Poly Organotin Acetates against DNA with Possible Implementation on Human Breast Cancer


 , ,
, , 

Abstract
:
1. Introduction
2. Results and Discussion
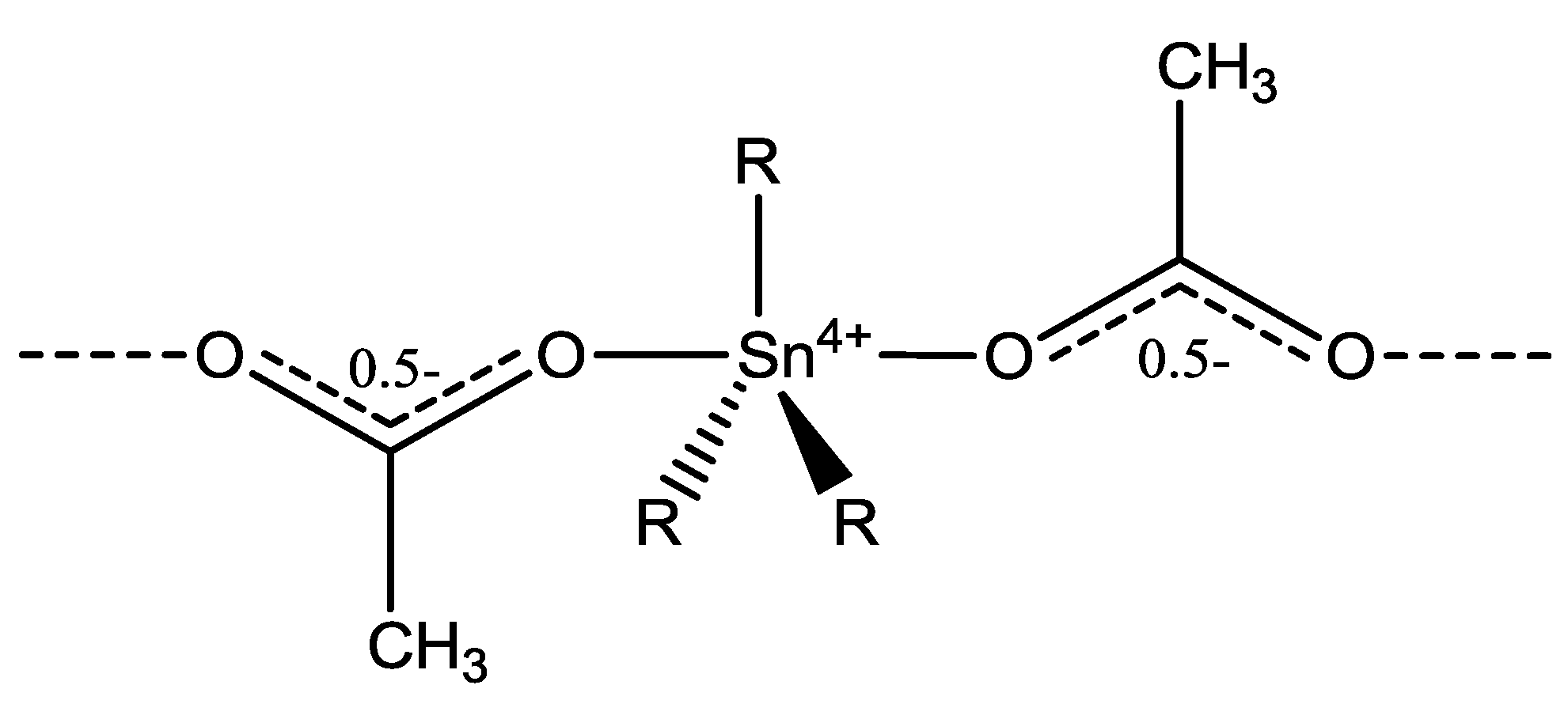
2.1. General Aspects
2.2. Solid State
2.2.1. Vibrational Spectroscopy
2.2.2. 119Sn Mössbauer Spectroscopy
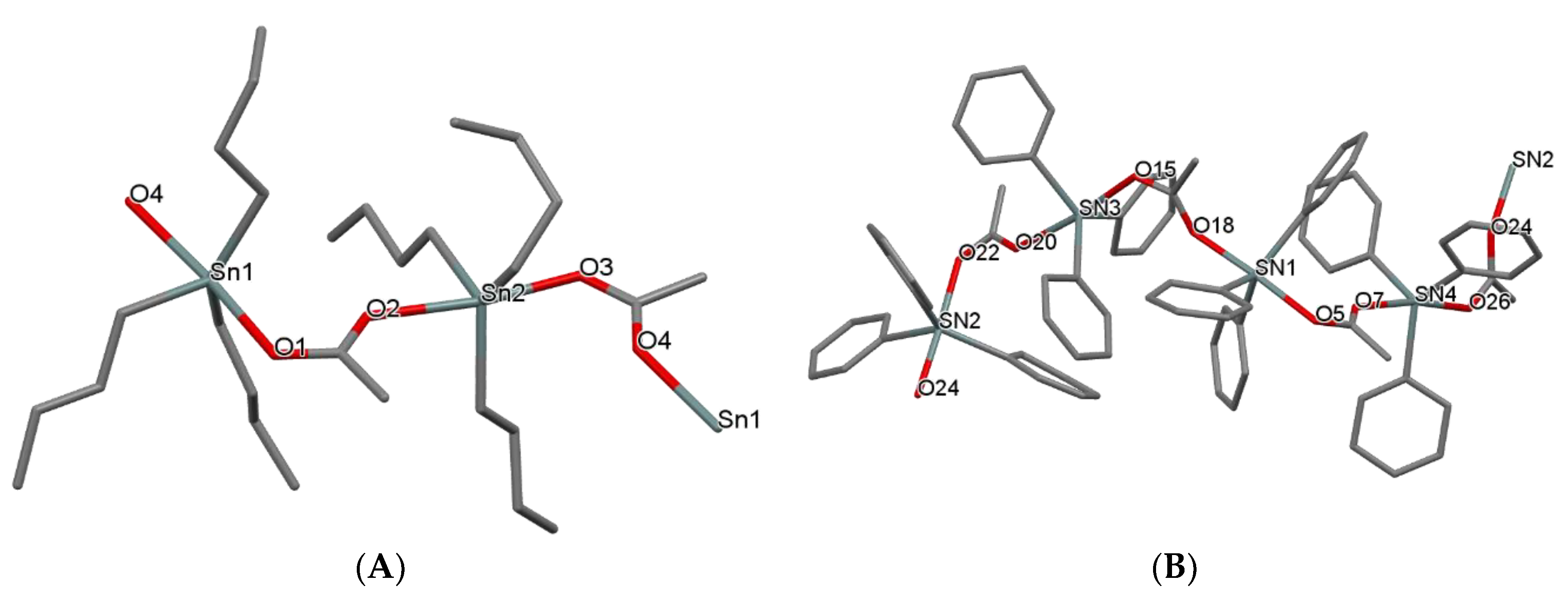
2.2.3. Crystal and Molecular Structures of [Bu3SnCH3COO]n (1) and [Ph3SnCH3COO]n (2)
2.3. Solution Studies
1H-NMR Spectrοscopy
2.4. Biological Tests
2.4.1. Anti-Proliferative Activity
2.4.2. Evaluation of Genotoxicity by Micronucleus Assay In Vitro
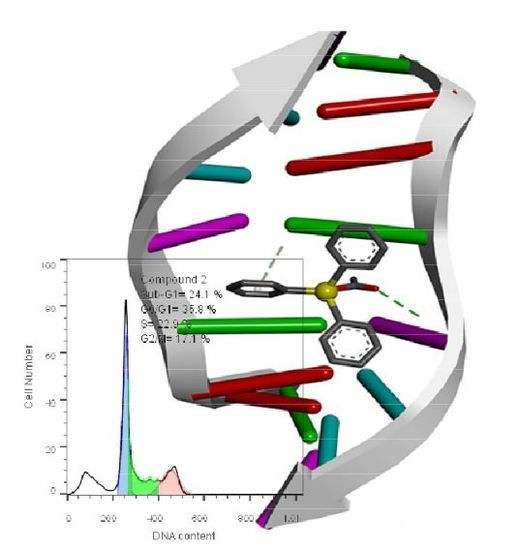
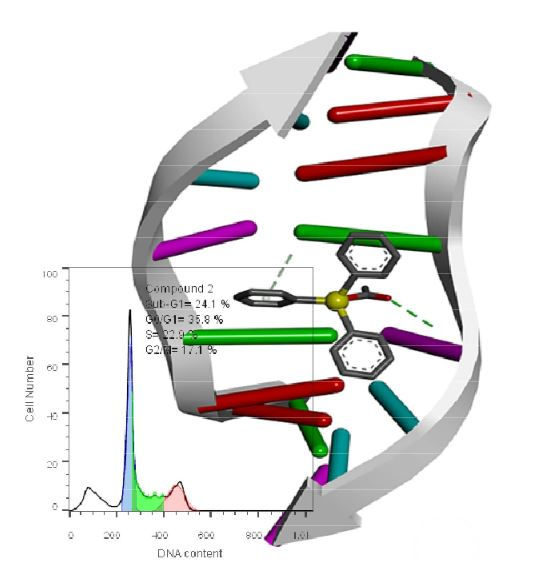
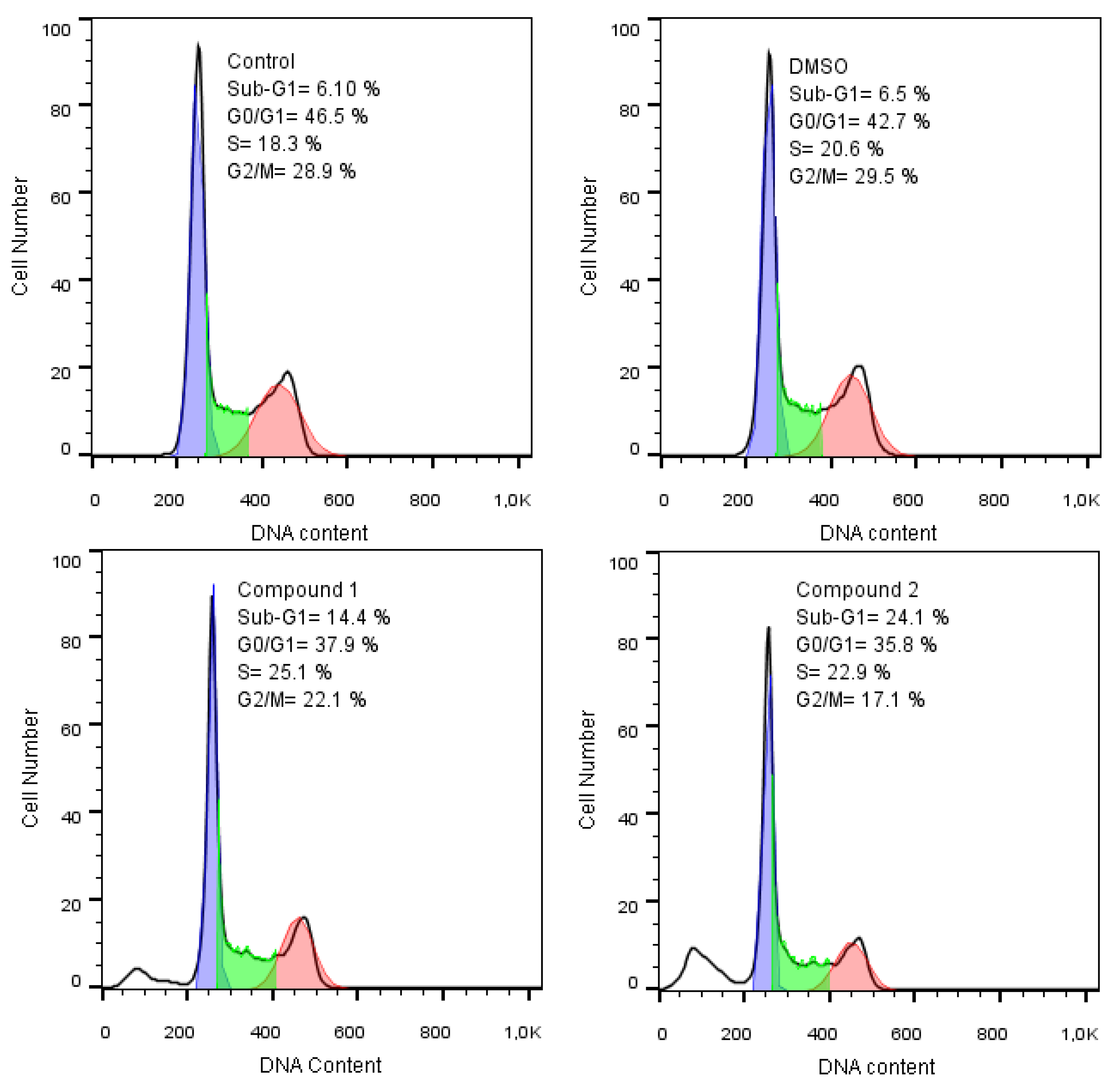
2.4.3. Cell Cycle Studies
2.4.4. Detection of the Loss of the Mitochondrial Membrane Permeabilization (MMP Assay)
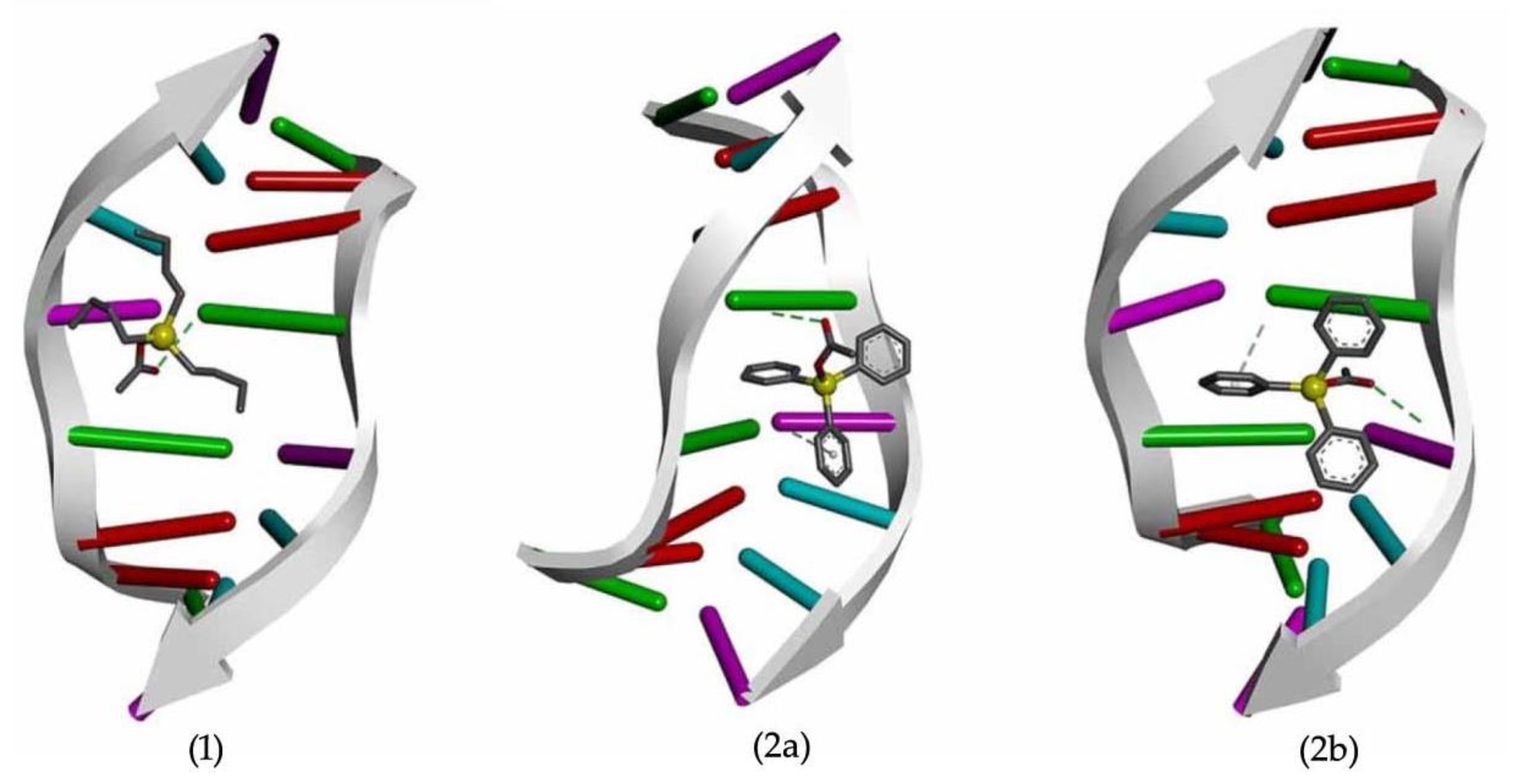
2.4.5. DNA Binding Studies
3. Experimental
3.1. Materials and Instruments
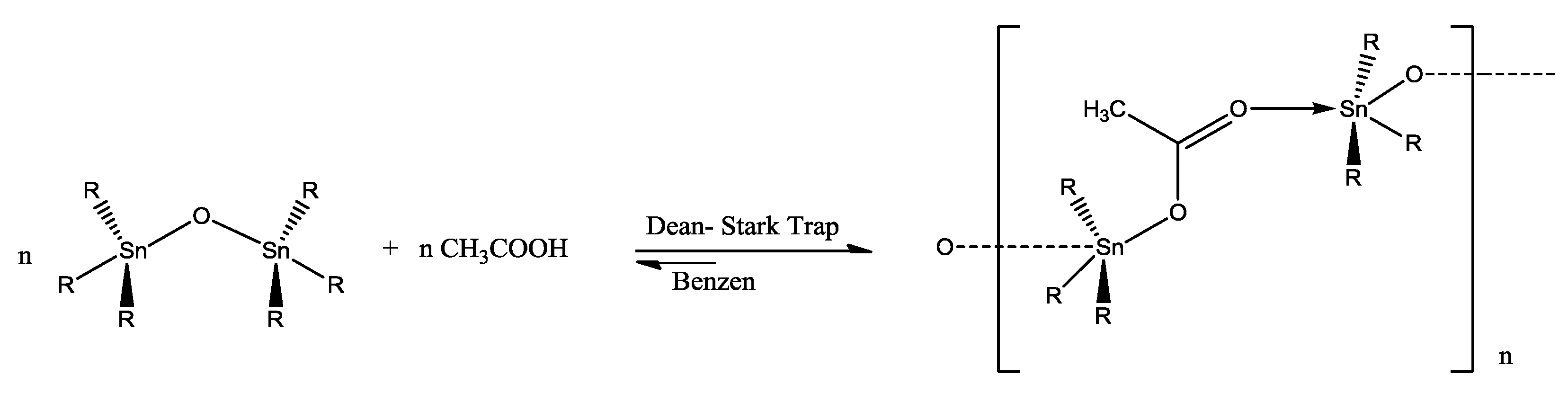
3.2. Synthesis and Crystallization of {[(n-Bu)3Sn(CH3COO)]n} (1) and {[Ph3Sn(CH3COO)]n} (2)
3.3. X-Ray Structure Determination
3.4. Biological Tests
3.5. Viscosity Measurements
3.6. Computational Studies—Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rosenberg, B. Platinum coordination complexes in cancer chemotherapy. Die Naturwissenschaften 1973, 60, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Lippert, B. Impact of Cisplatin on the recent development of Pt coordination chemistry: A case study. Coord. Chem. Rev. 1999, 182, 263–295. [Google Scholar] [CrossRef]
- Arnesano, F.; Natile, G. Mechanistic insight into the cellular uptake and processing of cisplatin 30 years after its approval by FDA. Coord. Chem. Rev. 2009, 253, 2070–2081. [Google Scholar] [CrossRef]
- Arnesano, F.; Pannunzio, A.; Natile, G. Effect of chirality in platinum drugs. Coord. Chem. Rev. 2015, 284, 286–297. [Google Scholar] [CrossRef]
