Generation of Immortalised But Unstable Cells after hTERT Introduction in Telomere-Compromised and p53-Deficient vHMECs


Abstract
:1. Introduction
2. Results
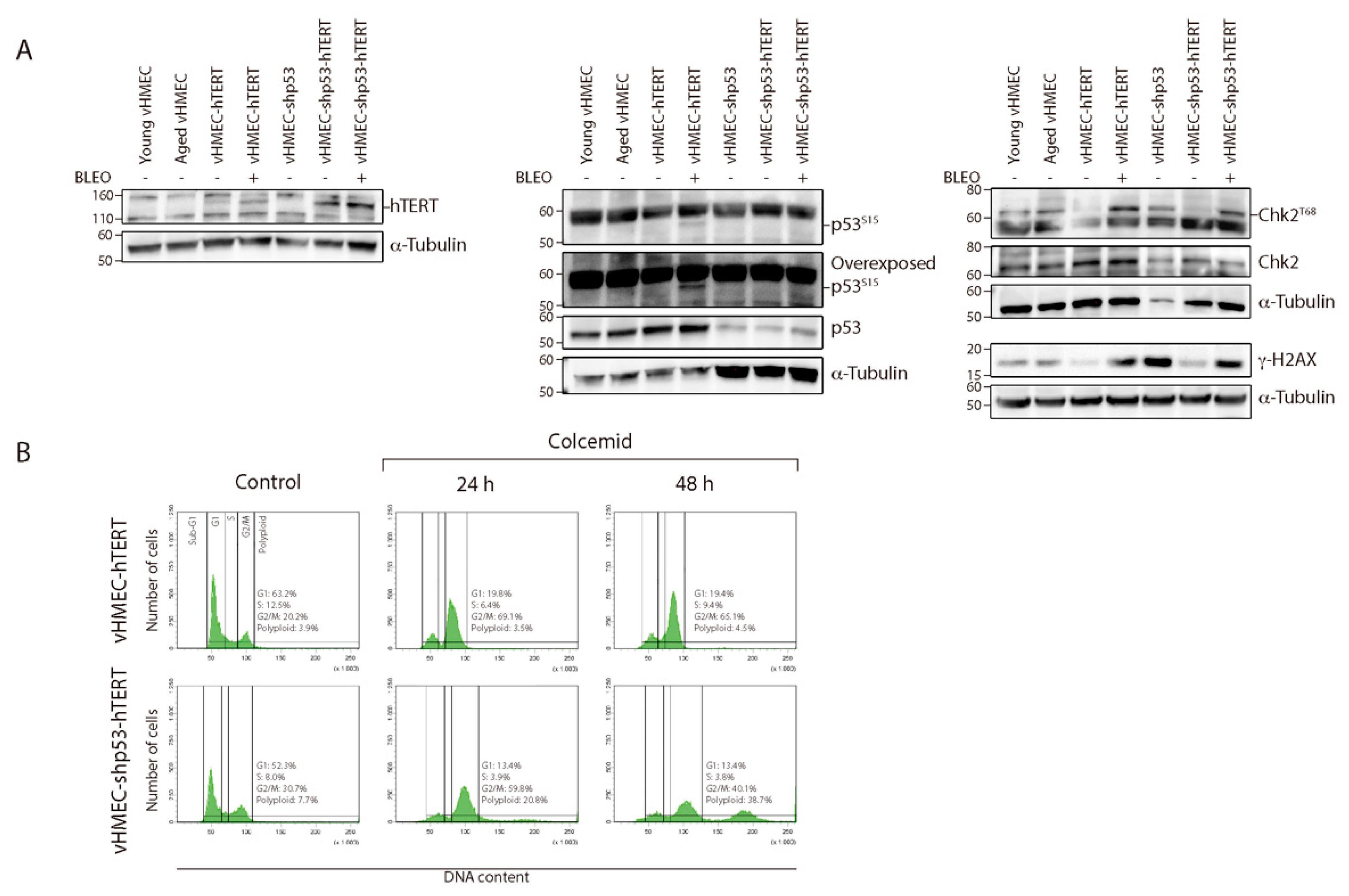
2.1. Establishment of Immortalised and Non-Immortalised vHMECs with Different Cell Cycle Settings
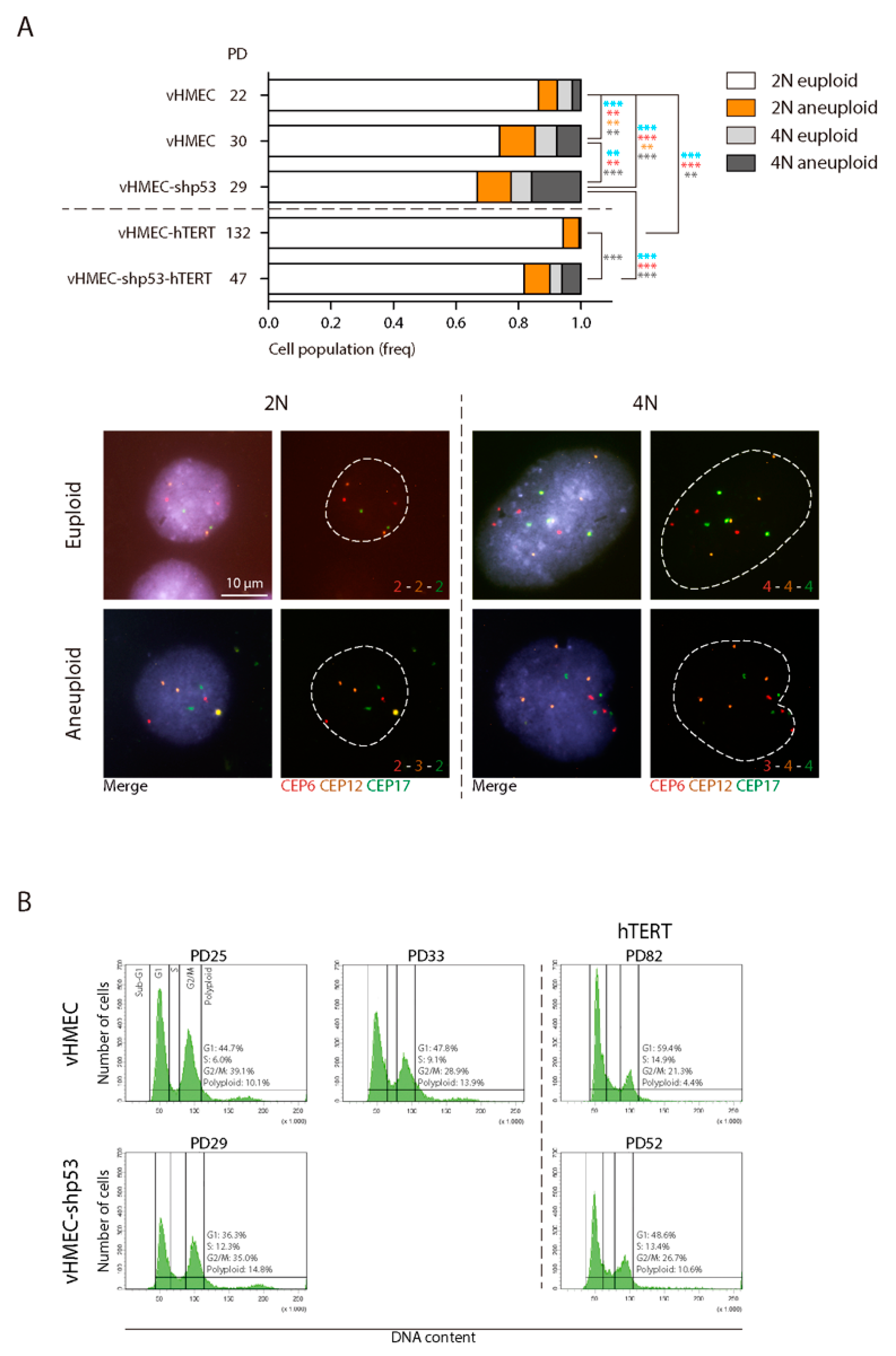
2.2. The Negative Impact of Telomere-Erosion on the Karyotype of vHMECs Is Enhanced by Targeted p53 Inactivation
2.3. Absence of CIN When hTERT Is Ectopically Expressed in p53-Proficient Young vHMECs
2.4. Reduced But Persistent CIN in p53-Deficient vHMECs Immortalised with hTERT
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Lentiviral Vectors, Lentivirus Production and Transduction
4.3. Western Blotting
4.4. Drug Treatments
4.5. Obtaining Metaphase Cells and End-to-End Fusion Scoring Criteria
4.6. In Situ Fluorescence Hybridisation
4.7. DAPI and Texas Red-X Phalloidin Staining
4.8. Fluorescent Microscopy and Fluorescent Images
4.9. Flow Cytometry
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Michor, F.; Iwasa, Y.; Vogelstein, B.; Lengauer, C.; Nowak, M.A. Can chromosomal instability initiate tumorigenesis? Semin. Cancer Biol. 2005, 15, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fu, L.; Zhang, L.Y.; Kwong, D.L.; Yan, L.; Guan, X.Y. Tumor suppressor genes on frequently deleted chromosome 3p in nasopharyngeal carcinoma. Chin. J. Cancer 2012, 31, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kops, G.J.; Weaver, B.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005, 5, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. Human cancers express mutator phenotypes: Origin, consequences and targeting. Nat. Rev. Cancer 2011, 11, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Cleveland, D.W.; Putnam, C.D. Aneuploidy drives a mutator phenotype in cancer. Science 2011, 333, 942–943. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of chromosomal instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, A.M. A theory of marginotomy: The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Daniali, L.; Benetos, A.; Susser, E.; Kark, J.D.; Labat, C.; Kimura, M.; Desai, K.; Granick, M.; Aviv, A. Telomeres shorten at equivalent rates in somatic tissues of adults. Nat. Commun. 2013, 4, 1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Smogorzewska, A.; De Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef]
- Kaul, Z.; Cesare, A.J.; Huschtscha, L.I.; Neumann, A.A.; Reddel, R.R. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep. 2011, 13, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Hayashi, M.T.; Crabbe, L.; Karlseder, J. The telomere deprotection response is functionally distinct from the genomic DNA damage response. Mol. Cell 2013, 51, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Artandi, S.E.; Shen, Q.; Tam, A.; Lee, S.L.; Gottlieb, G.J.; Greider, C.W.; Depinho, R.A. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999, 97, 527–538. [Google Scholar] [CrossRef]
