 6, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warn-Cramer, B.J.; Lau, A.F. Regulation of gap junctions by tyrosine protein kinases. Biochim. Biophys. Acta 2004, 1662, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Warn-Cramer, B.J.; Cottrell, G.T.; Burt, J.M.; Lau, A.F. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 1998, 273, 9188–9196. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.F.; Kanemitsu, M.Y.; Kurata, W.E.; Danesh, S.; Boynton, A.L. Epidermal growth factor disrupts gap-junctional communication and induces phosphorylation of connexin43 on serine. Mol. Biol. Cell 1992, 3, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.Z.; Ao, P.; Boynton, A.L. Platelet-derived growth factor-induced disruption of gap junctional communication and phosphorylation of connexin43 involves protein kinase C and mitogen-activated protein kinase. J. Cell Physiol. 1998, 176, 332–341. [Google Scholar] [CrossRef]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin43 C-terminus: A tail of many tales. Biochim. Biophys. Acta 2018, 1860, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, C.; Zhang, A.; Wang, K.; Jia, Z.; Wang, G.; Han, L.; Kang, C.; Pu, P. miR-221/222 is the regulator of Cx43 expression in human glioblastoma cells. Oncol. Rep. 2012, 27, 1504–1510. [Google Scholar] [PubMed]
- Jin, Z.; Xu, S.; Yu, H.; Yang, B.; Zhao, H.; Zhao, G. miR-125b inhibits Connexin43 and promotes glioma growth. Cell Mol. Neurobiol. 2013, 33, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pan, J.H.; Song, B.; Xiong, E.Q.; Chen, Z.W.; Zhou, Z.S.; Su, Y.P. Suppression of CX43 expression by miR-20a in the progression of human prostate cancer. Cancer Biol. Ther. 2012, 13, 890–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubina, M.V.; Iatckii, N.A.; Popov, D.E.; Vasiliev, S.V.; Krutovskikh, V.A. Connexin 43, but not connexin 32, is mutated at advanced stages of human sporadic colon cancer. Oncogene 2002, 21, 4992–4996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta 2018, 1860, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Litvin, O.; Tiunova, A.; Connell-Alberts, Y.; Panchin, Y.; Baranova, A. What is hidden in the pannexin treasure trove: The sneak peek and the guesswork. J. Cell Mol. Med. 2006, 10, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.P.; Bechberger, J.F.; Thompson, R.J.; MacVicar, B.A.; Bruzzone, R.; Naus, C.C. Tumor-suppressive effects of pannexin 1 in C6 glioma cells. Cancer Res. 2007, 67, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.P.; Bechberger, J.F.; Naus, C.C. Pannexin2 as a novel growth regulator in C6 glioma cells. Oncogene 2009, 28, 4402–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan, K.N.; Langlois, S.; Penuela, S.; Cowan, B.J.; Laird, D.W. Pannexin1 and Pannexin3 exhibit distinct localization patterns in human skin appendages and are regulated during keratinocyte differentiation and carcinogenesis. Cell Commun. Adhes. 2012, 19, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Celetti, S.J.; Cowan, K.N.; Penuela, S.; Shao, Q.; Churko, J.; Laird, D.W. Implications of pannexin 1 and pannexin 3 for keratinocyte differentiation. J. Cell Sci. 2010, 123, 1363–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, T.; Nakamura, T.; Doyle, A.; Ishikawa, M.; de Vega, S.; Fukumoto, S.; Yamada, Y. Pannexin 3 regulates intracellular ATP/cAMP levels and promotes chondrocyte differentiation. J. Biol. Chem. 2010, 285, 18948–18958. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Iwamoto, T.; Fukumoto, S.; Yamada, Y. Pannexin 3 inhibits proliferation of osteoprogenitor cells by regulating Wnt and p21 signaling. J. Biol. Chem. 2014, 289, 2839–2851. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Nakamura, T.; Ishikawa, M.; Yoshizaki, K.; Sugimoto, A.; Ida-Yonemochi, H.; Ohshima, H.; Saito, M.; Yamada, Y.; Fukumoto, S. Pannexin 3 regulates proliferation and differentiation of odontoblasts via its hemichannel activities. PLoS ONE 2017, 12, e0177557. [Google Scholar] [CrossRef] [PubMed]