- Astolfi, L.; Ghiselli, S.; Guaran, V.; Chicca, M.; Simoni, E.; Olivetto, E.; Lelli, G.; Martini, A. Correlation of adverse effects of cisplatin administration in patients affected by solid tumours: A retrospective evaluation. Oncol. Rep. 2013, 29, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-B.; Wang, F.-Y.; Tang, X.-M.; Feng, H.-W.; Chen, Z.-F.; Liu, Y.-C.; Liu, Y.-N.; Liang, H. Organometallic Gold(III) Complexes Similar to Tetrahydroisoquinoline Induce ER-Stress-Mediated Apoptosis and Pro-Death Autophagy in A549 Cancer Cells. J. Med. Chem. 2018, 61, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Haiduc, I.; Silvestru, C.; Gielen, M. Organotin compounds: New organometallic derivatives exhibiting anti-tuequr activity. Bull. Soc. Chim. Belg. 1983, 92, 187–189. [Google Scholar] [CrossRef]
- Gielen, M.; Melotte, M.; Atassi, G.; Wrllem, R. Synthesis, characterization and antitumour activity of 7,7-di-n-butyi-5,9-dioxo-6,6-dioxa-7-stanna-spiro[3,5]nonane, a di-n-butyltin(lV) analog of “paraplatin”, and of a series of di-n-butyitin(lV) derivatives of mono- and disubstituted malonic acids. Tetrahedron 1989, 45, 1219–1229. [Google Scholar] [CrossRef]
- Saxena, A.K.; Huber, F. Organotin compounds and cancer chemotherapy. Coord. Chem. Rev. 1989, 95, 109–112. [Google Scholar] [CrossRef]
- Gielen, M. Tin-based antitumour drugs. Coord. Chem. Rev. 1996, 151, 41–51. [Google Scholar] [CrossRef]
- Gielen, M. Organotin compounds and their therapeutic potential: A report from the Organometallic Chemistry Department of the Free University of Brussels. Appl. Organomet. Chem. 2002, 16, 481–494. [Google Scholar] [CrossRef]
- Hadjikakou, S.K.; Hadjiliadis, N. Antiproliferative and anti-tumor activity of organotin compounds. Coord. Chem. Rev. 2009, 253, 235–249. [Google Scholar] [CrossRef]
- Shpakovsky, D.B.; Banti, C.N.; Beaulieu-Houle, G.; Kourkoumelis, N.; Hadjikakou, S.K.; Manoli, M.; Manos, M.J.; Tasiopoulos, A.J.; Milaeva, E.R.; Charalabopoulos, K.; et al. Synthesis, structural characterization and in vitro inhibitory studies against humanagainst human breast cancer of the bis-(2,6-di-tert-butylphenol)tin(IV) dichloride andits complexes. Dalton Trans. 2012, 41, 14568–14582. [Google Scholar] [CrossRef] [PubMed]