- Artandi, S.E.; Chang, S.; Lee, S.L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wu, C.J.; Jaskelioff, M.; Ivanova, E.; Kost-Alimova, M.; Protopopov, A.; Chu, G.C.; Wang, G.; Lu, X.; Labrot, E.S.; et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell 2012, 148, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.; de Solorzano, C.O.; Knowles, D.; Jones, A.; Chou, W.; Rodriguez, E.G.; Kuo, W.-L.L.; Ljung, B.-M.M.; Chew, K.; Myambo, K.; et al. In situ analyses of genome instability in breast cancer. Nat. Genet. 2004, 36, 984–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeker, A.K.; Hicks, J.L.; Gabrielson, E.; Strauss, W.M.; De Marzo, A.M.; Argani, P. Telomere shortening occurs in subsets of normal breast epithelium as well as in situ and invasive carcinoma. Am. J. Pathol. 2004, 164, 925–935. [Google Scholar] [CrossRef]
- Meeker, A.K.; Argani, P. Telomere shortening occurs early during breast tumorigenesis: A cause of chromosome destabilization underlying malignant transformation? J. Mammary Gland Biol. Neoplasia 2004, 9, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Abe, S.; Huda, N.; Tu, L.; Beam, M.J.; Grimes, B.; Gilley, D. Telomere fusions in early human breast carcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 14098–14103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellsworth, R.E.; Blackburn, H.L.; Shriver, C.D.; Soon-Shiong, P.; Ellsworth, D.L. Molecular heterogeneity in breast cancer: State of the science and implications for patient care. Semin. Cell Dev. Biol. 2017, 64, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Sugino, T.; Yoshida, K.; Bolodeoku, J.; Tahara, H.; Buley, I.; Manek, S.; Wells, C.; Goodison, S.; Ide, T.; Suzuki, T.; et al. Telomerase activity in human breast cancer and benign breast lesions: Diagnostic applications in clinical specimens, including fine needle aspirates. Int. J. Cancer 1996, 69, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Bednarek, A.K.; Sahin, A.; Brenner, A.J.; Johnston, D.A.; Aldaz, C.M. Analysis of telomerase activity levels in breast cancer: Positive detection at the in situ breast carcinoma stage. Clin. Cancer Res. 1997, 3, 11–16. [Google Scholar] [PubMed]
- Poremba, C.; Shroyer, K.R.; Frost, M.; Diallo, R.; Fogt, F.; Schäfer, K.L.; Bürger, H.; Shroyer, A.L.; Dockhorn-Dworniczak, B.; Boecker, W. Telomerase is a highly sensitive and specific molecular marker in fine-needle aspirates of breast lesions. J. Clin. Oncol. 1999, 17, 2020–2026. [Google Scholar] [CrossRef] [PubMed]
