Impaired Photic Entrainment of Spontaneous Locomotor Activity in Mice Overexpressing Human Mutant α-Synuclein

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
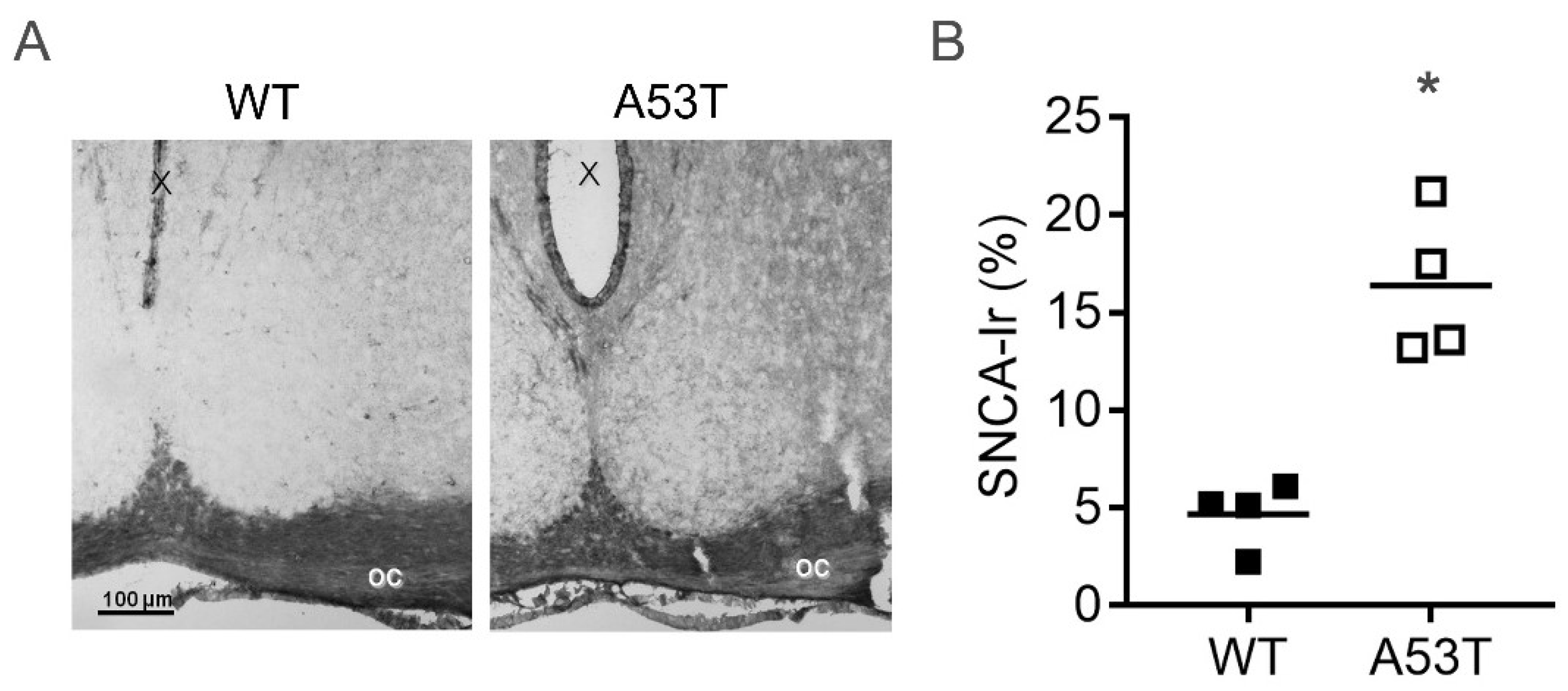
2.1. SNCA Immunoreaction Is Increased in the SCN of A53T Mice
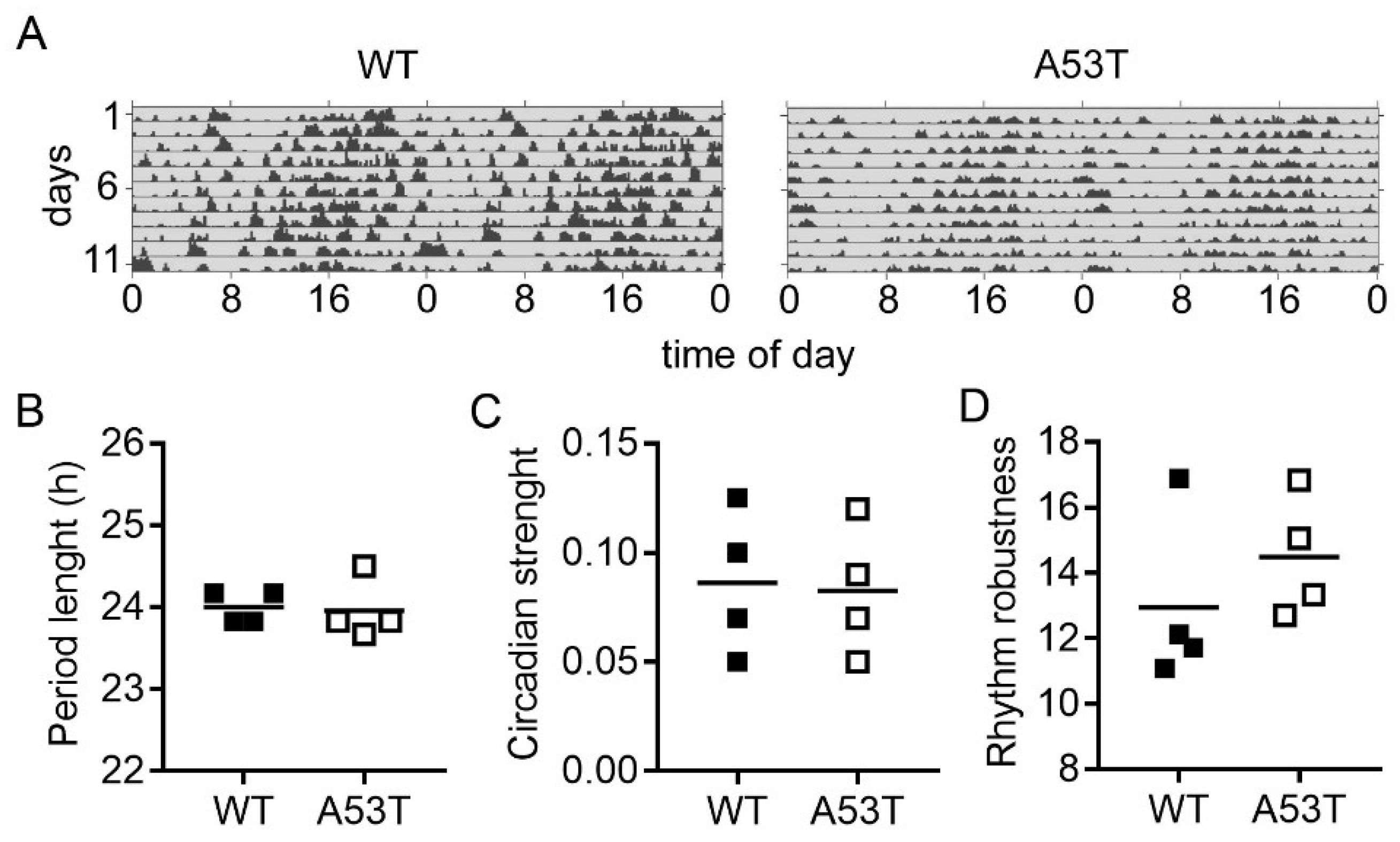
2.2. Rhythmic Spontaneous Locomotor Activity under Constant Darkness Is Not Affected in A35T Mice
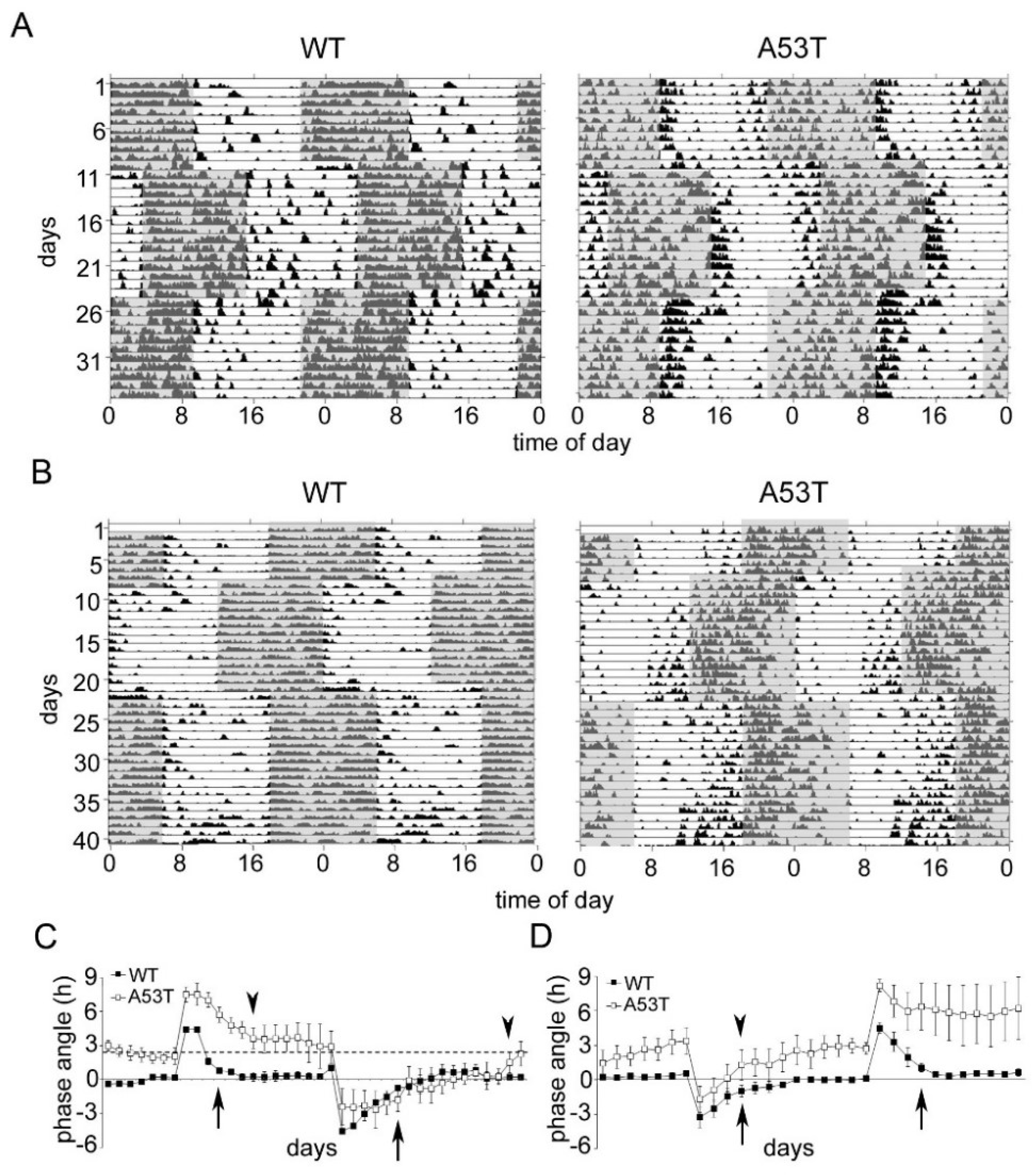
2.3. A35T Mice Show a Higher Daytime Activity and an Advanced Phase Angle of Entrainment under LD 12:12 Conditions
2.4. Re-Entrainment after Experimental Jet Lag Is Affected in A53T Mice
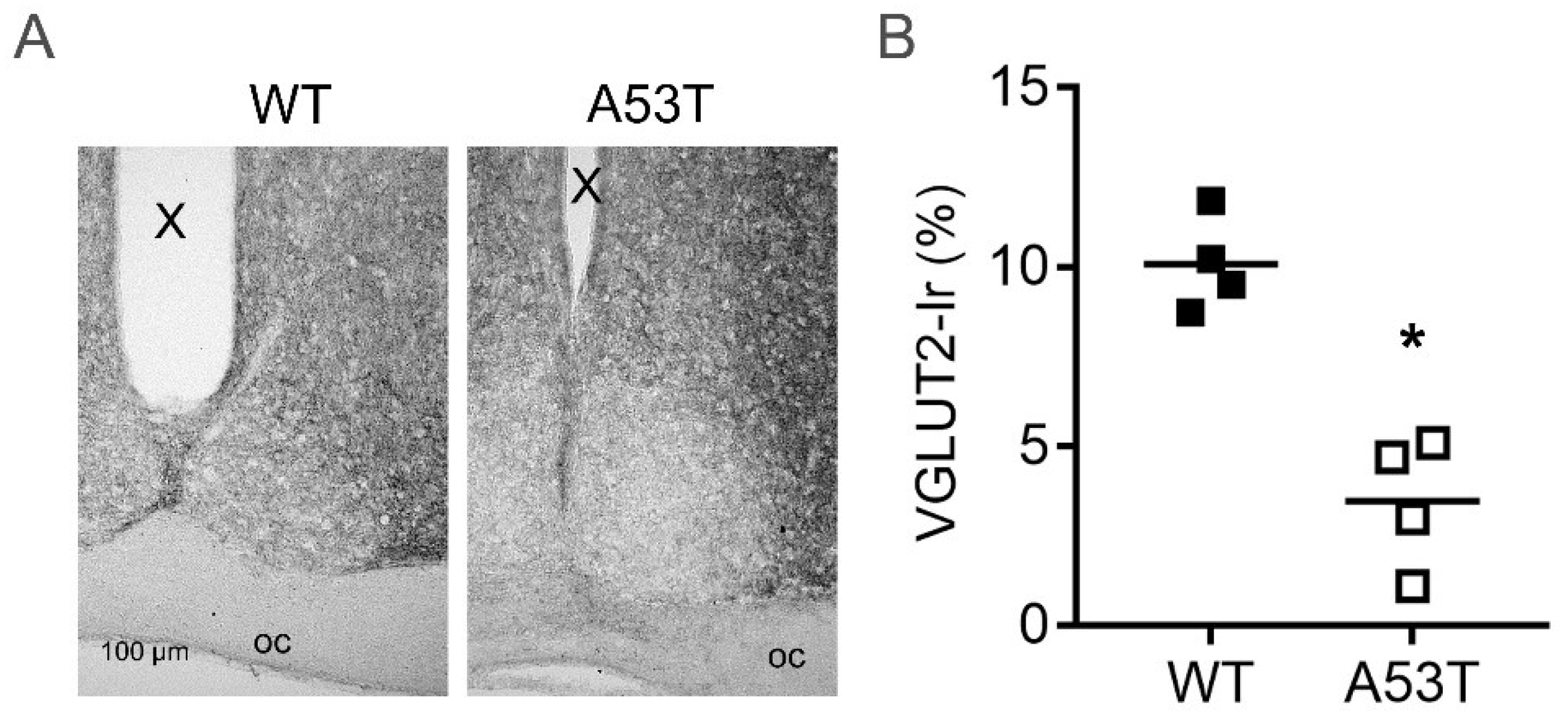
2.5. Vesicular Glutamate Transporter Type 2 Immunoreaction Is Reduced in A53T-SNCA Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Jet Lag
4.3. Data Analysis of Locomotor Activity Rhythms
4.4. Immunohistochemistry
4.5. Quantitative Analyses of Immunohistochemistry
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Saper, C.B.; Scammell, T.E.; Lu, J. Hypothalamic regulation of sleep and circadian rhythms. Nature 2005, 437, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Korf, H.W.; von Gall, C. Circadian Physiology. In Neuroscience in the 21st Century; Pfaff, D.W., Ed.; Springer Science + Business Media: Berlin, Germany, 2012. [Google Scholar]
- Dagan, Y. Circadian rhythm sleep disorders (CRSD). Sleep Med. Rev. 2002, 6, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Hogl, B.; Stefani, A.; Videnovic, A. Idiopathic REM sleep behaviour disorder and neurodegeneration—An update. Nat. Rev. Neurol. 2018, 14, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Chokroverty, S. Sleep and neurodegenerative diseases. Semin. Neurol. 2009, 29, 446–467. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, M.H.; Podder, N.; Chokroverty, S. Sleep and neurodegenerative diseases. Semin. Neurol. 2005, 25, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Menza, M.; Dobkin, R.D.; Marin, H.; Bienfait, K. Sleep disturbances in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25 (Suppl. 1), S117–S122. [Google Scholar] [CrossRef] [PubMed]