- Shpakovsky, D.B.; Banti, C.N.; Mukhatova, E.M.; Gracheva, Y.A.; Osipova, V.P.; Berberova, N.T.; Albov, D.V.; Antonenko, T.A.; Aslanov, L.А.; Milaeva, E.R.; et al. Synthesis, antiradical activity and in vitro cytotoxicity of novel organotin complexes based on 2,6-di-tert-butyl-4-mercaptophenol. Dalton Trans. 2014, 43, 6880–6890. [Google Scholar] [CrossRef] [PubMed]
- Balas, V.Ι.; Banti, C.N.; Kourkoumelis, N.; Hadjikakou, S.K.; Geromichalos, G.D.; Sahpazidou, D.; Male, L.; Hursthouse, M.B.; Bednarz, B.; Kubicki, M.; et al. Structural and in vitro biological studies of organotin(IV) precursors; selective inhibitory activity against human breast cancer cells, positive to estrogen receptors. Austr. J. Chem. 2012, 65, 1625–1637. [Google Scholar] [CrossRef]
- Balas, V.I.; Verginadis, I.I.; Geromichalos, G.D.; Kourkoumelis, N.; Male, L.; Hursthouse, M.B.; Repana, K.H.; Yiannaki, E.; Charalabopoulos, K.; Bakas, T.; et al. Synthesis, structural characterization and biological studies of the triphenyltin(IV)complex with 2-thiobarbituric acid. Eur. J. Med. Chem. 2011, 46, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Verginadis, I.I.; Karkabounas, S.; Simos, Y.; Kontargiris, E.; Hadjikakou, S.K.; Batistatou, A.; Evangelou, A.; Charalabopoulos, K. Anticancer and cytotoxic effects of a triorganotin compound with 2-mercapto-nicotinic acid in malignant cell lines and tumor bearing Wistar rats. Eur. J. Pharm. Sci. 2011, 42, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Agiorgiti, M.S.; Evangelou, A.; Vezyraki, P.; Hadjikakou, S.K.; Kalfakakou, V.; Tsanaktsidis, I.; Batistatou, A.; Zelovitis, J.; Simos, Y.V.; Ragos, V.; et al. Cytotoxic effect, antitumour activity and toxicity of organotin derivatives with ortho- or para-hydroxy-benzoic acids. Med. Chem. Res. 2018, 27, 1122–1130. [Google Scholar] [CrossRef]
- Abdellah, M.A.; Hadjikakou, S.K.; Hadjiliadis, N.; Kubicki, M.; Bakas, T.; Kourkoumelis, N.; Simos, Y.V.; Karkabounas, S.; Barsan, M.M.; Butler, I.S. Synthesis, characterization, and biological studies of organotin(IV) derivatives with o- or p-hydroxybenzoic acids. Bioinorg. Chem. Appl. 2009, 12. [Google Scholar] [CrossRef] [PubMed]