- Penuela, S.; Gyenis, L.; Ablack, A.; Churko, J.M.; Berger, A.C.; Litchfield, D.W.; Lewis, J.D.; Laird, D.W. Loss of pannexin 1 attenuates melanoma progression by reversion to a melanocytic phenotype. J. Biol. Chem. 2012, 287, 29184–29193. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Hingorani, S.R.; Lampe, P.D. Changes in Connexin43 Expression and Localization during Pancreatic Cancer Progression. J. Membr. Biol. 2012, 245, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Hodgins, M.B.; Edward, M.; Graham, S.V. The relationship between connexins, gap junctions, tissue architecture and tumour invasion, as studied in a novel in vitro model of HPV-16-associated cervical cancer progression. Oncogene 2003, 22, 7969–7980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanczuga-Koda, L.; Sulkowski, S.; Lenczewski, A.; Koda, M.; Wincewicz, A.; Baltaziak, M.; Sulkowska, M. Increased expression of connexins 26 and 43 in lymph node metastases of breast cancer. J. Clin. Pathol. 2006, 59, 429–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanczuga-Koda, L.; Koda, M.; Sulkowski, S.; Wincewicz, A.; Zalewski, B.; Sulkowska, M. Gradual loss of functional gap junction within progression of colorectal cancer—A shift from membranous Cx32 and Cx43 expression to cytoplasmic pattern during colorectal carcinogenesis. In Vivo 2010, 24, 101–107. [Google Scholar] [PubMed]
- Lowenstein, W.R. Junctional intercellular communication by phosphorylation. Biochem. Soc. Symp. 1985, 50, 43–58. [Google Scholar] [PubMed]
- Defamie, N.; Chepied, A.; Mesnil, M. Connexins, gap junctions and tissue invasion. FEBS Lett. 2014, 588, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- McNutt, N.; Hershberg, R.; Weinstein, R. Further observations on the occurrence of nexuses in benign and malignant human cervical epithelium. J. Cell Biol. 1971, 51, 805–825. [Google Scholar] [CrossRef] [PubMed]
- King, T.; Fukushima, L.; Hieber, A.; Shimabukuro, A.; Sakr, W.; Bertram, J. Reduced levels of connexin 43 in cervical dysplasia: Inducible expression in a cervical carcinoma line decreases neoplastic potential with implications for tumour progression. Carcinogenesis 2000, 21, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Graham, S.V.; Edward, M.; Hodgins, M.B. Reduced expression of multiple gap junction proteins is a feature of cervical dysplasia. Mol. Cancer 2005, 4, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, S.; Going, J.J.; D’Arcy, R.; George, W.D. Expression of gap junction proteins connexin 26 and connexin 43 in normal human breast and in breast tumours. J. Pathol. 1998, 184, 37–43. [Google Scholar] [CrossRef]
- Laird, D.W.; Fistouris, P.; Batist, G.; Alpert, L.; Huynh, H.T.; Carysrinos, G.; Alaoui-Jamali, M.A. Deficiency of connexin43 gap junctions is an independent marker for breast tumors. Cancer Res. 1999, 59, 4104–4110. [Google Scholar] [PubMed]
- Singal, R.; Tu, Z.; Vanwert, J.; Ginder, G.; Kiang, D. Modulation of the connexin26 tumour suppressor gene expression through methylation in human mammary epithelial cell lines. Anticancer Res. 2000, 20, 59–64. [Google Scholar] [PubMed]
- Naoi, Y.; Miyoshi, Y.; Taguchi, T.; Kim, S.J.; Arai, T.; Tamaki, Y.; Noguchi, S. Connexin26 expression is associated with lymphatic vessel invasion and poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2007, 106, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Habermann, H.; Ray, V.; Habermann, W.; Prins, G.S. Alterations in gap junction protein expression in human benign prostatic hyperplasia and prostate cancer. J. Urol. 2002, 167, 655–660. [Google Scholar] [CrossRef]
- Haass, N.K.; Ripperger, D.; Wladykowski, E.; Dawson, P.; Gimotty, P.A.; Blome, C.; Fischer, F.; Schmage, P.; Moll, I.; Brandner, J.M. Melanoma progression exhibits a significant impact on connexin expression patterns in the epidermal tumor microenvironment. Histochem. Cell Biol. 2009, 133, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Elzarrad, M.K.; Haroon, A.; Willecke, K.; Dobrowolski, R.; Gillespie, M.N.; Al-Mehdi, A.-B. Connexin-43 upregulation in micrometastases and tumor vasculature and its role in tumor cell attachment to pulmonary endothelium. BMC Med. 2008, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, P.; Saunders, M.M.; Li, Z.; Zhou, Z.; Sheaffer, N.; Kunze, E.L.; Samant, R.S.; Welch, D.R.; Donahue, H.J. Breast cancer metastatic potential: Correlation with increased heterotypic gap junctional intercellular communication between breast cancer cells and osteoblastic cells. Int. J. Cancer 2004, 111, 693–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhou, Z.; Donahue, H.J. Alterations in Cx43 and OB-cadherin affect breast cancer cell metastatic potential. Clin. Exp. Metastasis 2008, 25, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, Z.; Welch, D.R.; Donahue, H.J. Expressing connexin 43 in breast cancer cells reduces their metastasis to lungs. Clin. Exp. Metastasis 2008, 25, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, Y.; Wu, Q.; Acquafondata, M.; Dhir, R.; Wells, A. Partial Mesenchymal to Epithelial Reverting Transition in Breast and Prostate Cancer Metastases. Cancer Microenviron. 