- Raggi, A.; Ferri, R. Sleep disorders in neurodegenerative diseases. Eur. J. Neurol. 2010, 17, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Videnovic, A.; Golombek, D. Circadian Dysregulation in Parkinson’s Disease. Neurobiol. Sleep Circadian Rhythms 2017, 2, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.W.; Chahine, L.M.; Caspell-Garcia, C.; Long, J.D.; Coffey, C.; Hogl, B.; Videnovic, A.; Iranzo, A.; Mayer, G.; Foldvary-Schaefer, N.; et al. Longitudinal assessment of excessive daytime sleepiness in early Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Arnulf, I. Sleep and wakefulness disturbances in Parkinson’s disease. In Parkinson’s Disease and Related Disorders; Springer: Vienna, Austria, 2006; pp. 357–360. [Google Scholar]
- Bargiotas, P.; Schuepbach, M.W.; Bassetti, C.L. Sleep-wake disturbances in the premotor and early stage of Parkinson’s disease. Curr. Opin. Neurol. 2016, 29, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Claassen, D.O.; Kutscher, S.J. Sleep disturbances in Parkinson's disease patients and management options. Nat. Sci. Sleep 2011, 3, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.H.; Millman, R.P. Sleep disturbances and Parkinson’s disease. CNS Spectr. 2008, 13, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Yousaf, T.; Pagano, G.; Niccolini, F.; Politis, M. Excessive daytime sleepiness may be associated with caudate denervation in Parkinson disease. J. Neurol. Sci. 2018, 387, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, T.; Pagano, G.; Niccolini, F.; Politis, M. Increased dopaminergic function in the thalamus is associated with excessive daytime sleepiness. Sleep Med. 2018, 43, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Molloy, S.; Bain, P.G.; Rabiner, E.A.; Chaudhuri, K.R.; Brooks, D.J.; Pavese, N. Sleep problems and hypothalamic dopamine D3 receptor availability in Parkinson disease. Neurology 2016, 87, 2451–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fronczek, R.; Overeem, S.; Lee, S.Y.; Hegeman, I.M.; van Pelt, J.; van Duinen, S.G.; Lammers, G.J.; Swaab, D.F. Hypocretin (orexin) loss and sleep disturbances in Parkinson’s Disease. Brain J. Neurol. 