- Xanthopoulou, M.N.; Hadjikakou, S.K.; Hadjiliadis, N.; Milaeva, E.R.; Gracheva, J.A.; Tyurin, V.-Y.; Kourkoumelis, N.; Christoforidis, K.C.; Metsios, A.K.; Karkabounas, S.; et al. Biological studies of new organotin(IV) complexes ofthioamide ligands. Eur. J. Med. Chem. 2008, 43, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Balas, V.I.; Hadjikakou, S.K.; Hadjiliadis, N.; Kourkoumelis, N.; Light, M.E.; Hursthouse, M.; Metsios, A.K.; Karkabounas, S. Crystal structure and antitumor activity of the novel zwitterionic complex of tri-n-butyltin(IV) with 2-thiobarbituric acid. Bioinorg. Chem. Appl. 2008, 654137. [Google Scholar] [CrossRef] [PubMed]
- Xanthopoulou, M.N.; Hadjikakou, M.N.; Hadjiliadis, N.; Kourkoumelis, N.; Milaeva, E.R.; Gracheva, J.A.; Tyurin, V.-Y.; Verginadis, I.I.; Karkabounas, S.; Baril, M.; et al. Biological studies of organotin(IV) complexes with 2-mercaptopyrimidine. Russ. Chem. B 2007, 56, 767–773. [Google Scholar] [CrossRef]
- Xanthopoulou, M.N.; Hadjikakou, S.K.; Hadjiliadis, N.; Kubicki, M.; Karkabounas, S.; Charalabopoulos, K.; Kourkoumelis, N.; Bakas, T. Synthesis and characterization of a new chloro-di-phenyltin(IV) complex with 2-mercapto-nicotinic acid: Study of its influence upon the catalytic oxidation of linoleic acid to hydroperoxylinoleic acid by the enzyme lipoxygenase. J. Organomet. Chem. 2006, 691, 1780–1789. [Google Scholar] [CrossRef]
- Xanthopoulou, M.N.; Hadjikakou, S.K.; Hadjiliadis, N.; Schurmann, M.; Jurkschat, K.; Michaelides, A.; Skoulika, S.; Bakas, T.; Binolis, J.; Karkabounas, S.; et al. Synthesis, structural characterization and in vitro cytotoxicity of organotin(IV) derivatives of heterocyclic thioamides, 2-mercaptobenzothiazole, 5-chloro-2-mercaptobenzothiazole, 3-methyl-2-mercaptobenzothiazole and 2-mercaptonicotinic acid. J. Inorg. Biochem. 2003, 96, 425–434. [Google Scholar] [CrossRef]
- Banti, C.N.; Gkaniatsou, E.I.; Kourkoumelis, N.; Manos, M.J.; Tasiopoulos, A.J.; Bakas, T.; Hadjikakou, S.K. Assessment of organotins against the linoleic acid, glutathione and CT-DNA. Inorg. Chim. Acta 2014, 423, 98–106. [Google Scholar] [CrossRef]
