A Current Overview of the Biological and Cellular Effects of Nanosilver


Abstract
{kind=link}
{kind=link}
1. Introduction
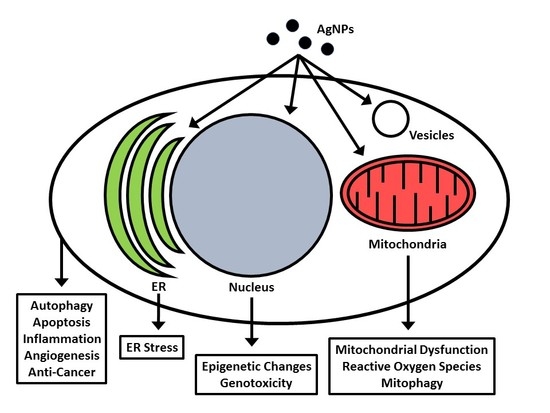
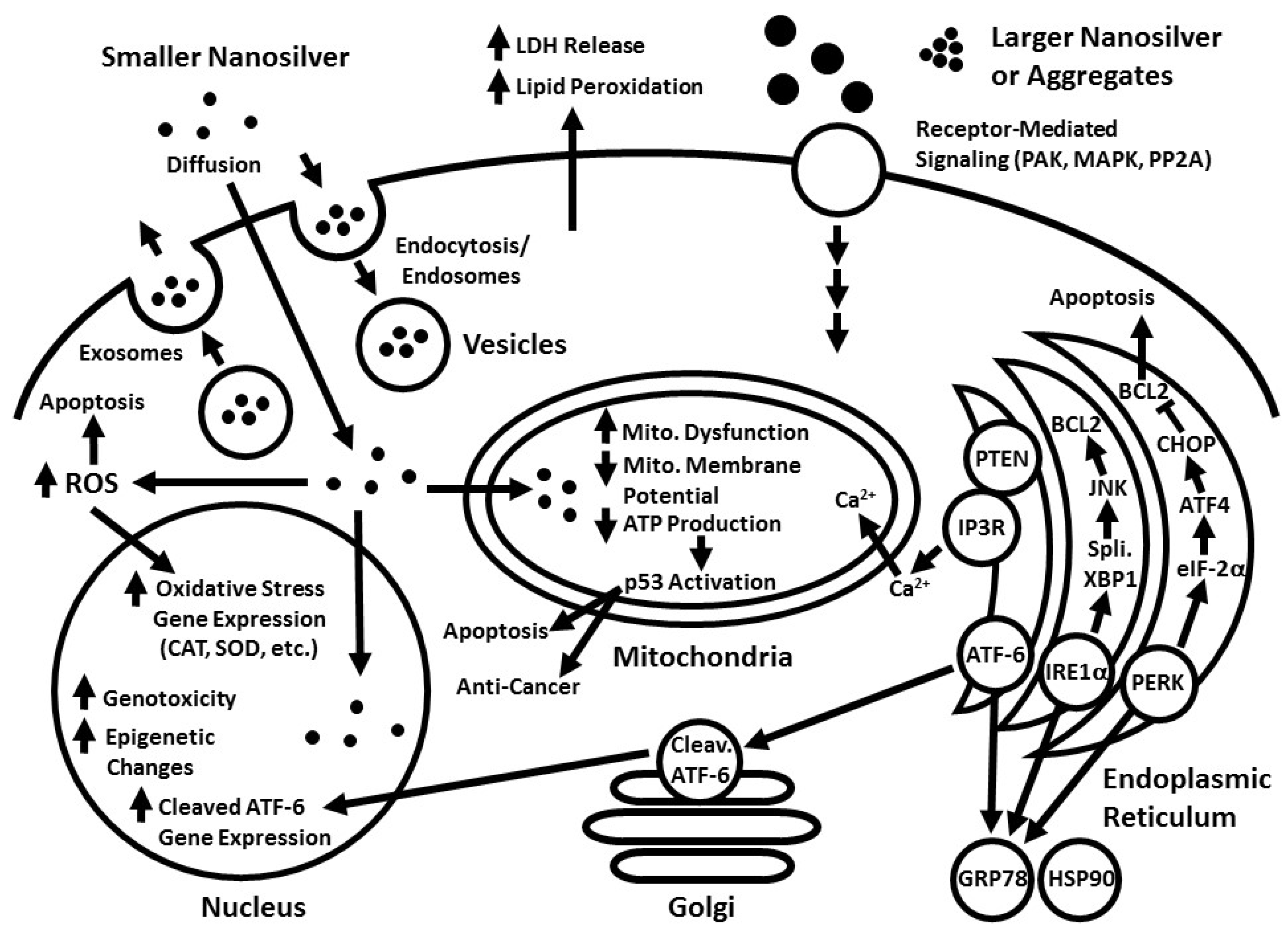
2. Cellular Uptake and Localization of Nanosilver
3. Nanosilver and Oxidative Stress (Reactive Oxygen Species)
3.1. Fluorescent Dyes Used to Evaluate ROS in Nanosilver Studies
3.2. Increase in ROS with Nanosilver Treatment
3.3. No Change or a Decrease in ROS with Nanosilver Treatment
4. Nanosilver and Inflammation
4.1. Nanosilver and Inflammation In Vitro
4.2. Nanosilver and Inflammation In Vivo
4.3. Anti-inflammatory Properties of Nanosilver in Wound Healing
5. Nanosilver and Hypoxia Stress
6. Nanosilver and the Mitochondria
7. Nanosilver and Endoplasmic Reticulum Stress (Unfolded Protein Response)
ER and Mitochondrial Interactions with Nanosilver
8. Effects of Nanosilver on Autophagy
8.1. Studies where Nanosilver Induces Autophagy
8.2. Studies where Nanosilver Blocks Autophagic Flux
9. Nanosilver and Angiogenesis
10. Nanosilver and Epigenetics
10.1. Nanosilver and DNA Methylation
10.2. Nanosilver and Histone Tail Modifications
10.3. Nanosilver and Non-Coding RNA Regulation
11. Nanosilver and Genotoxicity
11.1. Nanosilver and Genotoxicity In Vitro Studies
11.2. Nanosilver and Genotoxicity In Vivo Studies
12. Nanosilver and Cancer
12.1. Response of Cancer vs. Non-Cancer Cells to Nanosilver Treatment
12.2. Mechanisms Involved in the Effect of Nanosilver on Cancer Cells In Vitro
12.3. Cancer In Vivo Studies with Nanosilver Treatment
12.4. Nanosilver and Radiation Treatment
12.5. Nanosilver in Combination with other Drug Treatments
13. Interactions with, or Effects on, other Pathways
13.1. Nanosilver and the Cell Cycle
13.2. Effects of Nanosilver on DNA Polymerase and Transcription
13.3. Effects of Nanosilver on Pathways Involving Nrf2 and the Antioxidant Response
13.4. Nanosilver and the Insulin Signalling Pathway
13.5. Nanosilver Effects on Copper Homeodynamics
13.6. Effects of Nanosilver on Brain Function
14. Conclusions
Conflicts of Interest
References
- McShan, D.; Ray, P.C.; Yu, H. Molecular toxicity mechanism of nanosilver. J. Food Drug Anal. 2014, 22, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Luther, E.M.; Schmidt, M.M.; Diendorf, J.; Epple, M.; Dringen, R. Upregulation of metallothioneins after exposure of cultured primary astrocytes to silver nanoparticles. Neurochem. Res. 2012, 37, 1639–1648. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Son, M.Y.; Choi, M.S.; Kim, S.; Choi, A.-Y.; Lee, H.A.; Kim, K.S.; Kim, J.; Song, C.W.; Yoon, S. Integrative analysis of genes and miRNA alterations in human embryonic stem cells-derived neural cells after exposure to silver nanoparticles. Toxicol. Appl. Pharmacol. 2016, 299, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Villeret, B.; Dieu, A.; Straube, M.; Solhonne, B.; Miklavc, P.; Hamadi, S.; Le Borgne, R.; Mailleux, A.; Norel, X.; Aerts, J.; et al. Silver Nanoparticles Impair Retinoic Acid-Inducible Gene I-Mediated Mitochondrial Antiviral Immunity by Blocking the Autophagic Flux in Lung Epithelial Cells. ACS Nano 2018, 12, 1188–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, L.; Chen, Q.; Chen, C. Cytotoxic potential of silver nanoparticles. Yonsei Med. J. 2014, 55, 283–291. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Han, C.; Wang, X.; Zheng, Y.; Li, Q.; Hu, X.; Sun, H. The progress of silver nanoparticles in the antibacterial mechanism, clinical application and cytotoxicity. Mol. Biol. Rep. 2012, 39, 9193–9201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Park, J.-H.; Choi, Y.; Kang, M.-H.; Gurunathan, S.; Kim, J. Silver nanoparticles cause complications in pregnant mice Silver nanoparticles cause complications in pregnant mice. Int. J. Nanomed. 2015, 10, 7057–7071. [Google Scholar]
- Lu, Z.; Rong, K.; Li, J.; Yang, H.; Chen, R. Size-dependent antibacterial activities of silver nanoparticles against oral anaerobic pathogenic bacteria. J. Mater. Sci. Mater. Med. 2013, 24, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.W.-Y.; Chen, R.; Chung, N.P.-Y.; Ho, C.-M.; Lin, C.-L.S.; Che, C.-M. Silver nanoparticles fabricated in Hepes buffer exhibit cytoprotective activities toward HIV-1 infected cells. Chem. Commun. 2005, 40, 5059–5061. [Google Scholar] [CrossRef] [PubMed]
- Elechiguerra, J.L.; Burt, J.L.; Morones, J.R.; Camacho-Bragado, A.; Gao, X.; Lara, H.H.; Yacaman, M.J. Interaction of silver nanoparticles with HIV-1. J. Nanobiotechnol. 2005, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Goswami, K.; Sharma, R.D.; Reddy, M.V.R.; Dash, D. Novel microfilaricidal activity of nanosilver. Int. J. Nanomed. 2012, 7, 1023–1030. [Google Scholar]
- Saini, P.; Saha, S.K.; Roy, P.; Chowdhury, P.; Sinha Babu, S.P. Evidence of reactive oxygen species (ROS) mediated apoptosis in Setaria cervi induced by green silver nanoparticles from Acacia auriculiformis at a very low dose. Exp. Parasitol. 2016, 160, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, S.; Ma, J.; Qu, G.; Wang, X.; Yu, S.; He, J.; Liu, J.; Xia, T.; Jiang, G.-B. Silver nanoparticles induced RNA polymerase-silver binding and RNA transcription inhibition in erythroid progenitor cells. ACS Nano 2013, 7, 4171–4186. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Z.; Xu, M.; Wang, X.; Liu, R.; Liu, Q.; Zhang, Z.; Xia, T.; Zhao, J.; Jiang, G.; et al. Nanosilver incurs an adaptive shunt of energy metabolism mode to glycolysis in tumor and nontumor cells. ACS Nano 2014, 8, 5813–5825. [Google Scholar] [CrossRef] [PubMed]
- Cronholm, P.; Karlsson, H.L.; Hedberg, J.; Lowe, T.A.; Winnberg, L.; Elihn, K.; Wallinder, I.O.; Möller, L. Intracellular uptake and toxicity of Ag and CuO nanoparticles: A comparison between nanoparticles and their corresponding metal ions. Small 2013, 9, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Buttacavoli, M.; Albanese, N.N.; Di Cara, G.; Alduina, R.; Faleri, C.; Gallo, M.; Pizzolanti, G.; Gallo, G.; Feo, S.; Baldi, F.; et al. Anticancer activity of biogenerated silver nanoparticles: An integrated proteomic investigation. Oncotarget 2018, 9, 9685–9705. [Google Scholar] [CrossRef] [PubMed]