- Shpitz, B.; Zimlichman, S.; Zemer, R.; Bomstein, Y.; Zehavi, T.; Liverant, S.; Bernehim, J.; Kaufman, Z.; Klein, E.; Shapira, Y.; et al. Telomerase activity in ductal carcinoma in situ of the breast. Breast Cancer Res. Treat. 1999, 58, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.J.; Stampfer, M.R.; Aldaz, C.M. Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene 1998, 17, 199–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanov, S.R.; Kozakiewicz, B.K.; Holst, C.R.; Stampfer, M.R.; Haupt, L.M.; Tlsty, T.D. Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature 2001, 409, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Soler, D.; Genescà, A.; Arnedo, G.; Egozcue, J.; Tusell, L. Telomere dysfunction drives chromosomal instability in human mammary epithelial cells. Genes Chromosom. Cancer 2005, 44, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Genescà, A.; Pampalona, J.; Frías, C.; Domínguez, D.; Tusell, L. Role of telomere dysfunction in genetic intratumor diversity. Adv. Cancer Res. 2011, 112, 11–41. [Google Scholar] [PubMed]
- Garbe, J.C.; Holst, C.R.; Bassett, E.; Tlsty, T.D.; Stampfer, M.R. Inactivation of p53 function in cultured human mammary epithelial cells turns the telomere-length dependent senescence barrier from agonescence into crisis. Cell Cycle 2007, 6, 1927–1936. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Rieder, C.L.; Schultz, A.; Cole, R.; Sluder, G. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J. Cell Biol. 1994, 127, 1301–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieder, C.L.; Cole, R.W.; Khodjakov, A.; Sluder, G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 1995, 130, 941–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell 2001, 12, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Borel, F.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9819–9824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casenghi, M.; Mangiacasale, R.; Tuynder, M.; Caillet-Fauquet, P.; Elhajouji, A.; Lavia, P.; Mousset, S.; Kirsch-Volders, M.; Cundari, E. p53-independent apoptosis and p53-dependent block of DNA rereplication following mitotic spindle inhibition in human cells. Exp. Cell Res. 1999, 250, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.M.; Sanchez, C.A.; Morgan, C.A.; Schimke, M.K.; Ramel, S.; Idzerda, R.L.; Raskind, W.H.; Reid, B.J. A p53-dependent mouse spindle checkpoint. Science 1995, 267, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Kienitz, A.; Hofmann, I.; Müller, R.; Bastians, H. Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene 2004, 23, 6845–6853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Tsao, S.W.; Guan, X.Y.; Lucas, J.N.; Cheung, A.L.M. Role of short telomeres in inducing preferential chromosomal aberrations in human ovarian surface epithelial cells: A combined telomere quantitative fluorescence in situ hybridization and whole-chromosome painting study. Genes Chromosom. Cancer 2003, 37, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Tsao, S.W.; Guan, X.Y.; Lucas, J.N.; Si, H.X.; Leung, C.S.; Mak, P.; Wang, L.D.; Cheung, A.L.M. Distinct profiles of critically short telomeres are a key determinant of different chromosome aberrations in immortalized human cells: Whole-genome evidence from multiple cell lines. Oncogene 2004, 23, 9090–9101. [Google Scholar] [CrossRef] [PubMed]
- Der-Sarkissian, H.; Bacchetti, S.; Cazes, L.; Londoño-Vallejo, J.A. The shortest telomeres drive karyotype evolution in transformed cells. Oncogene 2004, 23, 1221–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plug-DeMaggio, A.W.; Sundsvold, T.; Wurscher, M.A.; Koop, J.I.; Klingelhutz, A.J.; McDougall, J.K. Telomere erosion and chromosomal instability in cells expressing the HPV oncogene 16E6. Oncogene 2004, 23, 3561–3571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pampalona, J.; Soler, D.; Genescà, A.; Tusell, L. Whole chromosome loss is promoted by telomere dysfunction in primary cells. Genes Chromosom. Cancer 2010, 49, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Denchi, E.L.; de Lange, T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010, 141, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Pampalona, J.; Frías, C.; Genescà, A.; Tusell, L. Progressive telomere dysfunction causes cytokinesis failure and leads to the accumulation of polyploid cells. PLoS Genet. 2012, 8, e1002679. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Coquelle, A.; Vivet, S.; Vitale, I.; Kauffmann, A.; Dessen, P.; Pequignot, M.O.; Casares, N.; Valent, A.; Mouhamad, S.; et al. Apoptosis regulation in tetraploid cancer cells. EMBO J. 2006, 25, 2584–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senovilla, L.; Vitale, I.; Galluzzi, L.; Vivet, S.; Joza, N.; Younes, A.B.; Rello-Varona, S.; Castedo, M.; Kroemer, G. p53 represses the polyploidization of primary mammary epithelial cells by activating apoptosis. Cell Cycle 2009, 8, 1380–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silkworth, W.T.; Nardi, I.K.; Scholl, L.M.; Cimini, D. Multipolar Spindle Pole Coalescence is a Major Source of Kinetochore Mis-Attachment and Chromosome Mis-Segregation in Cancer Cells. PLoS ONE 2009, 4, e6564. [Google Scholar] [CrossRef] [PubMed]
