Molecular Mechanisms of Disease Progression in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type during Ibrutinib Therapy


Abstract
Author Contributions
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Pham-Ledard, A.; Prochazkova-Carlotti, M.; Andrique, L.; Cappellen, D.; Vergier, B.; Martinez, F.; Grange, F.; Petrella, T.; Beylot-Barry, M.; Merlio, J.P. Multiple genetic alterations in primary cutaneous large B-cell lymphoma, leg type support a common lymphomagenesis with activated B-cell-like diffuse large B-cell lymphoma. Mod. Pathol. 2014, 27, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Pham-Ledard, A.; Cappellen, D.; Martinez, F.; Vergier, B.; Beylot-Barry, M.; Merlio, J.P. MYD88 Somatic Mutation Is a Genetic Feature of Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type. J. Investig. Dermatol. 2012, 132, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Pham-Ledard, A.; Beylot-Barry, M.; Barbe, C.; Leduc, M.; Petrella, T.; Vergier, B.; Martinez, F.; Cappellen, D.; Merlio, J.P.; Grange, F. High Frequency and Clinical Prognostic Value of MYD88 L265P Mutation in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg-Type. JAMA Dermatol. 2014, 150, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Mareschal, S.; Pham-Ledard, A.; Viailly, P.J.; Dubois, S.; Bertrand, P.; Maingonnat, C.; Fontanilles, M.; Bohers, E.; Ruminy, P.; Tournier, I.; et al. Identification of somatic mutations in primary cutaneous diffuse large B-cell lymphoma, leg-type by massive parallel sequencing. J. Investig. Dermatol. 2017, 137, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Doerre, S.; Corley, R.B. Constitutive nuclear translocation of NF-κB in B cells in the absence of IκB degradation. J. Immunol. 1999, 163, 269–277. [Google Scholar] [PubMed]
- Tinguely, M.; Thies, S.; Frigerio, S.; Reineke, T.; Korol, D.; Zimmermann, D.R. IRF8 is associated with germinal center B-cell-like type of diffuse large B-cell lymphoma and exceptionally involved in translocation t(14;16)(q32.33;q24.1). Leuk. Lymphoma 2014, 55, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Choi, Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Front. Immunol. 2014, 5, 511. [Google Scholar] [CrossRef] [PubMed]
- Alhejaily, A.; Day, A.G.; Feilotter, H.E.; Baetz, T.; LeBrun, D.P. Inactivation of the CDKN2A tumor-suppressor gene by deletion or methylation is common at diagnosis in follicular lymphoma and associated with poor clinical outcome. Clin. Cancer Res. 2014, 20, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Deng, A.L.; Kim, Y.R.; Lichtenstein, E.A.; O’Connor, O.A.; Deng, C. Combination of ibrutinib and chemotherapy produced a durable remission in multiply relapsed diffuse large B cell lymphoma leg type with mutant MYD88 and wildtype CD79. Haematologica 2017, 102, e275–e277. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