- Manshian, B.B.; Pfeiffer, C.; Pelaz, B.; Heimerl, T.; Gallego, M.; Möller, M.; Del Pino, P.; Himmelreich, U.; Parak, W.J.; Soenen, S.J. High-Content Imaging and Gene Expression Approaches to Unravel the Effect of Surface Functionality on Cellular Interactions of Silver Nanoparticles. ACS Nano 2015, 9, 10431–10444. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Huang, Z.; Wu, H.; Zhou, W.; Jin, P.; Wei, P.; Zhang, Y.; Zheng, F.; Zhang, J.; Xu, J.; et al. Inhibition of autophagy enhances the anticancer activity of silver nanoparticles. Autophagy 2014, 10, 2006–2020. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Guo, D.; Sun, L.; Huang, Z.; Zhang, X.; Ma, W.; Wu, J.; Xiao, L.; Zhao, Y.; Gu, N. Activation of autophagy by elevated reactive oxygen species rather than released silver ions promotes cytotoxicity of polyvinylpyrrolidone-coated silver nanoparticles in hematopoietic cells. Nanoscale 2017, 9, 5489–5498. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, H.L.; Gliga, A.R.; Calléja, F.M.G.R.; Gonçalves, C.S.A.G.; Wallinder, I.O.; Vrieling, H.; Fadeel, B.; Hendriks, G. Mechanism-based genotoxicity screening of metal oxide nanoparticles using the ToxTracker panel of reporter cell lines. Part. Fibre Toxicol. 2014, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, N.; Ramamoorthy, M.; Lyon, D.; Jones, K.; Duttaroy, A. Mechanism of Silver Nanoparticles Action on Insect Pigmentation Reveals Intervention of Copper Homeostasis. PLoS ONE 2013, 8, e53186. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Zhang, J.; Hu, Q.; Xu, M.; Chen, Y.; Hu, G.; Zhao, M.; Liu, S. Silver nanoparticle-induced hemoglobin decrease involves alteration of histone 3 methylation status. Biomaterials 2015, 70, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Durán, N.; Silveira, C.P.; Durán, M.; Martinez, D.S.T. Silver nanoparticle protein corona and toxicity: A mini-review. J. Nanobiotechnol. 2015, 13, 55. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.-H.; Tsai, J.-C.; Chen, C.-W.; Yan, S.-J.; Wang, Y.-J. Mechanisms of silver nanoparticle-induced toxicity and important role of autophagy. Nanotoxicology 2016, 10, 1021–1040. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, T.; Li, P.; Huang, W.; Tang, J.; Wang, P.; Liu, J.; Yuan, Q.; Bai, R.; Li, B.; et al. Use of Synchrotron Radiation Analytical Techniques to Reveal Chemical Origin of Silver Nanoparticle Cytotoxicity. ACS Nano 2015, 9, 6532–6547. [Google Scholar] [CrossRef] [PubMed]
- Lowry, G.V.; Espinasse, B.P.; Badireddy, A.R.; Richardson, C.J.; Reinsch, B.C.; Bryant, L.D.; Bone, A.J.; Deonarine, A.; Chae, S.; Therezien, M.; et al. Long-term transformation and fate of manufactured Ag nanoparticles in a simulated large scale freshwater emergent wetland. Environ. Sci. Technol. 2012, 46, 7027–7036. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.L.; Wamucho, A.; Tsyusko, O.V.; Unrine, J.M.; Crossley, A.; Svendsen, C.; Spurgeon, D.J. Multigenerational exposure to silver ions and silver nanoparticles reveals heightened sensitivity and epigenetic memory in Caenorhabditis elegans. Proc. R. Soc. B Biol. Sci. 2016, 283, 20152911. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, Z.; Wimmer, A.; Tian, Q.; Wang, X. New Insights into the Stability of Silver Sulfide Nanoparticles in Surface Water: Dissolution through Hypochlorite Oxidation. Environ. Sci. Technol. 2017, 51, 7920–7927. [Google Scholar] [CrossRef] [PubMed]
- Blaser, S.A.; Scheringer, M.; MacLeod, M.; Hungerbühler, K. Estimation of cumulative aquatic exposure and risk due to silver: Contribution of nano-functionalized plastics and textiles. Sci. Total Environ. 2008, 390, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Kaegi, R.; Voegelin, A.; Sinnet, B.; Zuleeg, S.; Hagendorfer, H.; Burkhardt, M.; Siegrist, H. Behavior of metallic silver nanoparticles in a pilot wastewater treatment plant. Environ. Sci. Technol. 2011, 45, 3902–3908. [Google Scholar] [CrossRef] [PubMed]
- Massarsky, A.; Abraham, R.; Nguyen, K.C.; Rippstein, P.; Tayabali, A.F.; Trudeau, V.L.; Moon, T.W. Nanosilver cytotoxicity in rainbow trout (Oncorhynchus mykiss) erythrocytes and hepatocytes. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2014, 159, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Wijnhoven, S.W.P.; Peijnenburg, W.J.G.M.; Herberts, C.A.; Hagens, W.I.; Oomen, A.G.; Heugens, E.H.W.; Roszek, B.; Bisschops, J.; Gosens, I.; Van De Meent, D.; et al. Nano-silver—A review of available data and knowledge gaps in human and environmental risk assessment. Nanotoxicology 2009, 3, 109–138. [Google Scholar] [CrossRef]
- Tang, J.; Xiong, L.; Wang, S.; Wang, J.; Liu, L.; Li, J.; Wan, Z.; Xi, T. Influence of silver nanoparticles on neurons and blood-brain barrier via subcutaneous injection in rats. Appl. Surf. Sci. 2008, 255, 502–504. [Google Scholar] [CrossRef]
- Wang, Z.; Qu, G.; Su, L.; Wang, L.; Yang, Z.; Jiang, J.; Liu, S.; Jiang, G. Evaluation of the Biological Fate and the Transport Through Biological Barriers of Nanosilver in Mice. Curr. Pharm. Des. 2013, 19, 6691–6697. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zande, M.; Vandebriel, R.J.; Van Doren, E.; Kramer, E.; Herrera Rivera, Z.; Serrano-Rojero, C.S.; Gremmer, E.R.; Mast, J.; Peters, R.J.B.; Hollman, P.C.H.; et al. Distribution, elimination, and toxicity of silver nanoparticles and silver ions in rats after 28-day oral exposure. ACS Nano 2012, 6, 7427–7442. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.; Rott, S.; Mantion, A.; Graf, P.; Plend, J.; Thünemann, A.F.; Meier, W.P.; Taubert, A.; Luch, A.; Reiser, G. Effects of silver nanoparticles on primary mixed neural cell cultures: Uptake, oxidative stress and acute calcium responses. Toxicol. Sci. 2012, 126, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Kim, J.S.; Cho, H.S.; Rha, D.S.; Kim, J.M.; Park, J.D.; Choi, B.S.; Lim, R.; Chang, H.K.; Chung, Y.H.; et al. Twenty-Eight-Day Oral Toxicity, Genotoxicity, and Gender-Related Tissue Distribution of Silver Nanoparticles in Sprague Dawley Rats. Inhal. Toxicol. 2008, 20, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-Y.; Kim, J.; Park, J.D.; Ryu, H.Y.; Yu, I.J. Histological Study of Gender Differences in Accumulation of Silver Nanoparticles in Kidneys of Fischer 344 Rats. J. Toxicol. Environ. Health Part A 2009, 72, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Song, M.Y.; Park, J.D.; Song, K.S.; Ryu, H.R.; Chung, Y.H.; Chang, H.K.; Lee, J.H.; Oh, K.H.; Kelman, B.J.; et al. Subchronic oral toxicity of silver nanoparticles. Part. Fibre Toxicol. 2010, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Ilyechova, E.Y.; Saveliev, A.N.; Skvortsov, A.N.; Babich, P.S.; Zatulovskaia, Y.A.; Pliss, M.G.; Korzhevskii, D.E.; Tsymbalenko, N.V.; Puchkova, L.V. The effects of silver ions on copper metabolism in rats. Metallomics 2014, 6, 1970–1987. [Google Scholar] [CrossRef] [PubMed]