- Garbe, J.C.; Vrba, L.; Sputova, K.; Fuchs, L.; Novak, P.; Brothman, A.R.; Jackson, M.; Chin, K.; LaBarge, M.A.; Watts, G.; et al. Immortalization of normal human mammary epithelial cells in two steps by direct targeting of senescence barriers does not require gross genomic alterations. Cell Cycle 2014, 13, 3423–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toouli, C.; Huschtscha, L.; Neumann, A. Comparison of human mammary epithelial cells immortalized by simian virus 40 T-Antigen or by the telomerase catalytic subunit. Oncogene 2002, 21, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, K.; Bryant, E.; O’Hara Larivee, S.; McDougall, J.K. Production of spindle cell carcinoma by transduction of H-Ras 61L into immortalized human mammary epithelial cells. Cancer Lett. 2003, 201, 79–88. [Google Scholar] [CrossRef]
- Haga, K.; Ohno, S.; Yugawa, T.; Narisawa-Saito, M.; Fujita, M.; Sakamoto, M.; Galloway, D.A.; Kiyono, T. Efficient immortalization of primary human cells by p16INK4a-specific short hairpin RNA or Bmi-1, combined with introduction of hTERT. Cancer Sci. 2007, 98, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.S.; Modur, V.; Cheng, J.; Robinson, K.; Rao, K. Characterization of immortalized human mammary epithelial cell line HMEC 2.6. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, U.; Sommer, A.; Beckmann, G.; Lutzenberger, M.; Seidel, H.; Kreft, B.; Toschi, L. Isogenic human mammary epithelial cell lines: Novel tools for target identification and validation. Comprehensive characterization of an isogenic human mammary epithelial cell model provides evidence for epithelial-mesenchymal transition. Breast Cancer Res. Treat. 2013, 138, 437–456. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Fauth, C.; Speicher, M.R.; Dutriaux, A.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer Res. 2002, 62, 1129–1133. [Google Scholar] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Roake, C.M.; Artandi, S.E. Control of Cellular Aging, Tissue Function, and Cancer by p53 Downstream of Telomeres. Cold Spring Harb. Perspect. Med. 2017, 7, a026088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Holst, C.R.; Nuovo, G.J.; Esteller, M.; Chew, K.; Baylin, S.B.; Herman, J.G.; Tlsty, T.D. Methylation of p16INK4a promoters occurs in vivo in histologically normal human mammary epithelia. Cancer Res. 2003, 63, 1596–1601. [Google Scholar] [PubMed]
- Shackney, S.E.; Silverman, J.F. Molecular evolutionary patterns in breast cancer. Adv. Anat. Pathol. 2003, 10, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Hui, R.; Macmillan, R.D.; Kenny, F.S.; Musgrove, E.A.; Blamey, R.W.; Nicholson, R.I.; Robertson, J.F.; Sutherland, R.L. INK4a gene expression and methylation in primary breast cancer: Overexpression of p16INK4a messenger RNA is a marker of poor prognosis. Clin. Cancer Res. 2000, 6, 2777–2787. [Google Scholar] [PubMed]