- Xanthopoulou, M.N.; Kourkoumelis, N.; Hadjikakou, S.K.; Hadjiliadis, N.; Kubicki, M.; Karkabounas, S.; Bakas, T. Structural and biological studies of organotin(IV) derivatives with 2-mercapto-benzoic acid and -mercapto-4-methyl-pyrimidine. Polyhedron 2008, 27, 3318–3324. [Google Scholar] [CrossRef]
- Hadjikakou, S.K.; Jurkschat, K.; Schurmann, M. Novel organotin(IV) compounds derived from bis(organostannyl)methanes: Synthesis and crystal structures of bis[diphenyl(pyridin-2-onato)stannyl]methane and bis[bromophenyl(pyrimidine-2-thionato)stannyl]methane C7H8. J. Organomet. Chem. 2006, 691, 1637–1642. [Google Scholar] [CrossRef]
- Xanthopoulou, M.N.; Hadjikakou, S.K.; Hadjiliadis, N.; Kubicki, M.; Skoulika, S.; Bakas, T.; Baril, M.; Butler, I.S. Synthesis, structural characterization, and biological studies of six and five-coordinate organotin(IV) Complexes with the Thioamides 2-Mercaptobenzothiazole, 5-Chloro-2-mercaptobenzothiazole, and 2-Mercaptobenzoxazole. Inorg. Chem. 2007, 46, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Carraher, C.E., Jr.; Roner, M.R. Organotin polymers as anticancer and antiviral agents. J. Org. Chem. 2014, 751, 67–82. [Google Scholar] [CrossRef]
- Jones, L.H.; McLaren, E. Infrared Spectra of CH3COONa and CD3COONa and Assignments of Vibrational Frequencies. J. Chem. Phys. 1954, 22, 1796–1800. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Gibb, T.C. Mossbauer Spectroscopy; Chapman and Hall: London, UK, 1971. [Google Scholar]
- Saeed, M.A.; Badshah, A.; Rauf, M.K.; Craig, D.C.; Ali, S. catena-Poly[tributyltin(IV)-l-acetato]. Acta Cryst. 2006, E62, m469–m471. [Google Scholar] [CrossRef]
- Molloy, K.C.; Purcell, T.G.; Quill, K. Organotin biocides 1. The structure of triphenyltin acetate. J. Organomet. Chem. 1984, 267, 237–241. [Google Scholar] [CrossRef]
- Lee, A.V.; Oesterreich, S.; Davidson, N.E. MCF-7 Cells—Changing the Course of Breast Cancer Research and Care for 45 Years. JNCI J. Nat. Cancer Inst. 2015, 107, Djv073. [Google Scholar] [CrossRef] [PubMed]