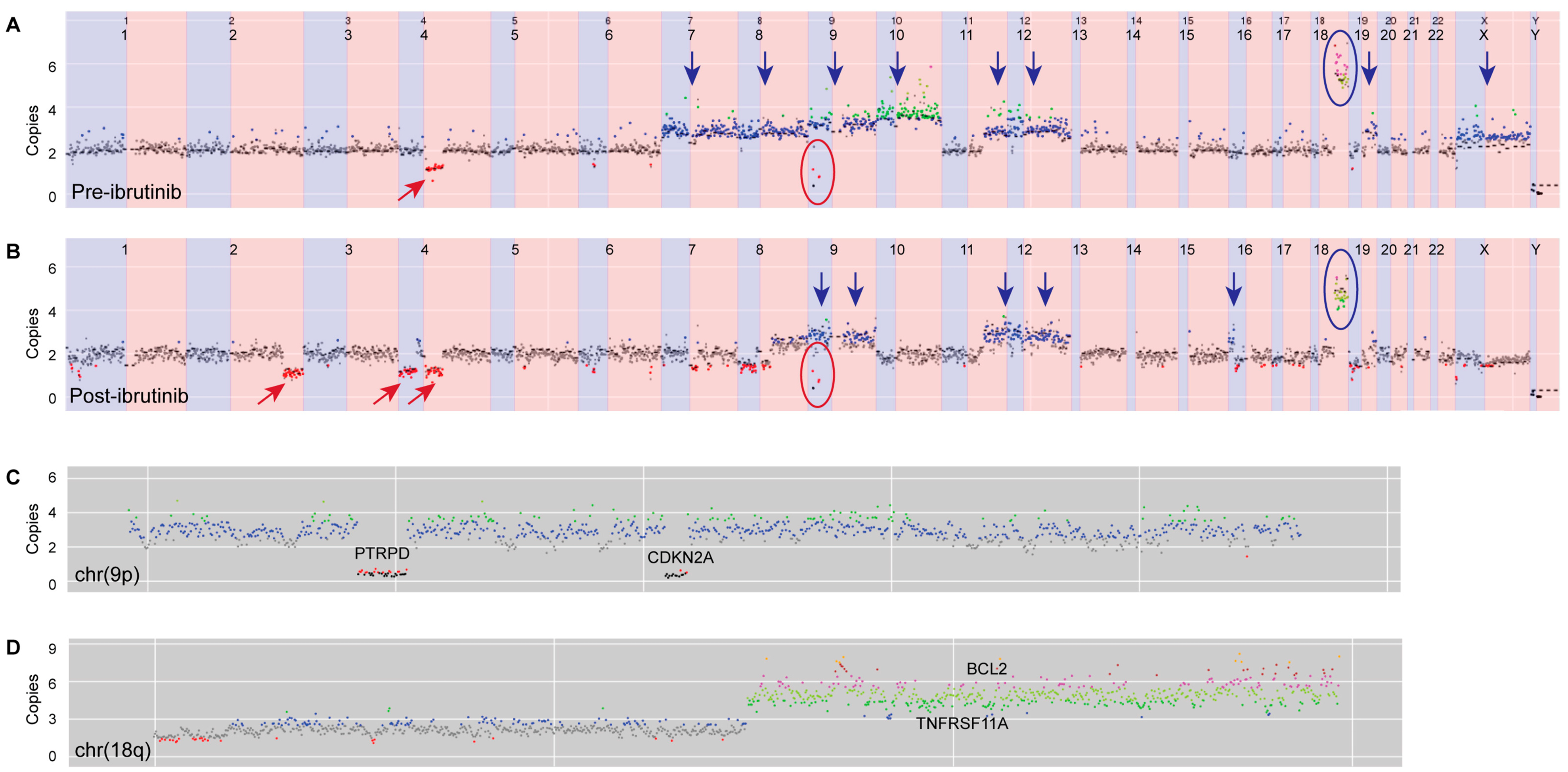
| Genomic Alteration | Pre-Ibrutinib (Skin) | Post-Ibrutinib (Node) |
|---|---|---|
| Variants | MYD88 c.794T > C;p.L265P | MYD88 c.794T > C;p.L265P |
| CD79B c.586T > C;p.Y196H | CD79B c.586T > C;p.Y196H | |
| CARD11c.367G > T;p.G123C | ||
| CARD11c.644A > T;p.K215M | ||
| NFKBIEc.1379G > C;p.G460A | ||
| Copy-number changes | Gain chr7, chr8, chr9, chr10, 11q, chr12, 19q, chrX | Gain 8q, chr9, 11q, chr12, 16p |
| Amplification 18q (BCL2/TNFRSF11A) | Amplification 18q (BCL2/TNFRSF11A) | |
| Del 4q | Del 2q, 4p, 4q, 6p, 7q, 8p, 8q, 16p, 16q, 17p, 17q, 19p, | |
| Homozygous deletion 9p containing CDKN2A Second homozygous deletion 9p containing PTRPD | Homozygous deletion 9p containing CDKN2A Second homozygous deletion 9p containing PTRPD | |
| Translocation | Not detected | t(14;16)(q32.33;q24.1) IgH-IRF8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fox, L.C.; Yannakou, C.K.; Ryland, G.; Lade, S.; Dickinson, M.; Campbell, B.A.; Prince, H.M. Molecular Mechanisms of Disease Progression in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type during Ibrutinib Therapy. Int. J. Mol. Sci. 2018, 19, 1758. https://doi.org/10.3390/ijms19061758
Fox LC, Yannakou CK, Ryland G, Lade S, Dickinson M, Campbell BA, Prince HM. Molecular Mechanisms of Disease Progression in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type during Ibrutinib Therapy. International Journal of Molecular Sciences. 2018; 19(6):1758. https://doi.org/10.3390/ijms19061758
Chicago/Turabian StyleFox, Lucy C., Costas K. Yannakou, Georgina Ryland, Stephen Lade, Michael Dickinson, Belinda A. Campbell, and Henry Miles Prince. 2018. "Molecular Mechanisms of Disease Progression in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type during Ibrutinib Therapy" International Journal of Molecular Sciences 19, no. 6: 1758. https://doi.org/10.3390/ijms19061758
APA StyleFox, L. C., Yannakou, C. K., Ryland, G., Lade, S., Dickinson, M., Campbell, B. A., & Prince, H. M. (2018). Molecular Mechanisms of Disease Progression in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type during Ibrutinib Therapy. International Journal of Molecular Sciences, 19(6), 1758. https://doi.org/10.3390/ijms19061758