- Tusell, L.; Soler, D.; Agostini, M.; Pampalona, J.; Genescà, A. The number of dysfunctional telomeres in a cell: One amplifies; more than one translocate. Cytogenet. Genome Res. 2008, 122, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Pampalona, J.; Roscioli, E.; Silkworth, W.T.; Bowden, B.; Genescà, A.; Tusell, L.; Cimini, D. Chromosome Bridges Maintain Kinetochore-Microtubule Attachment throughout Mitosis and Rarely Break during Anaphase. PLoS ONE 2016, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Seewaldt, V.L.; Mrózek, K.; Sigle, R.; Dietze, E.C.; Heine, K.; Hockenbery, D.M.; Hobbs, K.B.; Caldwell, L.E. Suppression of p53 function in normal human mammary epithelial cells increases sensitivity to extracellular matrix-induced apoptosis. J. Cell Biol. 2001, 155, 471–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; de Lange, T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 2012, 21, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Duelli, D.M.; Padilla-Nash, H.M.; Berman, D.; Murphy, K.M.; Ried, T.; Lazebnik, Y. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr. Biol. 2007, 17, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.G.; Makitalo, M.; Yang, D.; Chinnappan, D.; Hilaire, C.S.; Ravid, K.; St Hilaire, C.; Ravid, K. Deregulated Aurora-B induced tetraploidy promotes tumorigenesis. FASEB J. 2009, 23, 2741–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaharski, A.J.; Sotelo, R.; Solorza-Luna, G.; Gonsebatt, M.E.; Guzman, P.; Mohar, A.; Eastmond, D.A. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis 2006, 27, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, P.C.; Cowan, D.S.; Sanchez, C.A.; Barrett, M.T.; Emond, M.J.; Levine, D.S.; Rabinovitch, P.S.; Reid, B.J. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett’s esophagus. Proc. Natl. Acad. Sci. USA 1996, 93, 7081–7084. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef] [PubMed]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Gronroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of Whole-Genome Doubling Propagates Chromosomal Instability and Accelerates Cancer Genome Evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Hauser, S.H.; Liu, X.L.; Wazer, D.E.; Madoc-Jones, H.; Band, V. Mutant p53-induced immortalization of primary human mammary epithelial cells. Cancer Res. 1996, 56, 3129–3133. [Google Scholar] [PubMed]
- Gollahon, L.S.; Shay, J.W. Immortalization of human mammary epithelial cells transfected with mutant p53 (273his). Oncogene 1996, 12, 715–725. [Google Scholar] [PubMed]