- Rochefort, H.; Glondu, M.; Sahla, M.E.; Platet, N.; Garcia, M. How to target estrogen receptor-negative breast cancer? Endocr. Relat. Cancer 2003, 10, 261–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banti, C.N.; Papatriantafyllopoulou, C.; Manoli, M.; Tasiopoulos, A.J.; Hadjikakou, S.K. Nimesulide silver metallodrugs, containing the mitochondriotropic, triaryl derivatives of pnictogen; Anticancer activity against human breast cancer cells. Inorg. Chem. 2016, 55, 8681–8696. [Google Scholar] [CrossRef] [PubMed]
- HCilião, L.; Ribeiro, D.L.; Camargo-Godoy, R.B.O.; Specian, A.F.L.; Serpeloni, J.M.; Cólus, I.M.S. Cytotoxic and genotoxic effects of high concentrations of the immunosuppressive drugs cyclosporine and tacrolimus in MRC-5 cells. Exp. Toxicol. Pathol. 2015, 67, 179–187. [Google Scholar]
- Gao, E.; Lin, L.; Liu, L.; Zhu, M.; Wang, B.; Gao, X. Two new palladium(II) complexes: Synthesis, characterization and their interaction with HeLa cells. Dalton Trans. 2012, 41, 11187–11194. [Google Scholar] [CrossRef] [PubMed]
- Boer, D.R.; Canals, A.; Coll, M. DNA-binding drugs caught in action: The latest 3D pictures of drug-DNA complexes. Dalton Trans. 2009, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Gilad, Y.; Hanoch, S. Docking studies on DNA intercalators. J. Chem. Inf. Model. 2013, 54, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.; Robinson, H.; Wang, A.-H. Structure of actinomycin D bound with (GAAGCTTC)2 and (GATGCTTC)2 and its binding to the (CAG)n:(CTG)n triplet sequence as determined by NMR analysis. J. Am. Chem. Soc. 1996, 118, 8791–8801. [Google Scholar] [CrossRef]
- Icsel, C.; Yilmaz, V.T.; Kaya, Y.; Samli, H.; Harrison, W.T.A.; Buyukgungor, O. New palladium (II) and platinum (II) 5, 5-diethylbarbiturate complexes with 2-phenylpyridine, 2, 2′-bipyridine and 2, 2′-dipyridylamine: Synthesis, structures, DNA binding, molecular docking, cellular uptake, antioxidant activity and cytotoxicity. Dalton Trans. 2015, 44, 6880–6895. [Google Scholar] [CrossRef] [PubMed]