2008, 131 Pt 1, e88. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, T.C.; Lai, Y.Y.; Siegel, J.M. Hypocretin (orexin) cell loss in Parkinson’s disease. Brain J. Neurol. 2007, 130 Pt 6, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.; Plenzig, S.; Gispert, S.; Wada, K.; Korf, H.W.; Von Gall, C. Disturbed sleep/wake rhythms and neuronal cell loss in lateral hypothalamus and retina of mice with a spontaneous deletion in the ubiquitin carboxyl-terminal hydrolase L1 gene. Neurobiol. Aging 2012, 33, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Winkler, S.; Hagenah, J.; Lincoln, S.; Heckman, M.; Haugarvoll, K.; Lohmann-Hedrich, K.; Kostic, V.; Farrer, M.; Klein, C. α-Synuclein and Parkinson disease susceptibility. Neurology 2007, 69, 1745–1750. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129. [Google Scholar] [CrossRef]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ninan, I.; Antonova, I.; Battaglia, F.; Trinchese, F.; Narasanna, A.; Kolodilov, N.; Dauer, W.; Hawkins, R.D.; Arancio, O. α-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004, 23, 4506–4516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suedhof, T.C.; Rizo, J. Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol 2011, 3, a005637. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.; Mallory, M.; Hashimoto, M.; Song, D.; Shults, C.W.; Lang, I.; Masliah, E. Differential neuropathological alterations in transgenic mice expressing α-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res. 2002, 68, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Brehm, N.; Weil, J.; Seidel, K.; Rub, U.; Kern, B.; Walter, M.; Roeper, J.; Auburger, G. Potentiation of neurotoxicity in double-mutant mice with Pink1 ablation and A53T-SNCA overexpression. Hum. Mol. Genet. 2015, 24, 1061–1076. [Google Scholar] [CrossRef] [PubMed]