- Stampfer, M.R.; Garbe, J.; Nijjar, T.; Wigington, D.; Swisshelm, K.; Yaswen, P. Loss of p53 function accelerates acquisition of telomerase activity in indefinite lifespan human mammary epithelial cell lines. Oncogene 2003, 22, 5238–5251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herr, A.J.; Ogawa, M.; Lawrence, N.A.; Williams, L.N.; Eggington, J.M.; Singh, M.; Smith, R.A.; Preston, B.D. Mutator suppression and escape from replication error-induced extinction in yeast. PLoS Genet. 2011, 7, e1002282. [Google Scholar] [CrossRef]
- Sniegowski, P.D.; Gerrish, P.J.; Johnson, T.; Shaver, A. The evolution of mutation rates: Separating causes from consequences. Bioessays 2000, 22, 1057–1066. [Google Scholar] [CrossRef]
- Nowak, M.; Schuster, P. Error thresholds of replication in finite populations mutation frequencies and the onset of Muller’s ratchet. J. Theor. Biol. 1989, 137, 375–395. [Google Scholar] [CrossRef]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic Instability in Cancer: Teetering on the Limit of Tolerance. Cancer Res. 2017, 77, 2179–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Jin, Y.; Chen, X.; Jin, C.; Law, S.; Tsao, S.W.; Kwong, Y.L. Cytogenetic aberrations in immortalization of esophageal epithelial cells. Cancer Genet. Cytogenet. 2006, 165, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhang, H.; Tsao, S.W.; Jin, C.; Lv, M.; Strömbeck, B.; Wiegant, J.; Wan, T.S.; Yuen, P.W.; Kwong, Y.L. Cytogenetic and molecular genetic characterization of immortalized human ovarian surface epithelial cell lines: Consistent loss of chromosome 13 and amplification of chromosome 20. Gynecol. Oncol. 2004, 92, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Coursen, J.D.; Bennett, W.P.; Gollahon, L.; Shay, J.W.; Harris, C.C. Genomic instability and telomerase activity in human bronchial epithelial cells during immortalization by human papillomavirus-16 E6 and E7 genes. Exp. Cell Res. 1997, 235, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Savelieva, E.; Belair, C.D.; Newton, M.A.; DeVries, S.; Gray, J.W.; Waldman, F.; Reznikoff, C.A. 20q gain associates with immortalization: 20q13.2 amplification correlates with genome instability in human papillomavirus 16 E7 transformed human uroepithelial cells. Oncogene 1997, 14, 551–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Hatton, M.P.; Khandelwal, P.; Sullivan, D.A. Culture, immortalization, and characterization of human meibomian gland epithelial cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3993–4005. [Google Scholar] [CrossRef] [PubMed]
- Garbe, J.C.; Bhattacharya, S.; Merchant, B.; Bassett, E.; Swisshelm, K.; Feiler, H.S.; Wyrobek, A.J.; Stampfer, M.R. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res. 2009, 69, 7557–7568. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | PD | Analysed Cells n | Abnormal Cells % (n) | Cells with fus/dic % (n) | Cells with nrt % (n) | Cells with Structural AA % (n) | Cells with Clonal Numerical AA % (n) |