2012, 5, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Arun, S.; Ravisankar, S.; Vanisree, A.J. Implication of connexin30 on the stemness of glioma: Connexin30 reverses the malignant phenotype of glioma by modulating IGF-1R, CD133 and cMyc. J. Neuro-Oncol. 2017, 135, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhou, X.; Wang, X.; Yang, Y.; Luo, J.; Liu, Y.; Mao, Q. Complex role of connexin 43 in astrocytic tumors and possible promotion of gliom-associated epileptic discharge. Mol. Med. Rep. 2017, 16, 7890–7900. [Google Scholar] [CrossRef] [PubMed]
- Crespin, S.; Fromont, G.; Wager, M.; Levillain, P.; Cronier, L.; Monvoisin, A.; Defamie, N.; Mesnil, M. Expression of a gap junction protein, connexin43, in a large panel of human gliomas: New insights. Cancer Med. 2016, 5, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Hitomi, M.; Bar-Shain, N.; Dalimov, Z.; Ellis, L.; Velpula, K.K.; Fraizer, G.C.; Gourdie, R.G.; Lathia, J.D. Connexin 43 expression is associated with increased malignancy in prostate cancer cell lines and functions to promote migration. Oncotarget 2015, 6, 11640–11651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, K.; Pitchakarn, P.; Suzuki, S.; Chewonarin, T.; Tang, M.; Takahashi, S.; Naiki-Ito, A.; Sato, S.; Takahashi, S.; Asamoto, M.; et al. Silencing of connexin 43 suppresses invasion, migration and lung metastasis of rat hepatocellular carcinoma cells. Cancer Sci. 2012, 103, 860–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezumi, K.; Yamamoto, H.; Murata, K.; Higashiyama, M.; Damdinsuren, B.; Nakamura, Y.; Kyo, N.; Okami, J.; Ngan, C.Y.; Takemasa, I.; et al. Aberrant Expression of Connexin 26 Is Associated with Lung Metastasis of Colorectal Cancer. Clin. Cancer Res. 2008, 14, 677–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito-Katsuragi, M.; Asada, H.; Niizeki, H.; Katoh, F.; Masuzawa, M.; Tsutsumi, M.; Kuniyasu, H.; Ito, A.; Nojima, H.; Miyagawa, S. Role for connexin 26 in metastasis of human malignant melanoma. Cancer 2007, 110, 1162–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, A.; Katoh, F.; Kataoka, T.R.; Okada, M.; Tsubota, N.; Asada, H.; Yoshikawa, K.; Maeda, S.; Kitamura, Y.; Yamasaki, H.; et al. A role for heterologous gap junctions between melanoma and endothelial cells in metastasis. J. Clin. Investig. 2000, 105, 1189–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongen, W.M.; Fitzgerald, D.J.; Asamoto, M.; Piccoli, C.; Slaga, T.J.; Gros, D.; Takeichi, M.; Yamasaki, H. Regulation of connexin 43-mediated gap junctional intercellular communication by Ca2+ in mouse epidermal cells is controlled by E-cadherin. J. Cell Biol. 1991, 114, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Ryszawy, D.; Sarna, M.; Rak, M.; Szpak, K.; Kędracka-Krok, S.; Michalik, M.; Siedlar, M.; Zuba-Surma, E.; Burda, K.; Korohoda, W.; et al. Functional links between Snail-1 and Cx43 account for the recruitment of Cx43-positive cells into the invasive front of prostate cancer. Carcinogenesis 2014, 35, 1920–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Zhang, C.; Li, L.; Dong, S.; Zhang, N.; Tong, X. Cx43 reverses the resistance of A549 lung adenocarcinoma cells to cisplatin by inhibiting EMT. Oncol. Rep. 2014, 31, 2751–2758. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.H.M.; Hülser, D.F. Connexin Transfection Induces Invasive Properties in HeLa Cells. Exp. Cell Res. 1998, 243, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.C.; Sin, W.C.; Aftab, Q.; Naus, C.C. Connexin43 enhances glioma invasion by a mechanism involving the carboxy terminus. Glia 2007, 55, 1554–1564. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kumar, A.; Tripathi, R.P.; Chandna, S. Connexin-43 regulates p38-mediated cell migration and invasion induced selectively in tumour cells by low doses of γ-radiation in an ERK-1/2-independent manner. Carcinogenesis 2014, 35, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.; Christov, C.; Guillamo, J.S.; de Boüard, S.; Palfi, S.; Venance, L.; Tardy, M.; Peschanski, M. Contribution of gap junctional communication between tumor cells and astroglia to the invasion of the brain parenchyma by human glioblastomas. BMC Cell Biol. 2005, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sabban, M.E.; Pauli, B.U. Adhesion-mediated gap junctional communication between lung metastatic cancer cells and endothelium. Invasion Metastasis 1994, 14, 164–176. [Google Scholar] [PubMed]
- Thuringer, D.; Berthenet, K.; Cronier, L.; Solary, E.; Garrido, C. Primary tumor- and metastasis-derived colon cancer cells differently modulate connexin expression and function in human capillary endothelial cells. Oncotarget 2015, 6, 28800–28815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollmann, M.-A.; Shao, Q.; Laird, D.W.; Sandig, M. Connexin 43 mediated gap junctional communication enhances breast tumor cell diapedesis in culture. Breast Cancer Res. 2005, 7, R522–R534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoletov, K.; Strnadel, J.; Zardouzian, E.; Momiyama, M.; Park, F.D.; Kelber, J.A.; Pizzo, D.P.; Hoffman, R.; VandenBerg, S.R.; Klemke, R.L. Role of connexins in metastatic breast cancer and melanoma brain colonization. J. Cell Sci. 2013, 126, 904–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plante, I.; Stewart, M.K.G.; Barr, K.; Allan, A.L.; Laird, D.W. Cx43 suppresses mammary tumor metastasis to the lung in a Cx43 mutant mouse model of human disease. Oncogene 2011, 30, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Zhang, W.; Wang, N.Q.; Bani-Yaqhoub, M.; Lin, Z.X.; Naus, C.C. Suppression of tumorigenicity of human lung carcinoma cells after transfection with connexin 43. Carcinogenesis 1998, 19, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.M.; Seraj, M.J.; Li, Z.; Zhou, Z.; Winter, C.R.; Welch, D.R.; Donahue, H.J. Breast Cancer Metastatic Potential Correlates with a Breakdown in Homospecific and Heterospecific Gap Junctional Intercellular Communication. Cancer Res. 2001, 61, 1765–1767. [Google Scholar] [PubMed]
- Shao, Q.; Wang, H.; McLachlan, E.; Veitch, G.I.L.; Laird, D.W. Down-regulation of Cx43 by Retroviral Delivery of Small Interfering RNA Promotes an Aggressive Breast Cancer Cell Phenotype. Cancer Res. 2005, 65, 2705–2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamiche, C.; Clarhaut, J.; Strale, P.-O.; Crespin, S.; Pedretti, N.; Bernard, F.X.; Naus, C.C.; Chen, V.C.; Foster, L.J.; Defamie, N.; et al. The gap junction protein Cx43 is involved in the bone-targeted metastatic behaviour of human prostate cancer cells. Clin. Exp. Metastasis 2012, 29, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramón y Cajal, S.; Capdevila, C.; Hernandez-Losa, J.; De Mattos-Arruda, L.; Ghosh, A.; Lorent, J.; Larsson, O.; Aasen, T.; Postovit, L.M.; Topisirovic, I. Cancer as an ecomolecular disease and a neoplastic consortium. Biochim. Biophys. Acta (BBA) Rev. Cancer 2017, 1868, 484–499. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.C.; Takano, T.; Cotrina, M.L.; Arcuino, G.; Kang, J.; Liu, S.; Gao, Q.; Jiang, L.; Li, F.; Lichtenberg-Frate, H.; et al. Connexin 43 Enhances the Adhesivity and Mediates the Invasion of Malignant Glioma Cells. J. Neurosci. 2002, 22, 4302–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miekus, K.; Czernik, M.; Sroka, J.; Czyz, J.; Madeja, Z. Contact stimulation of prostate cancer cell migration: The role of gap junctional coupling and migration stimulated by heterotypic cell-to-cell contacts in determination of the metastatic phenotype of Dunning rat prostate cancer cells. Biol. Cell 2005, 97, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmann, D.; Ale-Agha, N.; Reinehr, R.; Steinbrenner, H.; Ramos, M.C.; Sies, H.; Brenneisen, P. Modulation of homologous gap junctional intercellular communication of human dermal fibroblasts via a paracrine factor(s) generated by squamous tumor cells. Carcinogenesis 2003, 24, 1737–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, E.; Sato, H.; Nagashima, Y.; Negishi, E.; Shirai, S.; Fukumoto, K.; Hagiwara, H.; Hagiwara, K.; Ueno, K.; Yano, T. A Src family inhibitor (PP1) potentiates tumor-suppressive effect of connexin 32 gene in renal cancer cells. Life Sci. 2005, 76, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Naranjo, A.; Saéz, P.J.; Johansson, C.C.; Ramírez, M.; Mandaković, D.; Pereda, C.; López, M.N.; Kiessling, R.; Sáez, J.C.; Salazar-Onfray, F. Functional Gap Junctions Facilitate Melanoma Antigen Transfer and Cross-Presentation between Human Dendritic Cells. J. Immunol. 