- Brehm, N.; Rau, K.; Kurz, A.; Gispert, S.; Auburger, G. Age-Related Changes of 14-3-3 Isoforms in Midbrain of A53T-SNCA Overexpressing Mice. J. Parkinsons Dis. 2015, 5, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A.; Double, K.L.; Lastres-Becker, I.; Tozzi, A.; Tantucci, M.; Bockhart, V.; Bonin, M.; Garcia-Arencibia, M.; Nuber, S.; Schlaudraff, F.; et al. A53T-α-synuclein overexpression impairs dopamine signaling and striatal synaptic plasticity in old mice. PLoS ONE 2010, 5, e11464. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, A.; Costa, C.; Siliquini, S.; Tantucci, M.; Picconi, B.; Kurz, A.; Gispert, S.; Auburger, G.; Calabresi, P. Mechanisms underlying altered striatal synaptic plasticity in old A53T-α synuclein overexpressing mice. Neurobiol. Aging 2012, 33, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Platt, N.J.; Gispert, S.; Auburger, G.; Cragg, S.J. Striatal dopamine transmission is subtly modified in human A53Tα-synuclein overexpressing mice. PLoS ONE 2012, 7, e36397. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Loh, D.H.; Truong, D.; Wu, Y.; Colwell, C.S. Circadian dysfunction in a mouse model of Parkinson’s disease. Exp. Neurol. 2011, 232, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Dauvilliers, Y. Insomnia in patients with neurodegenerative conditions. Sleep Med. 2007, 8 (Suppl. 4), S27–S34. [Google Scholar] [CrossRef]
- Breen, D.P.; Vuono, R.; Nawarathna, U.; Fisher, K.; Shneerson, J.M.; Reddy, A.B.; Barker, R.A. Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 2014, 71, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Bolitho, S.J.; Naismith, S.L.; Rajaratnam, S.M.; Grunstein, R.R.; Hodges, J.R.; Terpening, Z.; Rogers, N.; Lewis, S.J. Disturbances in melatonin secretion and circadian sleep-wake regulation in Parkinson disease. Sleep Med. 2014, 15, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.; Gispert, S.; Auburger, G.; Wicht, H.; Korf, H.W. Impact of Ataxin-2 knock out on circadian locomotor behavior and PER immunoreaction in the SCN of mice. Chronobiol. Int. 2017, 34, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Del Turco, D.; Garrett, L.; Chen, A.; Bernard, D.J.; Hamm-Clement, J.; Korf, H.W.; Deller, T.; Braak, H.; Auburger, G.; et al. Transgenic mice expressing mutant A53T human α-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol. Cell. Neurosci. 2003, 24, 419–429. [Google Scholar] [CrossRef]