|---|---|---|---|---|---|---|---|
| vHMEC | 22 | 26 | 42.31 (11) | 23.08 (6) | 7.69 (2) | 42.31 (11) | 0.00 (0) |
| vHMEC | 32 | 20 | 100.00 (20) | 85.00 (17) | 20.00 (4) | 100.00 (20) | 0.00 (0) |
| vHMEC-shp53 | 29 | 23 | 100.00 (23) | 82.61 (19) | 21.74 (5) | 100.00 (23) | 0.00 (0) |
| vHMEC-hTERT | 76 | 46 | 91.30 (42) | 0.00 (0) | 6.52 (3) | 17.39 (8) | 84.78 (39) |
| vHMEC-shp53-hTERT | 47 | 54 | 62.96 (34) | 16.67 (9) | 50.00 (27) | 62.96 (34) | 0.00 (0) |
| Cell Line | PD | Analysed Cells n | Total AA/Cell Freq. (n) | UNSTABLE AA | STABLE AA | OTHER AA | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| fus/dic n | Ace n | AA/Cell Freq. | nrt/i/mar /del n | AA/Cell Freq. | csb/ctb n | AA/Cell Freq. | ||||
| vHMEC | 22 | 26 | 0.46 (12) | 6 | 1 | 0.27 | 2 | 0.08 | 3 | 0.12 |
| vHMEC | 32 | 20 | 1.65 (33) | 22 | 0 | 1.10 | 6 | 0.30 | 5 | 0.25 |
| vHMEC-shp53 | 29 | 23 | 3.65 (84) | 54 | 9 | 2.74 | 15 | 0.65 | 6 | 0.26 |
| vHMEC-hTERT | 76 | 46 | 0.17 (8) | 0 | 0 | 0.00 | 8 | 0.17 | 0 | 0.00 |
| vHMEC-shp53-hTERT | 47 | 54 | 1.20 (65) | 15 | 3 | 0.33 | 46 | 0.85 | 1 | 0.02 |
| Cell Line | PD | Cells Analysed n | 2N | 4N | 4N Fraction % | ||
|---|---|---|---|---|---|---|---|
| Cells n | Aneuploidy Freq. (n) | Cells n | Aneuploidy Freq. (n) | ||||
| vHMEC | 22 | 392 | 362 | 0.06 (23) | 30 | 0.37 (11) | 7.65 |
| vHMEC | 30 | 414 | 353 | 0.13 (47) | 61 | 0.52 (32) | 14.73 |
| vHMEC-shp53 | 29 | 430 | 334 | 0.14 (47) | 96 | 0.71 (68) | 22.33 |
| vHMEC-hTERT | 132 | 846 | 841 | 0.05 (44) | 5 | 0.00 (0) | 0.59 |
| vHMEC-shp53-hTERT | 47 | 391 | 352 | 0.09 (32) | 39 | 0.62 (24) | 9.97 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernal, A.; Zafon, E.; Domínguez, D.; Bertran, E.; Tusell, L. Generation of Immortalised But Unstable Cells after hTERT Introduction in Telomere-Compromised and p53-Deficient vHMECs. Int. J. Mol. Sci. 2018, 19, 2078. https://doi.org/10.3390/ijms19072078
Bernal A, Zafon E, Domínguez D, Bertran E, Tusell L. Generation of Immortalised But Unstable Cells after hTERT Introduction in Telomere-Compromised and p53-Deficient vHMECs. International Journal of Molecular Sciences. 2018; 19(7):2078. https://doi.org/10.3390/ijms19072078
Chicago/Turabian StyleBernal, Aina, Elisenda Zafon, Daniel Domínguez, Enric Bertran, and Laura Tusell. 2018. "Generation of Immortalised But Unstable Cells after hTERT Introduction in Telomere-Compromised and p53-Deficient vHMECs" International Journal of Molecular Sciences 19, no. 7: 2078. https://doi.org/10.3390/ijms19072078