2007, 178, 6949–6957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzini, E.; Massimiliano, L.; Penna, G.; Rescigno, M. Oral Tolerance Can Be Established via Gap Junction Transfer of Fed Antigens from CX3CR1+ Macrophages to CD103+ Dendritic Cells. Immunity. 2014, 40, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Aucher, A.; Rudnicka, D.; Davis, D.M. MicroRNAs Transfer from Human Macrophages to Hepato-Carcinoma Cells and Inhibit Proliferation. J. Immunol. 2013, 191, 6250–6260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, B.; Tang, J.-W.; Wang, B.; Cui, X.-N.; Hou, L.; Sun, L.; Mao, L.M.; Zhou, C.H.; Du, Y.; Wang, L.H.; et al. Identify lymphatic metastasis-associated genes in mouse hepatocarcinoma cell lines using gene chip. World J. Gastroenterol. WJG 2005, 11, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Furlow, P.W.; Zhang, S.; Soong, T.D.; Halberg, N.; Goodarzi, H.; Mangrum, C.; Wu, Y.G.; Elemento, O.; Tavazoie, S.F. Mechanosensitive pannexin-1 channels mediate microvascular metastatic cell survival. Nat. Cell Biol. 2015, 17, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Penuela, S. Connexin and pannexin channels in cancer. BMC Cell Biol. 2016, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Willebrords, J.; Maes, M.; Crespo Yanguas, S.; Vinken, M. Inhibitors of connexin and pannexin channels as potential therapeutics. Pharmacol. Ther. 2017, 180, 144–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trosko, J.E.; Chang, C.C. Mechanism of up-regulated gap junctional intercellular communication during chemoprevention and chemotherapy of cancer. Mutat. Res. 2001, 480–481, 219–229. [Google Scholar] [CrossRef]
- Fornelli, F.; Leone, A.; Verdesca, I.; Minervini, F.; Zacheo, G. The influence of lycopene on the proliferation of human breast cell line (MCF-7). Toxicol. In Vitro 2007, 21, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Lecci, R.M.; Durante, M.; Piraino, S. Extract from the zooxanthellate jellyfish Cotylorhiza tuberculata modulates gap junction intercellular communication in human cell cultures. Mar. Drugs 2013, 11, 1728–1762. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, H.; Katoh, F. Novel method for selective killing of transformed rodent cells through intercellular communication, with possible therapeutic applications. Cancer Res. 1988, 48, 3203–3207. [Google Scholar] [PubMed]
- Safwat, S.; Ishak, R.A.; Hathout, R.M.; Mortada, N.D. Statins anticancer targeted delivery systems: Re-purposing an old molecule. J. Pharm. Pharmacol. 2017, 69, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Cesen-Cummings, K.; Warner, K.A.; Ruch, R.J. Role of protein kinase C in the deficient gap junctional intercellular communication of K-ras-transformed murine lung epithelial cells. Anticancer Res. 1998, 18, 4343–4346. [Google Scholar] [PubMed]
- Touraine, R.L.; Vahanian, N.; Ramsey, W.J.; Blaese, R.M. Enhancement of the herpes simplex virus thymidine kinase/ganciclovir bystander effect and its antitumor efficacy in vivo by pharmacologic manipulation of gap junctions. Hum. Gene Ther. 1998, 9, 2385–2391. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fu, Y.; Peng, J.; Wu, D.; Yu, M.; Xu, C.; Wang, Q.; Tao, L. Simvastatin-induced up-regulation of gap junctions composed of connexin 43 sensitize Leydig tumor cells to etoposide: An involvement of PKC pathway. Toxicology 2013, 312, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Peng, J.; Huang, H.; Wang, Q.; Yu, M.; Tao, L. Simvastatin protects Sertoli cells against cisplatin cytotoxicity through enhanced gap junction intercellular communication. Oncol. Rep. 2015, 34, 2133–2141. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Leon-Paravic, C.G.; Ezquer, M.; Ezquer, F.; Rio, R.D.; Pupo, A.; Martinez, A.D.; Gonzalez, C. Carbon monoxide: A new player in the redox regulation of connexin hemichannels. IUBMB Life 2015, 67, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Leon-Paravic, C.G.; Figueroa, V.A.; Guzman, D.J.; Valderrama, C.F.; Vallejos, A.A.; Fiori, M.C.; Altenberg, G.A.; Reuss, L.; Retamal, M.A. Carbon monoxide (CO) is a novel inhibitor of connexin hemichannels. J. Biol. Chem. 2014, 289, 36150–36157. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Iwata, H.; Takano, Y.; Yamada, R.; Okuzawa, H.; Nagashima, Y.; Yamaura, K.; Ueno, K.; Yano, T. Enhanced effect of connexin 43 on cisplatin-induced cytotoxicity in mesothelioma cells. J. Pharmacol. Sci. 2009, 110, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Uzu, M.; Sato, H.; Yamada, R.; Kashiba, T.; Shibata, Y.; Yamaura, K.; Ueno, K. Effect of enhanced expression of connexin 43 on sunitinib-induced cytotoxicity in mesothelioma cells. J. Pharmacol. Sci. 2015, 128, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Uzu, M.; Sato, H.; Shimizu, A.; Shibata, Y.; Ueno, K.; Hisaka, A. Connexin 43 enhances Bax activation via JNK activation in sunitinib-induced apoptosis in mesothelioma cells. J. Pharmacol. Sci. 2017, 134, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Liu, J.; Sun, X.; Wang, N. Ganoderma lucidum inhibits proliferation of human ovarian cancer cells by suppressing VEGF expression and up-regulating the expression of connexin 43. BMC Complement. Altern. Med. 2014, 14, 434. [Google Scholar] [CrossRef] [PubMed]
- Forster, T.; Rausch, V.; Zhang, Y.; Isayev, O.; Heilmann, K.; Schoensiegel, F.; Liu, L.; Nessling, M.; Richter, K.; Labsch, S.; et al. Sulforaphane counteracts aggressiveness of pancreatic cancer driven by dysregulated Cx43-mediated gap junctional intercellular communication. Oncotarget 2014, 5, 1621–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghatnekar, G.S.; O’Quinn, M.P.; Jourdan, L.J.; Gurjarpadhye, A.A.; Draughn, R.L.; Gourdie, R.G. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 2009, 4, 205–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grek, C.L.; Rhett, J.M.; Bruce, J.S.; Abt, M.A.; Ghatnekar, G.S.; Yeh, E.S. Targeting connexin 43 with alpha-connexin carboxyl-terminal (ACT1) peptide enhances the activity of the targeted inhibitors, tamoxifen and lapatinib, in breast cancer: Clinical implication for ACT1. BMC Cancer 2015, 15, 296. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.F.; Varghese, R.T.; Lamouille, S.; Guo, S.; Pridham, K.J.; Kanabur, P.; Osimani, A.M.; Sharma, S.; Jourdan, J.; Rodgers, C.M.; et al. Connexin 43 Inhibition Sensitizes Chemoresistant Glioblastoma Cells to Temozolomide. Cancer Res. 2016, 76, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Jaraiz-Rodriguez, M.; Tabernero, M.D.; Gonzalez-Tablas, M.; Otero, A.; Orfao, A.; Medina, J.M.; Tabernero, A. A Short Region of Connexin43 Reduces Human Glioma Stem Cell Migration, Invasion, and Survival through Src, PTEN, and FAK. Stem Cell Rep. 2017, 9, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Calder, B.W.; Fann, S.A.; Bainbridge, H.; Gourdie, R.G.; Yost, M.J. Mechanism of action of the anti-inflammatory connexin43 mimetic peptide JM2. Am. J. Physiol. Cell Physiol. 2017, 313, C314–C326. [Google Scholar] [CrossRef] [PubMed]
- Baklaushev, V.P.; Yusbalieva, G.M.; Tsitrin, E.B.; Gurina, O.I.; Grinenko, N.P.; Victorov, I.V.; Chekhonin, V.P. Visualization of Connexin 43-positive cells of glioma and the periglioma zone by means of intercenously injected monoclonal antibodies. Drug Deliv. 2011, 18, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Chekhonin, V.P.; Baklaushev, V.P.; Yusubalieva, G.M.; Belorusova, A.E.; Gulyaev, M.V.; Tsitrin, E.B.; Grinenko, N.F.; Gurina, O.I.; Pirogov, Y.A. Targeted delivery of liposomal nanocontainers to the peritumoral zone of glioma by means of monoclonal antibodies against GFAP and the extracellular loop of Cx43. Nanomedicine 2012, 8, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Yusubalieva, G.M.; Baklaushev, V.P.; Gurina, O.I.; Gulyaev, M.V.; Pirogov, Y.A.; Chekhonin, V.P. Antitumor effects of monoclonal antibodies to connexin 43 extracellular fragment in induced low-differentiated glioma. Bull. Exp. Biol. Med. 2012, 153, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Nukolova, N.V.; Baklaushev, V.P.; Abakumova, T.O.; Mel’nikov, P.A.; Abakumov, M.A.; Yusubalieva, G.M.; Bychkov, D.A.; Kabanov, A.V.; Chekhonin, V.P. Targeted delivery of cisplatin by small es, Cyrilliconnexin 43 vector nanogels to the focus of experimental glioma C6. Bull. Exp. Biol. Med. 2014, 157, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Baklaushev, V.P.; Nukolova, N.N.; Khalansky, A.S.; Gurina, O.I.; Yusubalieva, G.M.; Grinenko, N.P.; Gubskiy, I.L.; Melnikov, P.A.; Kardashova, K.; Kabanov, A.V.; et al. Treatment of glioma by cisplatin-loaded nanogels conjugated with monoclonal antibodies against Cx43 and BSAT1. Drug Deliv. 