- Miyayama, T.; Arai, Y.; Suzuki, N.; Hirano, S. Mitochondrial electron transport is inhibited by disappearance of metallothionein in human bronchial epithelial cells following exposure to silver nitrate. Toxicology 2013, 305, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Bergin, I.L.; Witzmann, F.A. Nanoparticle toxicity by the gastrointestinal route: Evidence and knowledge gaps. Int. J. Biomed. Nanosci. Nanotechnol. 2013, 3, 163–210. [Google Scholar] [CrossRef] [PubMed]
- Verano-Braga, T.; Miethling-Graff, R.; Wojdyla, K.; Rogowska-Wrzesinska, A.; Brewer, J.R.; Erdmann, H.; Kjeldsen, F. Insights into the Cellular Response Triggered by Silver Nanoparticles Using Quantitative Proteomics. ACS Nano 2014, 8, 2161–2175. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Zhou, H.; Gao, J. Nanoparticles modulate autophagic effect in a dispersity-dependent manner. Sci. Rep. 2015, 5, 14361. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lin, J.; Liu, P.; Huang, Z.; Zhao, P.; Jin, H.; Wang, C.; Wen, L.; Gu, N. Is the autophagy a friend or foe in the silver nanoparticles associated radiotherapy for glioma? Biomaterials 2015, 62, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lin, J.; Liu, P.; Huang, Z.; Zhao, P.; Jin, H.; Ma, J.; Wen, L.; Gu, N. Reactive oxygen species acts as executor in radiation enhancement and autophagy inducing by AgNPs. Biomaterials 2016, 101, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Liu, Y.; Wu, H.; Huang, Z.; Ma, J.; Guo, C.; Gao, F.; Jin, P.; Wei, P.; Zhang, Y.; et al. Key Role of TFEB Nucleus Translocation for Silver Nanoparticle-Induced Cytoprotective Autophagy. Small 2018, 14, 1703711. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Z.H.; Li, M.; Feng, Y.X.; Shi, J.C.; Zhang, J.; Shao, B. Hormesis effects of silver nanoparticles at non-cytotoxic doses to human hepatoma cells. PLoS ONE 2014, 9, e102564. [Google Scholar] [CrossRef] [PubMed]
- Sthijns, M.M.J.P.E.; Thongkam, W.; Albrecht, C.; Hellack, B.; Bast, A.; Haenen, G.R.M.M.; Schins, R.P.F. Silver nanoparticles induce hormesis in A549 human epithelial cells. Toxicol. Vitr. 2017, 40, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.Y.; McGee, J.K.; Killius, M.G.; Suarez, D.A.; Blackman, C.F.; DeMarini, D.M.; Simmons, S.O. Investigating oxidative stress and inflammatory responses elicited by silver nanoparticles using high-throughput reporter genes in HepG2 cells: Effect of size, surface coating, and intracellular uptake. Toxicol. Vitr. 2013, 27, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Nallanthighal, S.; Chan, C.; Bharali, D.J.; Mousa, S.A.; Reliene, R.; States, U.; States, U.; Sciences, H.; States, U.; States, U.; et al. Particle coatings but not silver ions mediate genotoxicity of ingested silver nanoparticles in a mouse model. Nanolmpact 2017, 5, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Greulich, C.; Diendorf, J.; Simon, T.; Eggeler, G.; Epple, M.; Köller, M. Uptake and intracellular distribution of silver nanoparticles in human mesenchymal stem cells. Acta Biomater. 2011, 7, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Kawata, K.; Osawa, M.; Okabe, S. In vitro toxicity of silver nanoparticles at noncytotoxic doses to HepG2 human hepatoma cells. Environ. Sci. Technol. 2009, 43, 6046–6051. [Google Scholar] [CrossRef] [PubMed]
- Zielinska, E.; Zauszkiewicz-Pawlak, A.; Wojcik, M.; Inkielewicz-Stepniak, I. Silver nanoparticles of different sizes induce a mixed type of programmed cell death in human pancreatic ductal adenocarcinoma. Oncotarget 2018, 9, 4675–4697. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Reyero, N.; Kennedy, A.J.; Escalon, B.L.; Habib, T.; Laird, J.G.; Rawat, A.; Wiseman, S.; Hecker, M.; Denslow, N.; Steevens, A.; et al. Differential effects and potential adverse outcomes of ionic silver and silver nanoparticles in vivo and in vitro. Environ. Sci. Technol. 2014, 48, 4546–4555. [Google Scholar] [CrossRef] [PubMed]
- Srikar, S.K.; Giri, D.D.; Pal, D.B.; Mishra, P.K.; Upadhyay, S.N. Green Synthesis of Silver Nanoparticles: A Review. Green Sustain. Chem. 2016, 6, 34–56. [Google Scholar] [CrossRef]
- Hackenberg, S.; Scherzed, A.; Kessler, M.; Hummel, S.; Technau, A.; Froelich, K.; Ginzkey, C.; Koehler, C.; Hagen, R.; Kleinsasser, N. Silver nanoparticles: Evaluation of DNA damage, toxicity and functional impairment in human mesenchymal stem cells. Toxicol. Lett. 2011, 201, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.C.; Zheng, J.; Yourick, J.J.; Sprando, R.L.; Gao, X. Toxicogenomic responses of human liver HepG2 cells to silver nanoparticles. J. Appl. Toxicol. 2015, 35, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.J.; Kim, S.; Kim, J.S.; Choi, I.H. Inflammasome formation and IL-1β release by human blood monocytes in response to silver nanoparticles. Biomaterials 2012, 33, 6858–6867. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Yao, Q.; Cao, F.; Liu, Q.; Liu, B.; Wang, X.H. Silver nanoparticles inhibit the function of hypoxia-inducible factor-1 and target genes: Insight into the cytotoxicity and antiangiogenesis. Int. J. Nanomed. 2016, 11, 6679–6692. [Google Scholar] [CrossRef] [PubMed]
- Havelaar, A.C.; De Gast, I.L.; Snijders, S.; Beerens, C.E.M.T.; Mancini, G.M.S.; Verheijen, F.W. Characterization of a heavy metal ion transporter in the lysosomal membrane. FEBS Lett. 1998, 436, 223–227. [Google Scholar] [CrossRef]