- Gooley, J.J.; Lu, J.; Chou, T.C.; Scammell, T.E.; Saper, C.B. Melanopsin in cells of origin of the retinohypothalamic tract. Nat. Neurosci. 2001, 4, 1165. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, J.; Fahrenkrug, J. Melanopsin: A novel photopigment involved in the photoentrainment of the brain’s biological clock? Ann. Med. 2002, 34, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Hattar, S.; Liao, H.W.; Takao, M.; Berson, D.M.; Yau, K.W. Melanopsin-containing retinal ganglion cells: Architecture, projections, and intrinsic photosensitivity. Science 2002, 295, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Kornhauser, J.M.; Nelson, D.E.; Mayo, K.E.; Takahashi, J.S. Photic and circadian regulation of c-fos gene expression in the hamster suprachiasmatic nucleus. Neuron 1990, 5, 127–134. [Google Scholar] [CrossRef]
- Von Gall, C.; Duffield, G.E.; Hastings, M.H.; Kopp, M.D.; Dehghani, F.; Korf, H.W.; Stehle, J.H. CREB in the mouse SCN: A molecular interface coding the phase-adjusting stimuli light, glutamate, PACAP, and melatonin for clockwork access. J. Neurosci. 1998, 18, 10389–10397. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.M.; Faiman, L.E.; Hurst, W.J.; Kuriashkina, L.R.; Gillette, M.U. Resetting the biological clock: Mediation of nocturnal CREB phosphorylation via light, glutamate, and nitric oxide. J. Neurosci. 1997, 17, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Gau, D.; Lemberger, T.; von Gall, C.; Kretz, O.; Le Minh, N.; Gass, P.; Schmid, W.; Schibler, U.; Korf, H.W.; Schutz, G. Phosphorylation of CREB Ser142 regulates light-induced phase shifts of the circadian clock. Neuron 2002, 34, 245–253. [Google Scholar] [CrossRef]
- Ding, J.M.; Chen, D.; Weber, E.T.; Faiman, L.E.; Rea, M.A.; Gillette, M.U. Resetting the biological clock: Mediation of nocturnal circadian shifts by glutamate and NO. Science 1994, 266, 1713–1717. [Google Scholar] [CrossRef] [PubMed]
