FGF2 Dual Warhead Conjugate with Monomethyl Auristatin E and α-Amanitin Displays a Cytotoxic Effect towards Cancer Cells Overproducing FGF Receptor 1

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Dual Conjugation of α-Amanitin and Monomethyl Auristatin E to Fibroblast Growth Factor 2 (FGF2)
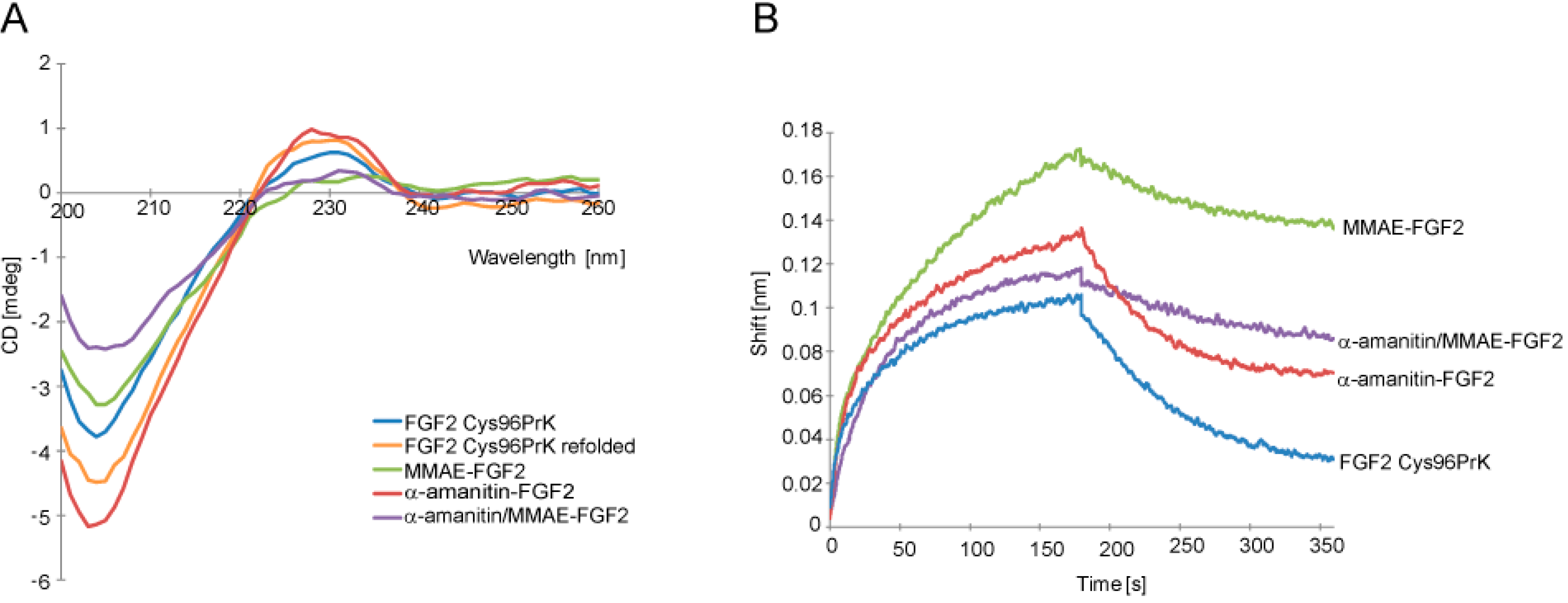
2.2. Characterization of α-Amainitin/Monomethyl Auristatin E (MMAE)-FGF2 Conjugate
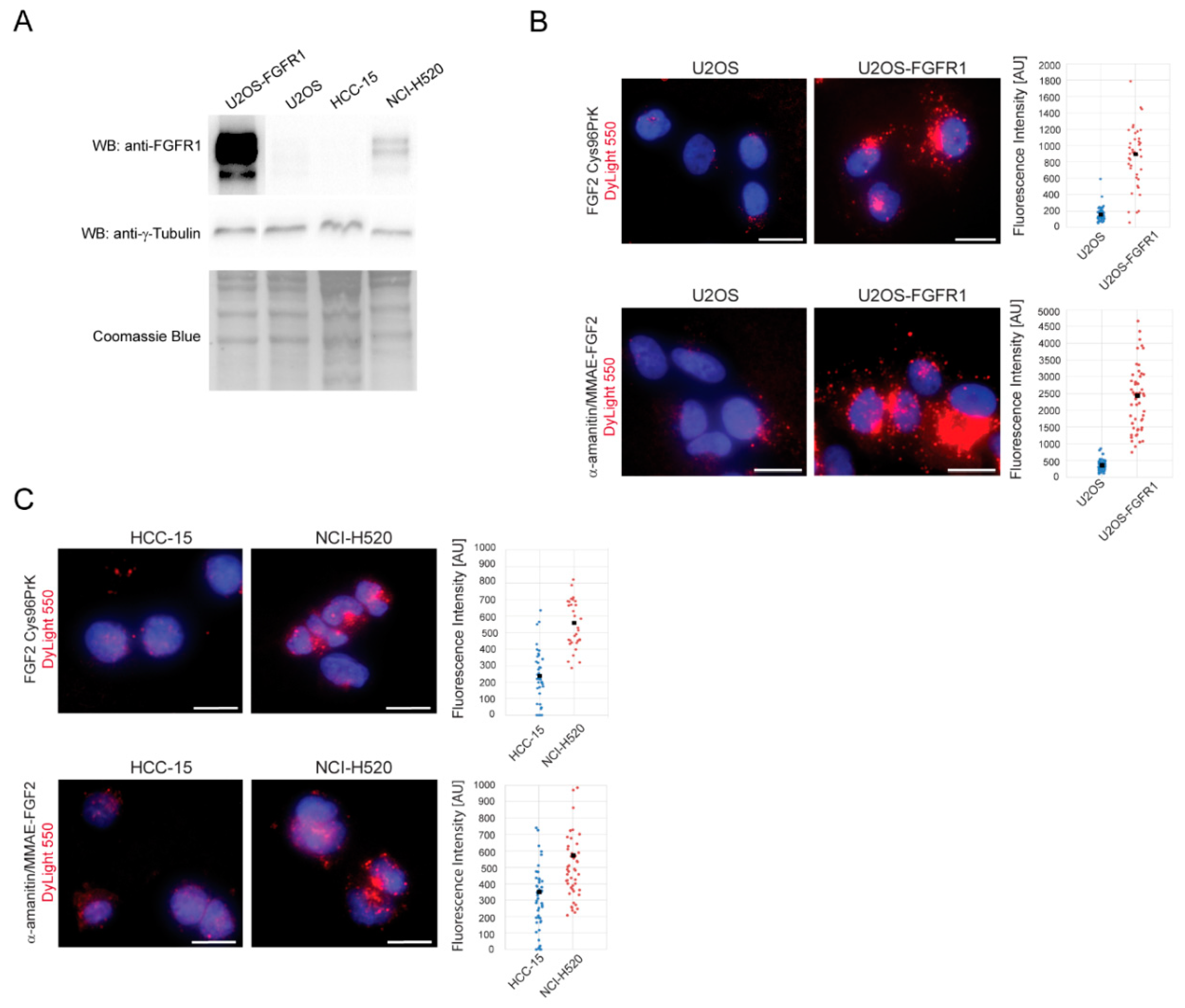
2.3. FGF2 Conjugates Are Selectively Internalized into Cancer Cells in the Fibroblast Growth Factor Receptor 1 (FGFR1)-Dependent Manner
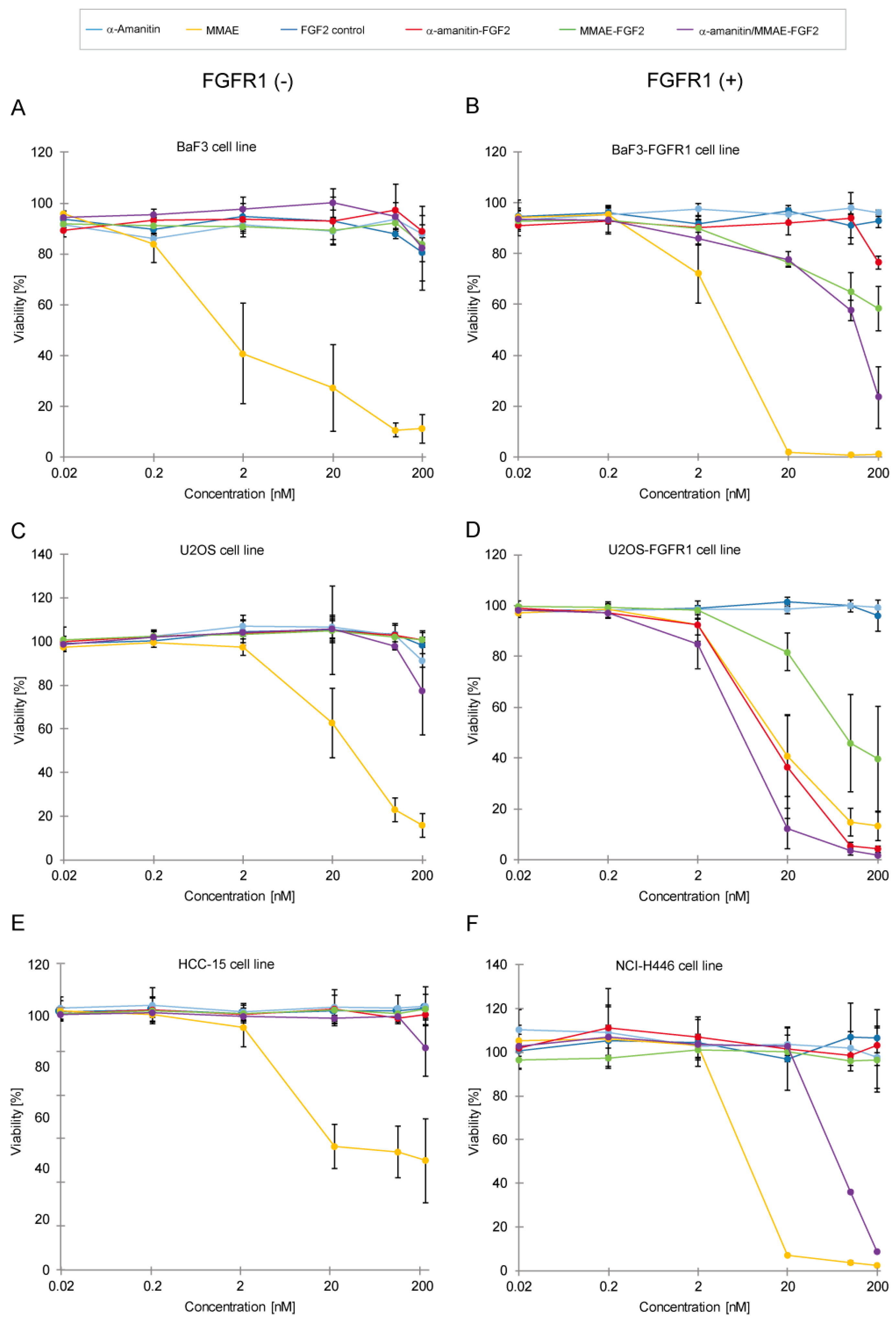
2.4. Cytotoxicity of α-Amainitin/MMAE-FGF2 Conjugate
3. Discussion
4. Materials and Methods
4.1. FGF2 Cys96PrK Variant Expression and Purification
4.2. FGF2 Cys78-α-Amanitin Conjugate Preparation
4.3. FGF2 PrK96-MMAE Conjugate Preparation
4.4. FGF2 Cys78-α-Amanitin/PrK96-MMAE Conjugate Preparation
4.5. Mass Spectrometry
4.6. Circular Dichroism Spectroscopy
4.7. BLI Assays
4.8. The Stability of α-Amanitin/MMAE-FGF2 Conjugate in Human Serum
4.9. Western Blotting
4.10. Fluorescence Microscopy
4.11. Cell Lines
4.12. Cytotoxicity Assay
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-Amanitin-FGF2 | FGF2 single conjugate with α-amanitin incorporated at Cys78 position |
| α-Amanitin/MMAE-FGF2 | FGF2 dual warheads conjugate with α-amanitin incorporated at Cys78 position and MMAE incorporated at PrK96 position |
| ADCs | Antibody drug conjugates |
| AR2G | Amine reactive second-generation biosensor |
| ATCC | American type culture collection |
| BLI | Bio-layer Interferometry technique |
| CuAAC | Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition |
| FBS | Fetal bovine serum |
| FGF2 | Fibroblast growth factor 2 |
| FGFR1 | Fibroblast growth factor receptor 1 |
| MMAD | Monomethyl auristatin D |
| MMAE | Monomethyl auristatin E |
| MMAE-FGF2 | FGF2 single conjugate with MMAE incorporated at PrK96 position |
| NBCS | Newborn bovine calf serum |
| PrK | N-ε-Propargyloxycarbonyl-l-lysine |