- Asharani, P.V.; Hande, M.P.; Valiyaveettil, S. Anti-proliferative activity of silver nanoparticles. BMC Cell Biol. 2009, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.R.; Zheng, J.; Tang, X.; Goering, P.L. Silver nanoparticle-induced autophagic-Lysosomal disruption and NLRP3-inflammasome activation in HepG2 cells is size-dependent. Toxicol. Sci. 2016, 150, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Foldbjerg, R.; Miclaus, T.; Wang, L.; Singh, R.; Hayashi, Y.; Sutherland, D.; Chen, C.; Autrup, H.; Beer, C. Multi-platform genotoxicity analysis of silver nanoparticles in the model cell line CHO-K1. Toxicol. Lett. 2013, 222, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Cheng, F.-Y.; Chiu, H.-W.; Tsai, J.-C.; Fang, C.-Y.; Chen, C.-W.; Wang, Y.-J. Cytotoxicity, oxidative stress, apoptosis and the autophagic effects of silver nanoparticles in mouse embryonic fibroblasts. Biomaterials 2014, 35, 4706–4715. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Jin, H.; Guo, Z.; Ma, J.; Zhao, J.; Li, D.; Wu, H.; Gu, N. Silver nanoparticles outperform gold nanoparticles in radiosensitizing U251 cells in vitro and in an intracranial mouse model of glioma. Int. J. Nanomed. 2016, 11, 5003–5014. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-Y.; Liu, M.-S.; Huang, L.-J.; Lue, S.-I.; Lin, L.-C.; Kwan, A.-L.; Yang, R.-C. Bioenergetic failure correlates with autophagy and apoptosis in rat liver following silver nanoparticle intraperitoneal administration. Part. Fibre Toxicol. 2013, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Choi, Y.J.; Han, J.W.; Kim, E.; Park, J.H.; Gurunathan, S.; Kim, J.H. Differential nanoreprotoxicity of silver nanoparticles in male somatic cells and spermatogonial stem cells. Int. J. Nanomed. 2015, 10, 1335–1357. [Google Scholar]
- Chorley, B.; Ward, W.; Simmons, S.O.; Vallanat, B.; Veronesi, B. The cellular and genomic response of rat dopaminergic neurons (N27) to coated nanosilver. Neurotoxicology 2014, 45, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2′,7′-Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: Forty years of application and controversy. Free Radic. Res. 2010, 44, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Oxy radicals, lipid peroxidation and DNA damage. Toxicology 2002, 181, 219–222. [Google Scholar] [CrossRef]
- Zhang, X.F.; Gurunathan, S. Combination of salinomycin and silver nanoparticles enhances apoptosis and autophagy in human ovarian cancer cells: An effective anticancer therapy. Int. J. Nanomed. 2016, 11, 3655–3675. [Google Scholar]
- Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant Properties Dedicated to Nanotechnologies. Antioxidants 2018, 7, 62–82. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.; Tomás-Hernández, S.; García, T.; Mulero, M.; Gómez, M.; Domingo, J.L.; Sánchez, D.J. Oral exposure to silver nanoparticles increases oxidative stress markers in the liver of male rats and deregulates the insulin signalling pathway and p53 and cleaved caspase 3 protein expression. Food Chem. Toxicol. 2018, 115, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zhao, B.; Chen, M.; Liu, Y.; Xu, M.; Wang, Z.; Liu, S.; Zhang, C. Nrf-2-driven long noncoding RNA ODRUL contributes to modulating silver nanoparticle-induced effects on erythroid cells. Biomaterials 2017, 130, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Takabayashi, F.; Ibuki, Y. Coexposure to silver nanoparticles and ultraviolet A synergistically enhances the phosphorylation of histone H2AX. J. Photochem. Photobiol. B Biol. 2016, 162, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Hoet, P.H.; Nemery, B.; Napierska, D. Intracellular oxidative stress caused by nanoparticles: What do we measure with the dichlorofluorescein assay? Nano Today 2013, 8, 223–227. [Google Scholar] [CrossRef]
- McBee, M.E.; Chionh, Y.H.; Sharaf, M.L.; Ho, P.; Cai, M.W.L.; Dedon, P.C. Production of superoxide in bacteria is stress- and cell state-dependent: A gating-optimized flow cytometry method that minimizes ROS measurement artifacts with fluorescent dyes. Front. Microbiol. 2017, 8, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Yang, Z.; Weisshaar, J.C. Single-cell, real-time detection of oxidative stress induced in Escherichia coli by the antimicrobial peptide CM15. Proc. Natl. Acad. Sci. USA 2015, 112, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Haskó, G.; Hawkins, B.J.; Madesh, M.; Pacher, P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat. Protoc. 2007, 2, 2295–2301. [Google Scholar] [CrossRef] [PubMed]
- Farah, M.A.; Ali, M.A.; Chen, S.-M.; Li, Y.; Al-Hemaid, F.M.; Abou-Tarboush, F.M.; Al-Anazi, K.M.; Lee, J. Silver nanoparticles synthesized from Adenium obesum leaf extract induced DNA damage, apoptosis and autophagy via generation of reactive oxygen species. Colloids Surf. B Biointerfaces 2016, 141, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Lee, Y.J.; Lee, E.K.; Kwak, M.K. Silver nanoparticles-mediated G2/M cycle arrest of renal epithelial cells is associated with NRF2-GSH signaling. Toxicol. Lett. 2012, 211, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.K.; Gurunathan, S.; Kang, M.H.; Han, J.W.; Das, J.; Choi, Y.J.; Kwon, D.N.; Cho, S.G.; Park, C.; Seo, H.G.; et al. Hypoxia-mediated autophagic flux inhibits silver nanoparticle-triggered apoptosis in human lung cancer cells. Sci. Rep. 2016, 6, 21688. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.C.; Zheng, J.; Graham, L.; Chen, L.; Ihrie, J.; Yourick, J.J.; Sprando, R.L. Comparative cytotoxicity of nanosilver in human liver HepG2 and colon Caco2 cells in culture. J. Appl. Toxicol. 2014, 34, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Gallorini, M.; di Giacomo, V.; Di Valerio, V.; Rapino, M.; Bosco, D.; Travan, A.; Di Giulio, M.; Di Pietro, R.; Paoletti, S.; Cataldi, A.; et al. Cell-protection mechanism through autophagy in HGFs/S. mitis co-culture treated with Chitlac-nAg. J. Mater. Sci. Mater. Med. 2016, 27, 186. [Google Scholar] [CrossRef] [PubMed]
- Poirier, M.; Simard, J.C.; Girard, D. Silver nanoparticles of 70 nm and 20 nm affect differently the biology of human neutrophils. J. Immunotoxicol. 2016, 13, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.C.; Pazin, M.; Franco-Bernardes, M.F.; da Martins, A.C.; Barcelos, G.R.M.; Pereira, M.C.; Mesquita, J.P.; Rodrigues, J.L.; Barbosa, F.; Dorta, D.J. A perspective of mitochondrial dysfunction in rats treated with silver and titanium nanoparticles (AgNPs and TiNPs). J. Trace Elem. Med. Biol. 2018, 47, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 2006, 8 (Suppl. 2). [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S. Why cancer and inflammation? Yale J. Biol. Med. 2006, 79, 123–130. [Google Scholar] [PubMed]
- Franková, J.; Pivodová, V.; Vágnerová, H.; Juránová, J.; Ulrichová, J. Effects of silver nanoparticles on primary cell cultures of fibroblasts and keratinocytes in a wound-healing model. J. Appl. Biomater. Funct. Mater. 2016, 14, e137–e142. [Google Scholar] [CrossRef] [PubMed]