- Lauria, A.; Patella, C.; Dattolo, G.; Almerico, A.M. Design and Synthesis of 4-Substituted Indolo [3, 2-e][1, 2, 3] triazolo [1, 5-a] pyrimidine Derivatives with Antitumor Activity. J. Med. Chem. 2008, 51, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Pages, B.J.; Ang, D.L.; Wright, E.P.; Aldrich-Wright, J.R. Metal complex interactions with DNA. Dalton Trans. 2015, 44, 3505–3526. [Google Scholar] [CrossRef] [PubMed]
- Oxford Diffraction. CrysAlis CCD and CrysAlis RED; Oxford Diffraction Ltd.: Abingdon, UK, 2008. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; de Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Cryst. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-2014/7, Program for Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Bevington, P.R.; Robinson, D.K. Data Reduction and Error Analysis for the Physical Sciences; McGraw Hill: New York, NY, USA, 1992; pp. 194–197. [Google Scholar]
- OTrott, O.; Olson, A.J. AutoDockVina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. GAUSSIAN 03 (Revision C.02); Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μΜ) | ||||
|---|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | MRC-5 | TPI * | Ref. | |
| 1 | 0.25 ± 0.02 | 0.20 ± 0.01 | 0.22 ± 0.01 | 0.88 | [present] |
| 2 | 0.21 ± 0.01 | 0.12 ± 0.01 | 0.11 ± 0.01 | 0.52 | [present] |
| {[Ph3Sn]2(mna)·[(CH3)2CO]} | 0.030 | >0.200 | [18] | ||
| [Me2Sn(Sal)2] | 0.142 ± 0.043 | 0.0975 ± 0.00015 | 0.69 | [19] | |
| [(n-Bu)2Sn(Sal)2] | 0.108 ± 0.0026 | 0.1041 ± 0.0002 | 0.96 | [19] | |
| [(n-Bu)3Sn(Sal)] | 0.724 ± 0.0054 | 0.0981 ± 0.0001 | 0.14 | [19] | |
| [Ph3Sn(Sal)] | 0.121 ± 0.0037 | 0.0945 ± 0.000.2 | 0.78 | [19] | |
| [(n-Bu)3Sn(pHbza)] | 0.325 ± 0.0023 | 0.0784 ± 0.0002 | 0.24 | [19] | |
| {[Ph3Sn(o-HTBA)]}n | 0.103 | 0.203 | 0.130 | 1.26 | [17] |
| (n-Bu)3Sn(o-HTBA)(H2O) | 0.068 | 0.106 | 0.108 | 1.59 | [17] |
| [(tert-Bu–)2(HO–Ph)]2SnCl2 | 3.12 ± 0.38 | [14] | |||
| [(tert-Bu–)2(HO–Ph)]2Sn(PMT)2 | 7.86 ± 0.87 | [14] | |||
| [(tert-Bu–)2(HO–Ph)]2Sn(MPMT)2 | 0.58 ± 0.1 | [14] | |||
| {[(tert-Bu–)2(HO–Ph)]2SnCl(PYT)} | >30 | [14] | |||
| [(tert-Bu–)2(HO–Ph)]2SnCl(MBZT)} | >30 | [14] | |||
| Ph3SnCl | 0.130 | 0.166 | 0.141 | 1.08 | [16] |
| [Ph3SnOH]n | 0.070 | 0.165 | 0.090 | 1.29 | [16] |