- Land, P.W.; Kyonka, E.; Shamalla-Hannah, L. Vesicular glutamate transporters in the lateral geniculate nucleus: Expression of VGLUT2 by retinal terminals. Brain Res. 2004, 996, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Wassle, H.; Regus-Leidig, H.; Haverkamp, S. Expression of the vesicular glutamate transporter vGluT2 in a subset of cones of the mouse retina. J. Comp. Neurol. 2006, 496, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Fremeau, R.T., Jr.; Duncan, J.L.; Renteria, R.C.; Yang, H.; Hua, Z.; Liu, X.; LaVail, M.M.; Edwards, R.H.; Copenhagen, D.R. Vesicular glutamate transporter 1 is required for photoreceptor synaptic signaling but not for intrinsic visual functions. J. Neurosci. 2007, 27, 7245–7255. [Google Scholar] [CrossRef] [PubMed]
- Gompf, H.S.; Fuller, P.M.; Hattar, S.; Saper, C.B.; Lu, J. Impaired circadian photosensitivity in mice lacking glutamate transmission from retinal melanopsin cells. J. Biol. Rhythms 2015, 30, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Purrier, N.; Engeland, W.C.; Kofuji, P. Mice deficient of glutamatergic signaling from intrinsically photosensitive retinal ganglion cells exhibit abnormal circadian photoentrainment. PLoS ONE 2014, 9, e111449. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with CSPα in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A.; May, C.; Schmidt, O.; Muller, T.; Stephan, C.; Meyer, H.E.; Gispert, S.; Auburger, G.; Marcus, K. A53T-α-synuclein-overexpression in the mouse nigrostriatal pathway leads to early increase of 14-3-3 epsilon and late increase of GFAP. J. Neural Transm. (Vienna) 2012, 119, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.Y.; Bullock, C.M.; Li, C.; Lee, A.G.; Bermak, J.C.; Belluzzi, J.; Weaver, D.R.; Leslie, F.M.; Zhou, Q.Y. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachiasmatic nucleus. Nature 2002, 417, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Witkovsky, P. Dopamine and retinal function. Doc. Ophthalmol. 2004, 108, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Gajula Balija, M.B.; Griesinger, C.; Herzig, A.; Zweckstetter, M.; Jackle, H. Pre-fibrillar α-synuclein mutants cause Parkinson’s disease-like non-motor symptoms in Drosophila. PLoS ONE 2011, 6, e24701. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.; Rauch, A.; Korf, H.W.; von Gall, C. The endogenous melatonin (MT) signal facilitates reentrainment of the circadian system to light-induced phase advances by acting upon MT2 receptors. Chronobiol. Int. 2012, 29, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Saavedra, M.; Antoun, G.; Yanagiya, A.; Oliva-Hernandez, R.; Cornejo-Palma, D.; Perez-Iratxeta, C.; Sonenberg, N.; Cheng, H.Y. miRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum. Mol. Genet. 2011, 20, 731–751. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, J.; Ding, J.M.; Chen, D.; Fahrenkrug, J.; Larsen, P.J.; Gillette, M.U.; Mikkelsen, J.D. Pituitary adenylate cyclase activating peptide (PACAP) in the retinohypothalamic tract: A daytime regulator of the biological clock. Ann. N. Y. Acad. Sci. 1998, 865, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, J.; Ding, J.M.; Chen, D.; Fahrenkrug, J.; Larsen, P.J.; Gillette, M.U.; Mikkelsen, J.D. Pituitary adenylate cyclase-activating peptide (PACAP) in the retinohypothalamic tract: A potential daytime regulator of the biological clock. J. Neurosci. 1997, 17, 2637–2644. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Korkmaz, A.; Erren, T.C.; Piekarski, C.; Tamura, H.; Manchester, L.C. Light at night, chronodisruption, melatonin suppression, and cancer risk: A review. Crit. Rev. Oncog. 2007, 13, 303–328. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, M.; Patton, A.P.; Chesham, J.E.; Maywood, E.S.; Hastings, M.H. Astrocytes Control Circadian Timekeeping in the Suprachiasmatic Nucleus via Glutamatergic Signaling. Neuron 2017, 93, 1420–1435.e5. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.; Muller, C.M.; Mordel, J.; Meissl, H.; Ansari, N.; Deller, T.; Korf, H.W.; von Gall, C. The mammalian molecular clockwork controls rhythmic expression of its own input pathway components. J. Neurosci. 2009, 29, 6114–6123. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.; Korf, H.W.; von Gall, C. Chronotype and stability of spontaneous locomotor activity rhythm in BMAL1-deficient mice. Chronobiol. Int. 2015, 32, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Refinetti, R. Non-stationary time series and the robustness of circadian rhythms. J. Theor. Biol. 2004, 227, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Van der Putten, H.; Wiederhold, K.H.; Probst, A.; Barbieri, S.; Mistl, C.; Danner, S.; Kauffmann, S.; Hofele, K.; Spooren, W.P.; Ruegg, M.A.; et al. Neuropathology in mice expressing human α-synuclein. J. Neurosci. 