- Manshian, B.B.; Jimenez, J.; Himmelreich, U.; Soenen, S.J. Presence of an immune system increases anti-tumor effect of Ag nanoparticle treated mice. Adv. Healthc. Mater. 2017, 6, 1601099. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, H.; Saura, R.; Harada, T.; Doita, M.; Mizuno, K. The role of cyclooxygenase-2 and inflammatory cytokines in pain induction of herniated lumbar intervertebral disc. Kobe J. Med. Sci. 2000, 46, 13–28. [Google Scholar] [PubMed]
- Gazon, H.; Barbeau, B.; Mesnard, J.M.; Peloponese, J.M. Hijacking of the AP-1 signaling pathway during development of ATL. Front. Microbiol. 2018, 8, 2686. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Bae, E.; Yi, J.; Kim, Y.; Choi, K.; Lee, S.H.; Yoon, J.; Lee, B.C.; Park, K. Repeated-dose toxicity and inflammatory responses in mice by oral administration of silver nanoparticles. Environ. Toxicol. Pharmacol. 2010, 30, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wong, K.K.Y.; Ho, C.M.; Lok, C.N.; Yu, W.Y.; Che, C.M.; Chiu, J.F.; Tam, P.K.H. Topical delivery of silver nanoparticles promotes wound healing. ChemMedChem 2007, 2, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.Y.; Cheung, S.O.F.; Huang, L.; Niu, J.; Tao, C.; Ho, C.M.; Che, C.M.; Tam, P.K.H. Further evidence of the anti-inflammatory effects of silver nanoparticles. ChemMedChem 2009, 4, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Pothireddy, S.; Kaliki, A.; Mekapogu, A.R.; Yegireddy, M.; Pagadala, E.P.; Prasad, T.N.V.K.V. Evaluation of the Wound Healing Efficacy of Chemical and Phytogenic Silver Nanoparticles. IET Nanobiotechnol. 2016, 10, 340–348. [Google Scholar] [PubMed]
- Pourali, P.; Razavian Zadeh, N.; Yahyaei, B. Silver nanoparticles production by two soil isolated bacteria, Bacillus thuringiensis and Enterobacter cloacae, and assessment of their cytotoxicity and wound healing effect in rats. Wound Repair Regen. 2016, 24, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Pourali, P.; Yahyaei, B. Biological production of silver nanoparticles by soil isolated bacteria and preliminary study of their cytotoxicity and cutaneous wound healing efficiency in rat. J. Trace Elem. Med. Biol. 2016, 34, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, P.; Zmigrodzka, M.; Tomaszewska, E.; Ranoszek-Soliwoda, K.; Czupryn, M.; Antos-Bielska, M.; Szemraj, J.; Celichowski, G.; Grobelny, J.; Krzyzowska, M. Tannic acid-modified silver nanoparticles for wound healing: The importance of size. Int. J. Nanomed. 2018, 13, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Li, Q.; Wang, X.; Wu, P.; Ho, J.K.; Jin, R.; Zhang, L.; Shao, H.; Han, C. Silver nanoparticle loaded collagen/chitosan scaffolds promote wound healing via regulating fibroblast migration and macrophage activation. Sci. Rep. 2017, 7, 10489. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Ahn, S.; Kang, J.P.; Veronika, S.; Huo, Y.; Singh, H.; Chokkaligam, M.; El-Agamy Farh, M.; Aceituno, V.C.; Kim, Y.J.; et al. In vitro anti-inflammatory activity of spherical silver nanoparticles and monodisperse hexagonal gold nanoparticles by fruit extract of Prunus serrulata: A green synthetic approach. Artif. Cells Nanomed. Biotechnol. 2017, 30, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ozer, A.; Bruick, R.K. Non-heme dioxygenases: Cellular sensors and regulators jelly rolled into one? Nat. Chem. Biol. 2007, 3, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Vorrink, S.U.; Severson, P.L.; Kulak, M.V.; Futscher, B.W.; Domann, F.E. Hypoxia perturbs aryl hydrocarbon receptor signaling and CYP1A1 expression induced by PCB 126 in human skin and liver-derived cell lines. Toxicol. Appl. Pharmacol. 2014, 274, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.K.; Kim, J.H.; Kim, H.J.; Ji, Y.H.; Kim, J.H.; Son, S.W. Silver nanoparticle-induced hmsc proliferation is associated with HIF-1-mediated upregulation of il-8 expression. J. Investig. Dermatol. 2014, 134, 3003–3007. [Google Scholar] [CrossRef] [PubMed]
- Eom, H.J.; Ahn, J.M.; Kim, Y.; Choi, J. Hypoxia inducible factor-1 (HIF-1)-flavin containing monooxygenase-2 (FMO-2) signaling acts in silver nanoparticles and silver ion toxicity in the nematode, Caenorhabditis elegans. Toxicol. Appl. Pharmacol. 2013, 270, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Roh, J.Y.; Eom, H.J.; Choi, J.Y.; Hyun, J.; Choi, J. Oxidative stress-related PMK-1 P38 MAPK activation as a mechanism for toxicity of silver nanoparticles to reproduction in the nematode Caenorhabditis elegans. Environ. Toxicol. Chem. 2012, 31, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Park, J.W.; Cha, H.R.; Jung, S.Y.; Lee, J.E.; Jung, S.S.; Kim, J.O.; Kim, S.Y.; Lee, C.S.; Park, H.S. Silver nanoparticles modify VEGF signaling pathway and mucus hypersecretion in allergic airway inflammation. Int. J. Nanomed. 2012, 7, 1329–1343. [Google Scholar]
- Giorgi, C.; De Stefani, D.; Bononi, A.; Rizzuto, R.; Pinton, P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int. J. Biochem. Cell Biol. 2009, 41, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Sukhorukov, V.M.; Dikov, D.; Reichert, A.S.; Meyer-Hermann, M. Emergence of the Mitochondrial Reticulum from Fission and Fusion Dynamics. PLoS Comput. Biol. 2012, 8, e1002745. [Google Scholar] [CrossRef] [PubMed]
- Murley, A.; Nunnari, J. The emerging network of mitochondria-organelle contacts. Mol. Cell 2016, 61, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Hadrup, N.; Loeschner, K.; Mortensen, A.; Sharma, A.K.; Qvortrup, K.; Larsen, E.H.; Lam, H.R. The similar neurotoxic effects of nanoparticulate and ionic silver in vivo and in vitro. Neurotoxicology 2012, 33, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Ziemińska, E.; Stafiej, A.; Struzyńska, L. The role of the glutamatergic NMDA receptor in nanosilver-evoked neurotoxicity in primary cultures of cerebellar granule cells. Toxicology 2014, 315, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Lazarewicz, J.W.; Rybkowski, W.; Sadowski, M.; Ziembowicz, A.; Alaraj, M.; Wegiel, J.; Wisniewski, H.M. N-methyl-D-aspartate receptor-mediated, calcium-induced calcium release in rat dentate gyrus/CA4 in vivo. J. Neurosci. Res. 1998, 51, 76–84. [Google Scholar] [CrossRef]