2015, 22, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Yusubalieva, G.M.; Baklaushev, V.P.; Gurina, O.I.; Zorkina, Y.A.; Gubskii, I.L.; Kobyakov, G.L.; Golanov, A.V.; Goryainov, S.A.; Gorlachev, G.E.; Konovalov, A.N.; et al. Treatment of poorly differentiated glioma using a combination of monoclonal antibodies to extracellular connexin-43 fragment, temozolomide, and radiotherapy. Bull. Exp. Biol. Med. 2014, 157, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Abakumova, T.; Abakumov, M.; Shein, S.; Chelushkin, P.; Bychkov, D.; Mukhin, V.; Yusubalieva, G.; Grinenko, N.; Kabanov, A.; Nukolova, N.; et al. Connexin 43-targeted T1 contrast agent for MRI diagnosis of glioma. Contrast Media Mol. Imaging 2016, 11, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Guo, S.; Zhang, X.; Sun, A.; Tao, F.; Ju, H.; Qian, H. Effects of small interfering RNA interference of connexin 37 on subcutaneous gastric tumours in mice. Mol. Med. Rep. 2014, 10, 2955–2960. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, M.; Deleyrolle, L.P.; Mulkearns-Hubert, E.E.; Jarrar, A.; Li, M.; Sinyuk, M.; Otvos, B.; Brunet, S.; Flavahan, W.A.; Hubert, C.G.; et al. Differential connexin function enhances self-renewal in glioblastoma. Cell Rep. 2015, 11, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Rodriguez-Cruz, V.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Temozolomide resistance in glioblastoma cells occurs partly through epidermal growth factor receptor-mediated induction of connexin 43. Cell Death Dis. 2014, 5, e1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gielen, P.R.; Aftab, Q.; Ma, N.; Chen, V.C.; Hong, X.; Lozinsky, S.; Naus, C.C.; Sin, W.C. Connexin43 confers Temozolomide resistance in human glioma cells by modulating the mitochondrial apoptosis pathway. Neuropharmacology 2013, 75, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.W.; Huang, B.R.; Liu, Y.S.; Lin, H.Y.; Chen, C.C.; Tsai, C.F.; Lu, D.Y.; Lin, C. Differential Characterization of Temozolomide-Resistant Human Glioma Cells. Int. J. Mol. Sci. 2018, 19, E127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; O’Carroll, S.J.; Henare, K.; Ching, L.M.; Ormonde, S.; Nicholson, L.F.; Danesh-Meyer, H.V.; Green, C.R. Connexin hemichannel induced vascular leak suggests a new paradigm for cancer therapy. FEBS Lett. 2014, 588, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danesh-Meyer, H.V.; Kerr, N.M.; Zhang, J.; Eady, E.K.; O’Carroll, S.J.; Nicholson, L.F.; Johnson, C.S.; Green, C.R. Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischaemia. Brain 2012, 135, 506–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd-Tressler, A.; Penuela, S.; Laird, D.W.; Dubyak, G.R. Chemotherapeutic drugs induce ATP release via caspase-gated pannexin-1 channels and a caspase/pannexin-1- independent mechanism. J. Biol. Chem. 2014, 289, 27246–27263. [Google Scholar] [CrossRef] [PubMed]
- Boyd-Tressler, A.M.; Lane, G.S.; Dubyak, G.R. Up-regulated Ectonucleotidases in Fas-Associated Death Domain Protein- and Receptor-Interacting Protein Kinase 1-Deficient Jurkat Leukemia Cells Counteract Extracellular ATP/AMP Accumulation via Pannexin-1 Channels during Chemotherapeutic Drug-Induced Apoptosis. Mol. Pharmacol. 2017, 92, 30–47. [Google Scholar] [PubMed]