| THPTA | Tris(3-hydroxypropyltriazolylmethyl)amine |
References
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. WIREs Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korc, M.; Friesel, R.E. The Role of Fibroblast Growth Factors in Tumor Growth. Curr. Cancer Drug Targets 2009, 9, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Khalid, A.; Javaid, M.A. Fibroblast Growth Factors and their Emerging Cancer-Related Aspects. J. Cancer Sci. Ther. 2016, 8, 190–205. [Google Scholar] [CrossRef]
- Hojjat-Farsangi, M. Small-Molecule Inhibitors of the Receptor Tyrosine Kinases: Promising Tools for Targeted Cancer Therapies. Int. J. Mol. Sci. 2014, 15, 13768–13801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGregor, M.J. A pharmacophore map of small molecule protein kinase inhibitors. J. Chem. Inf. Model. 2007, 6, 2374–2382. [Google Scholar] [CrossRef] [PubMed]
- McCombs, J.R.; Owen, S.C. Antibody Drug Conjugates: Design and Selection of Linker, Payload and Conjugation Chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Chari, R.V.J. Ado-trastuzumab Emtansine (T-DM1): An Antibody—Drug Conjugate (ADC) for HER2-Positive Breast Cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef] [PubMed]
- Godwin, C.D.; Gale, R.P.; Walter, R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855–1868. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Dougher, M.M.; Kalyandrug, L.B.; Armellino, D.C.; Boghaert, E.R.; Hamann, P.R.; Moran, J.K.; Damle, N.K. Antitumor efficacy of a combination of CMC-544 (inotuzumab ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against non-Hodgkin’s B-cell lymphoma. Clin. Cancer Res. 2006, 12, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Kline, T.; Steiner, A.R.; Penta, K.; Sato, A.K.; Hallam, T.J.; Yin, G. Methods to Make Homogenous Antibody Drug Conjugates. Pharm. Res. 2015, 32, 3480–3493. [Google Scholar] [CrossRef] [PubMed]