- Zhang, R.; Piao, M.J.; Kim, K.C.; Kim, A.D.; Choi, J.Y.; Choi, J.; Hyun, J.W. Endoplasmic reticulum stress signaling is involved in silver nanoparticles-induced apoptosis. Int. J. Biochem. Cell Biol. 2012, 44, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cui, J.; Liu, Z.; Zhou, X.; Li, Z.; Yu, Y.; Jia, Y.; Zuo, D.; Wu, Y. Silver nanoparticles induce SH-SY5Y cell apoptosis via endoplasmic reticulum- and mitochondrial pathways that lengthen endoplasmic reticulum-mitochondria contact sites and alter inositol-3-phosphate receptor function. Toxicol. Lett. 2018, 285, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.C.; Durocher, I.; Girard, D. Silver nanoparticles induce irremediable endoplasmic reticulum stress leading to unfolded protein response dependent apoptosis in breast cancer cells. Apoptosis 2016, 21, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Huo, L.; Chen, R.; Zhao, L.; Shi, X.; Bai, R.; Long, D.; Chen, F.; Zhao, Y.; Chang, Y.Z.; Chen, C. Silver nanoparticles activate endoplasmic reticulum stress signaling pathway in cell and mouse models: The role in toxicity evaluation. Biomaterials 2015, 61, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Ziemińska, E.; Strużyńska, L. Zinc modulates nanosilver-induced toxicity in primary neuronal cultures. Neurotox. Res. 2016, 29, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Satapathy, S.R.; Mohapatra, P.; Das, D.; Siddharth, S.; Kundu, C.N. The apoptotic effect of plant based nanosilver in colon cancer cells is a p53 dependent process involving ROS and JNK cascade. Pathol. Oncol. Res. 2015, 21, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Hsin, Y.H.; Chen, C.F.; Huang, S.; Shih, T.S.; Lai, P.S.; Chueh, P.J. The apoptotic effect of nanosilver is mediated by a ROS- and JNK-dependent mechanism involving the mitochondrial pathway in NIH3T3 cells. Toxicol. Lett. 2008, 179, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Christen, V.; Fent, K. Silica nanoparticles and silver-doped silica nanoparticles induce endoplasmatic reticulum stress response and alter cytochrome P4501A activity. Chemosphere 2012, 87, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.C.; Vallieres, F.; De Liz, R.; Lavastre, V.; Girard, D. Silver nanoparticles induce degradation of the endoplasmic reticulum stress sensor activating transcription factor-6 Leading to activation of the NLRP-3 Inflammasome. J. Biol. Chem. 2015, 290, 5926–5939. [Google Scholar] [CrossRef] [PubMed]
- Christen, V.; Capelle, M.; Fent, K. Silver nanoparticles induce endoplasmatic reticulum stress response in zebrafish. Toxicol. Appl. Pharmacol. 2013, 272, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [PubMed]
- Rozpędek, W.; Pytel, D.; Mucha, B.; Leszczyńska, H.; Diehl, J.A.; Majsterek, I. The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Layhadi, J.A.; Fountain, S.J. Influence of ER leak on resting cytoplasmic Ca2+ and receptor- mediated Ca2+ signalling in human macrophage. Biochem. Biophys. Res. Commun. 2017, 487, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I. Autophagy basics. Microbiol. Immunol. 2011, 55, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Miyayama, T.; Fujiki, K.; Matsuoka, M. Silver nanoparticles induce lysosomal-autophagic defects and decreased expression of transcription factor EB in A549 human lung adenocarcinoma cells. Toxicol. Vitr. 2018, 46, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Xavier, P.; Francisco, R.; Platini, F.; Pérez, R.; Ambrosio, S. LC3-I conversion to LC3-II does not necessarily result in complete autophagy. Int. J. Mol. Med. 2008, 22, 781–785. [Google Scholar] [PubMed]
- Xu, Y.; Wang, L.; Bai, R.; Zhang, T.; Chen, C. Silver nanoparticles impede phorbol myristate acetate-induced monocyte-macrophage differentiation and autophagy. Nanoscale 2015, 7, 16100–16109. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lü, X.; Ma, J. Toxicity of silver nanoparticles to human dermal fibroblasts on MicroRNA level. J. Biomed. Nanotechnol. 2014, 10, 3304–3317. [Google Scholar] [CrossRef] [PubMed]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar] [PubMed]
- Kanzawa, T.; Kondo, Y.; Ito, H.; Kondo, S.; Germano, I. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003, 63, 2103–2108. [Google Scholar] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, S.; Honda, S.; Yamaguchi, H.; Shimizu, S. Molecular mechanisms and physiological roles of Atg5/Atg7-independent alternative autophagy. Proc. Jpn. Acad. 2017, 93, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Mukherjee, P. Biological properties of “naked” metal nanoparticles. Adv. Drug Deliv. Rev. 2008, 60, 1289–1306. [Google Scholar] [CrossRef] [PubMed]
- Boisselier, E.; Astruc, D. Gold nanoparticles in nanomedicine: Preparations, imaging, diagnostics, therapies and toxicity. Chem. Soc. Rev. 2009, 38, 1759–1782. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Lee, K.J.; Kalishwaralal, K.; Sheikpranbabu, S.; Vaidyanathan, R.; Eom, S.H. Antiangiogenic properties of silver nanoparticles. Biomaterials 2009, 30, 6341–6350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-F.; Liu, Z.-G.; Shen, W.; Gurunathan, S. Silver nanoparticles: synthesis, characterization, properties, applications, and therapeutic approaches. Int. J. Mol. Sci. 2016, 17, 1534. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Kalishwaralal, K.; Banumathi, E.; Pandian, S.R.K.; Deepak, V.; Muniyandi, J.; Eom, S.H.; Gurunathan, S. Silver nanoparticles inhibit VEGF induced cell proliferation and migration in bovine retinal endothelial cells. Colloids Surf. B Biointerfaces 2009, 73, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Sheikpranbabu, S.; Kalishwaralal, K.; Venkataraman, D.; Eom, S.H.; Park, J.; Gurunathan, S. Silver nanoparticles inhibit VEGF-and IL-1beta-induced vascular permeability via Src dependent pathway in porcine retinal endothelial cells. J. Nanobiotechnol. 2009, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Zan, L.; Zhang, X.; Xi, Y.; Wu, H.; Song, Y.; Teng, G.; Li, H.; Qi, J.; Wang, J. Src regulates angiogenic factors and vascular permeability after focal cerebral ischemia-reperfusion. Neuroscience 2014, 262, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, S. Short- and long-term effects of silver nanoparticles on human microvascular endothelial cells. World J. Biol. Chem. 2014, 5, 457. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Razi, M.; Sadrkhanlou, R. Nanosilver particles increase follicular atresia: Correlation with oxidative stress and aromatization. Environ. Toxicol. 2017, 32, 2244–2255. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.M.; Kumar, A.; Mousa, S.; Dyskin, E.; Yalcin, M.; Ajayan, P.; Linhardt, R.J.; Mousa, S.A. Gold and silver nanoparticles conjugated with heparin derivative possess anti-angiogenesis properties. Nanotechnology 2009, 20, 455104. [Google Scholar] [CrossRef] [PubMed]