2000, 20, 6021–6029. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, D.; Chen, P.; Li, C. Conditional viral tracing reveals that steroidogenic factor 1-positive neurons of the dorsomedial subdivision of the ventromedial hypothalamus project to autonomic centers of the hypothalamus and hindbrain. J. Comp. Neurol. 2013, 521, 3167–3190. [Google Scholar] [CrossRef] [PubMed]
- Steinkellner, T.; Zell, V.; Farino, Z.J.; Sonders, M.S.; Villeneuve, M.; Freyberg, R.J.; Przedborski, S.; Lu, W.; Freyberg, Z.; Hnasko, T.S. Role for VGLUT2 in selective vulnerability of midbrain dopamine neurons. J. Clin. Investig. 2018, 128, 774–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Gall, C.; Garabette, M.L.; Kell, C.A.; Frenzel, S.; Dehghani, F.; Schumm-Draeger, P.M.; Weaver, D.R.; Korf, H.W.; Hastings, M.H.; Stehle, J.H. Rhythmic gene expression in pituitary depends on heterologous sensitization by the neurohormone melatonin. Nat. Neurosci. 2002, 5, 234–238. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pfeffer, M.; Zimmermann, Z.; Gispert, S.; Auburger, G.; Korf, H.-W.; Von Gall, C. Impaired Photic Entrainment of Spontaneous Locomotor Activity in Mice Overexpressing Human Mutant α-Synuclein. Int. J. Mol. Sci. 2018, 19, 1651. https://doi.org/10.3390/ijms19061651
Pfeffer M, Zimmermann Z, Gispert S, Auburger G, Korf H-W, Von Gall C. Impaired Photic Entrainment of Spontaneous Locomotor Activity in Mice Overexpressing Human Mutant α-Synuclein. International Journal of Molecular Sciences. 2018; 19(6):1651. https://doi.org/10.3390/ijms19061651
Chicago/Turabian StylePfeffer, Martina, Zuzana Zimmermann, Suzana Gispert, Georg Auburger, Horst-Werner Korf, and Charlotte Von Gall. 2018. "Impaired Photic Entrainment of Spontaneous Locomotor Activity in Mice Overexpressing Human Mutant α-Synuclein" International Journal of Molecular Sciences 19, no. 6: 1651. https://doi.org/10.3390/ijms19061651