- Krzyscik, M.; Zakrzewska, M.; Sørensen, V.; Sokolowska-Wedzina, A.; Lobocki, M.; Swiderska, K.W.; Krowarsch, D.; Wiedlocha, A.; Otlewski, J. Cytotoxic conjugates of fibroblast growth factor 2 with monomethyl auristatin E for effective killing of cells expressing FGF receptors. ACS Omega 2017, 2, 3792–3805. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Moldenhauer, G.; Salnikov, A.V.; Lüttgau, S.; Herr, I.; Anderl, J.; Faulstich, H. Therapeutic potential of amanitin-conjugated anti-epithelial cell adhesion molecule monoclonal antibody against pancreatic carcinoma. J. Natl. Cancer Inst. 2012, 104, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Lackner, M.R.; Wilson, T.R.; Settleman, J. Mechanisms of acquired resistance to targeted cancer therapies. Future Oncol. 2012, 8, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Neel, D.S.; Bivona, T.G. Resistance is futile: Overcoming resistance to targeted therapies in lung adenocarcinoma. NPJ Precis. Oncol. 2017, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Szlachcic, A.; Zakrzewska, M.; Lobocki, M.; Jakimowicz, P.; Otlewski, J. Design and characteristics of cytotoxic fibroblast growth factor 1 conjugate for fibroblast growth factor receptor-targeted cancer therapy. Drug Des. Dev. Ther. 2016, 10, 2547–2560. [Google Scholar] [CrossRef] [PubMed]
- Swiderska, K.W.; Szlachcic, A.; Czyrek, A.; Zakrzewska, M.; Otlewski, J. Site-specific conjugation of fibroblast growth factor 2 (FGF2) based on incorporation of alkyne-reactive unnatural amino acid. Bioorg. Med. Chem. 2017, 25, 3685–3693. [Google Scholar] [CrossRef] [PubMed]
- Herbert, C.; Lassalle, G.; Alcouffe, C.; Bono, F. Approaches targeting the FGF-FGFR system: A review of the recent patent literature and associated advanced therapeutic agents. Pharm. Pat. Anal. 2014, 3, 585–612. [Google Scholar] [CrossRef] [PubMed]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signaling in cancer. Nat. Rev. Cancer 2017, 5, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Arnedos, M.; Andre, F.; Soria, J.C. Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives. Cancer Discov. 