- Baharara, J.; Namvar, F.; Mousavi, M.; Ramezani, T.; Mohamad, R. Anti-angiogenesis effect of biogenic silver nanoparticles synthesized using Saliva officinalis on chick chorioalantoic membrane (CAM). Molecules 2014, 19, 13498–13508. [Google Scholar] [CrossRef] [PubMed]
- Baharara, J.; Namvar, F.; Ramezani, T.; Hosseini, N.; Mohamad, R. Green synthesis of silver nanoparticles using Achillea biebersteinii flower extract and its anti-angiogenic properties in the rat aortic ring model. Molecules 2014, 19, 4624–4634. [Google Scholar] [CrossRef] [PubMed]
- Satapathy, S.R.; Siddharth, S.; Das, D.; Nayak, A.; Kundu, C.N. Enhancement of Cytotoxicity and Inhibition of Angiogenesis in Oral Cancer Stem Cells by a Hybrid Nanoparticle of Bioactive Quinacrine and Silver: Implication of Base Excision Repair Cascade. Mol. Pharm. 2015, 12, 4011–4025. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Lim, D.H.; Choi, I.H.; Kang, T.; Lee, K.; Moon, E.Y.; Yang, Y.; Lee, M.S.; Lim, J.S. Vascular tube formation and angiogenesis induced by polyvinylpyrrolidone-coated silver nanoparticles. Toxicol. Lett. 2011, 205, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Stoccoro, A.; Karlsson, H.L.; Coppedè, F.; Migliore, L. Epigenetic effects of nano-sized materials. Toxicology 2013, 313, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.S.E.; Hu, Q.; Baeg, G.H. Epigenetic modulations in nanoparticle-mediated toxicity. Food Chem. Toxicol. 2017, 109, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; Leproust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.F.; Liang, R.P.; Wang, J.W.; Qiu, J.D. One-pot synthesis of GO/AgNPs/luminol composites with electrochemiluminescence activity for sensitive detection of DNA methyltransferase activity. Biosens. Bioelectron. 2015, 63, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.; Lafuente, D.; Gómez, M.; García, T.; Domingo, J.L.; Sánchez, D.J. Polyvinyl pyrrolidone-coated silver nanoparticles in a human lung cancer cells: Time- and dose-dependent influence over p53 and caspase-3 protein expression and epigenetic effects. Arch. Toxicol. 2017, 91, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Mytych, J.; Zebrowski, J.; Lewinska, A.; Wnuk, M. Prolonged effects of silver nanoparticles on p53/p21 pathway-mediated proliferation, DNA damage response, and methylation parameters in HT22 hippocampal neuronal cells. Mol. Neurobiol. 2017, 54, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.; Zhang, R.; Petridis, L.; Cheng, X.; Smith, J.C.; Langowski, J. The role of histone tails in the nucleosome: A computational study. Biophys. J. 2014, 107, 2911–2922. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Toyooka, T.; Ibuki, Y. Silver nanoparticle-induced phosphorylation of histone H3 at serine 10 is due to dynamic changes in actin filaments and the activation of Aurora kinases. Toxicol. Lett. 2017, 276, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ibuki, Y. Evaluating the toxicity of silver nanoparticles by detecting phosphorylation of histone H3 in combination with flow cytometry side-scattered light. Environ. Sci. Technol. 2015, 49, 5003–5012. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. Double-stranded brekas induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Eom, H.J.; Chatterjee, N.; Lee, J.; Choi, J. Integrated mRNA and micro RNA profiling reveals epigenetic mechanism of differential sensitivity of Jurkat T cells to AgNPs and Ag ions. Toxicol. Lett. 2014, 229, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Moss, E.G. RNA repair: Damage control. Curr. Biol. 2003, 13, 482–484. [Google Scholar] [CrossRef]
- Corvi, R.; Madia, F. In vitro genotoxicity testing–can the performance be enhanced? Food Chem. Toxicol. 2017, 106, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Magdolenova, Z.; Collins, A.; Kumar, A.; Dhawan, A.; Stone, V.; Dusinska, M. Mechanisms of genotoxicity. A review of in vitro and in vivo studies with engineered nanoparticles. Nanotoxicology 2013, 8, 233–278. [Google Scholar] [CrossRef] [PubMed]
- Albertini, R.J.; Anderson, D.; Douglas, G.R.; Hagmar, L.; Hemminki, K.; Merlo, F.; Natarajan, A.T.; Norppa, H.; Shuker, D.E.G.; Tice, R.; et al. IPCS guidelines for the monitoring of genotoxic effects of carcinogens in humans. Mutat. Res. Rev. Mutat. Res. 2000, 463, 111–172. [Google Scholar] [CrossRef]
- Johnson, G.E. Mammalian cell HPRT gene mutation assay: test methods. Methods Mol. Biol. 2012, 817, 55–67. [Google Scholar] [PubMed]
- Piao, M.J.; Kim, K.C.; Choi, J.Y.; Choi, J.; Hyun, J.W. Silver nanoparticles downregulate Nrf2-mediated 8-oxoguanine DNA glycosylase 1 through inactivation of extracellular regulated kinase and protein kinase B in human Chang liver cells. Toxicol. Lett. 2011, 207, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Park, Y.J.; Shin, D.Y.; Oh, S.M.; Chung, K.H. Appropriate in vitro methods for genotoxicity testing of silver nanoparticles. Environ. Health Toxicol. 2013, 28, e2013003. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Su, F.; Raj, P.; Dozmorov, I.; Mishra, R.; Wakeland, E.K.; Ghose, S.; Mukherjee, S.; et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017, 45, 4590–4605. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Senapati, D.; Wang, S.; Tovmachenko, O.; Singh, A.K.; Yu, H.; Ray, P.C. Effect of surface coating on the toxicity of silver nanomaterials on human skin keratinocytes. Chem. Phys. Lett. 2010, 487, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Park, M.V.D.Z.; Neigh, A.M.; Vermeulen, J.P.; de la Fonteyne, L.J.J.; Verharen, H.W.; Briedé, J.J.; van Loveren, H.; de Jong, W.H. The effect of particle size on the cytotoxicity, inflammation, developmental toxicity and genotoxicity of silver nanoparticles. Biomaterials 2011, 32, 9810–9817. [Google Scholar] [CrossRef] [PubMed]