- Saez, P.J.; Vargas, P.; Shoji, K.F.; Harcha, P.A.; Lennon-Dumenil, A.M.; Saez, J.C. ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X7 receptors. Sci. Signal 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Draganov, D.; Gopalakrishna-Pillai, S.; Chen, Y.R.; Zuckerman, N.; Moeller, S.; Wang, C.; Ann, D.; Lee, P.P. Modulation of P2X4/P2X7/Pannexin-1 sensitivity to extracellular ATP via Ivermectin induces a non-apoptotic and inflammatory form of cancer cell death. Sci. Rep. 2015, 5, 16222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwill, K.D.; Quigley, D.A.; Kang, H.C.; Del Rosario, R.; Ginzinger, D.; Balmain, A. Panx3 links body mass index and tumorigenesis in a genetically heterogeneous mouse model of carcinogen-induced cancer. Genome Med. 2016, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Romano, R.C.; Gardner, J.M.; Shalin, S.C.; Ram, R.; Govindarajan, R.; Montgomery, C.O.; Gilley, J.H.; Nicholas, R.W. High Relative Expression of Pannexin 3 (PANX3) in an Axillary Sweat Gland Carcinoma With Osteosarcomatous Transformation. Am. J. Dermatopathol. 2016, 38, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Marcoli, M.; Micheli, L.; Zanardelli, M.; Maura, G.; Ghelardini, C.; Cervetto, C. Oxaliplatin evokes P2X7-dependent glutamate release in the cerebral cortex: A pain mechanism mediated by Pannexin 1. Neuropharmacology 2015, 97, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.L.; Arandjelovic, S.; Brown Mendu, K.; Schappe, S.; Buckley, M.W.; Chiu, Y.H.; Shu, S.; Kim, J.K.; Chung, J.; Chung, J.; et al. Hematopoietic pannexin 1 function is critical for neuropathic pain. Sci. Rep. 2017, 7, 42550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Mesnil, M.; Aasen, T.; Boucher, J.; Chépied, A.; Cronier, L.; Defamie, N.; Kameritsch, P.; Laird, D.W.; Lampe, P.D.; Lathia, J.D.; et al. An update on minding the gap in cancer. Biochim. Biophys. Acta 2018, 1860, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Sin, W.C.; Crespin, S.; Mesnil, M. Opposing roles of connexin43 in glioma progression. Biochim. Biophys. Acta 2012, 1818, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Sin, W.C.; Aftab, Q.; Bechberger, J.F.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- Matsuuchi, L.; Naus, C.C. Gap junction proteins on the move: Connexins, the cytoskeleton and migration. Biochim. Biophys. Acta 2013, 1828, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Machtaler, S.; Choi, K.; Dang-Lawson, M.; Falk, L.; Pournia, F.; Naus, C.C.; Matsuuchi, L. The role of the gap junction protein connexin43 in B lymphocyte motility and migration. FEBS Lett. 2014, 588, 1249–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, L.A.; Wang, D.D.; Kriegstein, A.R. Gap junction adhesion is necessary for radial migration in the neocortex. Nature 2007, 448, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.; Aftab, Q.; Sin, W.C. Common mechanisms linking connexin43 to neural progenitor cell migration and glioma invasion. Semin. Cell Dev. Biol. 2016, 50, 59–66. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| TISSUE | ORGANISM | CONNEXIN | REGULATION | REFERENCE |
|---|---|---|---|---|
| PRECANCERS AND PRIMARY TUMOURS | ||||
| PANCREATIC DUCTAL ADENOCARCINOMA | Mouse | Cx43 | Increased levels Changes in phosphorylation | [89] |
| CERVICAL CANCER | Human | Cx26, Cx30, Cx43 | Loss of connexin expression | [95,96,97] |
| BREAST CANCER | Human | Cx26, Cx43 | Loss of Cx43 gap junctions | [98,99,100,101] |
| PROSTATE CANCER | Human | Cx32, Cx43 | Decreased expression | [102] |
| COLON CANCER | Human | Cx32, Cx43 | Gradual loss of expression | [92] |
| MELANOMA | Human | Cx26, Cx30 | Increased expression | [103] |
| PRIMARY TUMOUR TO METASTASIS | ||||
| BREAST CANCER | Human | Cx26, Cx43 | [101,104,105,106,107,108] | |
| BRAIN | Human, rat Human | Cx30 Cx43 | Reduced expression | [109] [110,111] |
| PROSTATE | Human cell lines | Cx43 | Increased Cx43 associated with increased invasion | [112] |
| LIVER | Rat cell lines Human | Cx43 Cx26 | Cx43 overexpression High expression | [113] [114] |
| MELANOMA | Human Human cell lines | Cx26 | Increased expression | [103,115] [116] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graham, S.V.; Jiang, J.X.; Mesnil, M. Connexins and Pannexins: Important Players in Tumorigenesis, Metastasis and Potential Therapeutics. Int. J. Mol. Sci. 2018, 19, 1645. https://doi.org/10.3390/ijms19061645
Graham SV, Jiang JX, Mesnil M. Connexins and Pannexins: Important Players in Tumorigenesis, Metastasis and Potential Therapeutics. International Journal of Molecular Sciences. 2018; 19(6):1645. https://doi.org/10.3390/ijms19061645
Chicago/Turabian StyleGraham, Sheila V., Jean X. Jiang, and Marc Mesnil. 2018. "Connexins and Pannexins: Important Players in Tumorigenesis, Metastasis and Potential Therapeutics" International Journal of Molecular Sciences 19, no. 6: 1645. https://doi.org/10.3390/ijms19061645