2013, 3, 264–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallinan, N.; Finn, S.; Cuffe, S.; Rafee, S.; O’Byrne, K.; Gately, K. Targeting the fibroblast growth factor receptor family in cancer. Cancer Treat. Rev. 2016, 46, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, C.; Giglione, P.; Liguigli, W.; Paglino, C. Dovitinib (CHIR258, TKI258): Structure, development and preclinical and clinical activity. Future Oncol. 2008, 11, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIB 1120: Triple Angiokinase Inhibitor with Sustained Receptor Blockade and Good Antitumor Efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [PubMed]
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: Biologic and clinical implications. Cancer Metastasis Rev. 2015, 34, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Baudino, T.A. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ibrahimi, O.A.; Olsen, S.K.; Umemori, H.; Mohammadi, M.; Ornitz, D.M. Receptor Specificity of the Fibroblast Growth Factor Family. The complete mammalian FGF family. J. Biol. Chem. 2006, 281, 15694–15700. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Narla, R.K.; Jun, X.; Zeren, T.; Venkatachalam, T.; Waddick, K.G.; Rostostev, A.; Myers, D.E. Cytotoxic activity of epidermal growth factor-genistein against breast cancer cells. Clin. Cancer Res. 1998, 4, 901–912. [Google Scholar] [PubMed]
- Schaffert, D.; Kiss, M.; Rodl, W.; Shir, A.; Levitzki, A.; Ogris, M.; Wagner, E. Poly(I:C)-mediated tumor growth suppression in EGF-receptor overexpressing tumors using EGF-polyethylene glycol-linear polyethylenimine as carrier. Pharm. Res. 2011, 4, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Jinno, H.; Ueda, M.; Ozawa, S.; Kikuchi, K.; Ikeda, T.; Enomoto, K.; Kitajima, M. Epidermal growth factor receptor-dependent cytotoxic effect by an EGF-ribonuclease conjugate on human cancer cell lines—A trial for less immunogenic chimeric toxin. Cancer Chemother. Pharmacol. 1996, 4, 303–308. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer Drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, J.; Yao, X.; Lin, S.; Gu, Y.; Xu, J.; Deng, Z.; Ma, W.; Zhang, H. FIGNL1 is overexpressed in small cell lung cancer patients and enhances NCI-H446 cell resistance to cisplatin and etoposide. Oncol. Rep. 2017, 4, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Puthenveetil, S.; Musto, S.; Loganzo, F.; Tumey, N.L.; O’Donnell, C.J.; Graziani, E. Development of Solid-Phase Site-Specific Conjugation and Its Application toward Generation of Dual Labeled Antibody and Fab Drug Conjugates. Bioconj. Chem. 2016, 27, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Okeley, N.M.; Alley, S.C.; Cerveny, C.G.; Andreyka, J.; Sanderson, J.R.; Anderson, M.; Benjamin, D.R.; Law, C.L.; Sievers, E.; Ihle, N.C. Specific tumor targeting and potent bystander killing with SNG-35, an anti-CD30 antibody drug conjugate. In Proceedings of the ASH Annual Meeting Abstracts, Orlando, FL, USA, 9–12 December 2006; Volume 108, p. 72a. [Google Scholar]
- Sokolowska-Wedzina, A.; Chodaczek, G.; Chudzian, J.; Borek, A.; Zakrzewska, M.; Otlewski, J. High-Affinity Internalizing Human scFv-Fc Antibody for Targeting FGFR1-Overexpressing Lung Cancer. Mol. Cancer Res. 2017, 8, 1040–1050. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Świderska, K.W.; Szlachcic, A.; Opaliński, Ł.; Zakrzewska, M.; Otlewski, J. FGF2 Dual Warhead Conjugate with Monomethyl Auristatin E and α-Amanitin Displays a Cytotoxic Effect towards Cancer Cells Overproducing FGF Receptor 1. Int. J. Mol. Sci. 2018, 19, 2098. https://doi.org/10.3390/ijms19072098
Świderska KW, Szlachcic A, Opaliński Ł, Zakrzewska M, Otlewski J. FGF2 Dual Warhead Conjugate with Monomethyl Auristatin E and α-Amanitin Displays a Cytotoxic Effect towards Cancer Cells Overproducing FGF Receptor 1. International Journal of Molecular Sciences. 2018; 19(7):2098. https://doi.org/10.3390/ijms19072098
Chicago/Turabian StyleŚwiderska, Karolina Weronika, Anna Szlachcic, Łukasz Opaliński, Małgorzata Zakrzewska, and Jacek Otlewski. 2018. "FGF2 Dual Warhead Conjugate with Monomethyl Auristatin E and α-Amanitin Displays a Cytotoxic Effect towards Cancer Cells Overproducing FGF Receptor 1" International Journal of Molecular Sciences 19, no. 7: 2098. https://doi.org/10.3390/ijms19072098