- Landsiedel, R.; Kapp, M.D.; Schulz, M.; Wiench, K.; Oesch, F. Genotoxicity investigations on nanomaterials: Methods, preparation and characterization of test material, potential artifacts and limitations-Many questions, some answers. Mutat. Res. Rev. Mutat. Res. 2009, 681, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Patlolla, A.K.; Hackett, D.; Tchounwou, P.B. Genotoxicity study of silver nanoparticles in bone marrow cells of Sprague Dawley rats. Food Chem. Toxicol. 2015, 85, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Nallanthighala, S.; Chana, C.; Murray, T.M.; Mosierd, A.P.; Cadyd, N.C.; Reliene, R. Differential effects of silver nanoparticles on DNA damage and DNA repair gene expression in Ogg1-deficient and wild type mice. Nanotoxicology 2017, 11, 996–1011. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Sung, J.H.; Ji, J.H.; Song, K.S.; Lee, J.H.; Kang, C.S.; Yu, I.J. In vivo Genotoxicity of silver nanoparticles after 90-day silver nanoparticle inhalation exposure. Saf. Health Work 2011, 2, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Dan, M.; Yang, Y.; Lyu, J.; Shao, A.; Cheng, X.; Chen, L.; Xu, L. Acute toxicity and genotoxicity of silver nanoparticle in rats. PLoS ONE 2017, 12, e0185554. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. (Lond.) 2010, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.-G.; Gurunathan, S. Combination of graphene oxide-silver nanoparticle nanocomposites and cisplatin enhances apoptosis and autophagy in human cervical cancer cells. Int. J. Nanomed. 2017, 12, 6537–6558. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, J.; Huang, Y.; Gao, X.; Kong, L.; Zhang, T.; Tang, M. Comparative cytotoxicity and apoptotic pathways induced by nanosilver in human liver HepG2 and L02 cells. Hum. Exp. Toxicol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Han, J.W.; Kim, E.; Choi, Y.-J.; Kwon, D.-N.; Kim, J.-H. Reduced graphene oxide—Silver nanoparticle nanocomposite: A potential anticancer nanotherapy. Int. J. Nanomed. 2015, 10, 6257–6276. [Google Scholar] [CrossRef] [PubMed]
- Foldbjerg, R.; Dang, D.A.; Autrup, H. Cytotoxicity and genotoxicity of silver nanoparticles in the human lung cancer cell line, A549. Arch. Toxicol. 2011, 85, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Sathishkumar, G.; Gobinath, C.; Wilson, A.; Sivaramakrishnan, S. Dendrophthoe falcata (Lf) Ettingsh (Neem mistletoe): A potent bioresource to fabricate silver nanoparticles for anticancer effect against human breast cancer cells (MCF-7). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 128, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Loutfy, S.A.; Al-Ansary, N.A.; Abdel-Ghani, N.T.; Hamed, A.R.; Mohamed, M.B.; Craik, J.D.; Salah Eldin, T.A.; Abdellah, A.M.; Hussein, Y.; Hasanin, M.T.M.; et al. Anti-proliferative activities of metallic nanoparticles in an in vitro breast cancer model. Asian Pac. J. Cancer Prev. 2015, 16, 6039–6046. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.G.; Fernández-Baldo, M.A.; Fernández, J.G.; Serrano, M.J.; Sanz, M.I.; Diaz-Mochón, J.J.; Lorente, J.A.; Raba, J. Study of antitumor activity in breast cell lines using silver nanoparticles produced by yeast. Int. J. Nanomed. 2015, 10, 2021–2031. [Google Scholar]
- Venugopal, K.; Rather, H.A.; Rajagopal, K.; Shanthi, M.P.; Sheriff, K.; Illiyas, M.; Rather, R.A.; Manikandan, E.; Uvarajan, S.; Bhaskar, M.; et al. Synthesis of silver nanoparticles (Ag NPs) for anticancer activities (MCF 7 breast and A549 lung cell lines) of the crude extract of Syzygium aromaticum. J. Photochem. Photobiol. B Biol. 2017, 167, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Tyner, A.L. Transcriptional regulation of the p21(WAF1/CIP1) gene. Exp. Cell Res. 1999, 246, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Sriram, M.I.; Kanth, S.B.M.; Kalishwaralal, K.; Gurunathan, S. Antitumor activity of silver nanoparticles in Dalton’s lymphoma ascites tumor model. Int. J. Nanomed. 2010, 5, 753–762. [Google Scholar]
- Jacob, J.A.; Shanmugam, A. Silver nanoparticles provoke apoptosis of Dalton’s ascites lymphoma in vivo by mitochondria dependent and independent pathways. Colloids Surf. B Biointerfaces 2015, 136, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, Y.; Guo, Q.; Wang, Z.; Wang, H.; Yang, Y.; Huang, Y. TAT-modified nanosilver for combating multidrug-resistant cancer. Biomaterials 2012, 33, 6155–6161. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Yang, H.; Wei, J.; Tong, J.L.; Shu, Y.Q. The role and mechanisms of nanoparticles to enhance radiosensitivity in hepatocellular cell. Biomed Pharmacother. 2013, 67, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Márquez, I.G.; Ghiyasvand, M.; Massarsky, A.; Babu, M.; Samanfar, B.; Omidi, K.; Moon, T.W.; Smith, M.L.; Golshani, A. Zinc oxide and silver nanoparticles toxicity in the baker’s yeast, Saccharomyces cerevisiae. PLoS ONE 2018, 13, e0193111. [Google Scholar]
- Wang, L.; He, X.; Szklarz, G.D.; Bi, Y.; Rojanasakul, Y.; Ma, Q. The aryl hydrocarbon receptor interacts with nuclear factor erythroid 2-related factor 2 to mediate induction of NAD(P)H: Quinoneoxidoreductase 1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch. Biochem. Biophys. 2013, 537, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonenn, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Ramshini, H.; Moghaddasi, A.S.; Aldaghi, L.S.; Mollania, N.; Ebrahim-Habibi, A. Silver nano particles ameliorate learning and spatial memory of male Wistar rats by prevention of amyloid fibril-induced neurotoxicity. Arch. Ital. Biol. 2017, 155, 131–141. [Google Scholar] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cameron, S.J.; Hosseinian, F.; Willmore, W.G. A Current Overview of the Biological and Cellular Effects of Nanosilver. Int. J. Mol. Sci. 2018, 19, 2030. https://doi.org/10.3390/ijms19072030
Cameron SJ, Hosseinian F, Willmore WG. A Current Overview of the Biological and Cellular Effects of Nanosilver. International Journal of Molecular Sciences. 2018; 19(7):2030. https://doi.org/10.3390/ijms19072030
Chicago/Turabian StyleCameron, Shana J., Farah Hosseinian, and William G. Willmore. 2018. "A Current Overview of the Biological and Cellular Effects of Nanosilver" International Journal of Molecular Sciences 19, no. 7: 2030. https://doi.org/10.3390/ijms19072030
APA StyleCameron, S. J., Hosseinian, F., & Willmore, W. G. (2018). A Current Overview of the Biological and Cellular Effects of Nanosilver. International Journal of Molecular Sciences, 19(7), 2030. https://doi.org/10.3390/ijms19072030