| [(Ph2Sn)4Cl2O2(OH)2] | >10 | >10 | >10 | [16] | |
| Me2Sn((tert-Bu–)2(HO–Ph–S))2 | 19.20 ± 1.70 | 19.50 ± 1.40 | 1.02 | [15] | |
| Et2Sn(((tert-Bu–)2(HO–Ph–S))2 | 6.20 ± 0.80 | 7.30 ± 0.60 | 1.18 | [15] | |
| (n-Bu)2Sn–(((tert-Bu–)2(HO–Ph–S))2 | 0.40 ± 0.06 | 0.61 ± 0.07 | 1.53 | [15] | |
| Ph2Sn(((tert-Bu–)2(HO–Ph–S))2 | 6.20 ± 0.80 | 12.40 ± 1.40 | 2.00 | [15] | |
| [(tert-Bu–)2(HO–Ph)]2Sn(((tert-Bu–)2(HO–Ph–S))2 | >30 | >30 | >30 | [15] | |
| Me3Sn((tert-Bu–)2(HO–Ph–S)) | 4.90 ± 0.50 | 3.36 ± 0.13 | 0.69 | [15] | |
| Ph3Sn(((tert-Bu–)2(HO–Ph–S)) | 0.25 ± 0.03 | 0.22 ± 0.01 | 0.88 | [15] | |
| Cisplatin | 5.5 ± 0.4 | 26.7 ± 1.1 | 1.1 ± 0.2 | 0.20 | [37] |
| Description | Phases of cell cycle | |||
|---|---|---|---|---|
| Sub-G1 | G0/G1 | S | G2/M | |
| Untreated cells | 6.1% | 46.5% | 18.3% | 28.9% |
| Treated cells with DMSO | 6.5 | 42.7 | 20.6 | 29.5 |
| 1 | 14.4 | 37.9% | 25.1 | 22.1 |
| 2 | 24.1 | 35.8%. | 22.9 | 17.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latsis, G.K.; Banti, C.N.; Kourkoumelis, N.; Papatriantafyllopoulou, C.; Panagiotou, N.; Tasiopoulos, A.; Douvalis, A.; Kalampounias, A.G.; Bakas, T.; Hadjikakou, S.K. Poly Organotin Acetates against DNA with Possible Implementation on Human Breast Cancer. Int. J. Mol. Sci. 2018, 19, 2055. https://doi.org/10.3390/ijms19072055
Latsis GK, Banti CN, Kourkoumelis N, Papatriantafyllopoulou C, Panagiotou N, Tasiopoulos A, Douvalis A, Kalampounias AG, Bakas T, Hadjikakou SK. Poly Organotin Acetates against DNA with Possible Implementation on Human Breast Cancer. International Journal of Molecular Sciences. 2018; 19(7):2055. https://doi.org/10.3390/ijms19072055
Chicago/Turabian StyleLatsis, George K., Christina N. Banti, Nikolaos Kourkoumelis, Constantina Papatriantafyllopoulou, Nikos Panagiotou, Anastasios Tasiopoulos, Alexios Douvalis, Angelos G. Kalampounias, Thomas Bakas, and Sotiris K. Hadjikakou. 2018. "Poly Organotin Acetates against DNA with Possible Implementation on Human Breast Cancer" International Journal of Molecular Sciences 19, no. 7: 2055. https://doi.org/10.3390/ijms19072055