A Naturally-Occurring Dominant-Negative Inhibitor of Keap1 Competitively against Its Negative Regulation of Nrf2


Abstract
:
1. Introduction
2. Results and Discussion
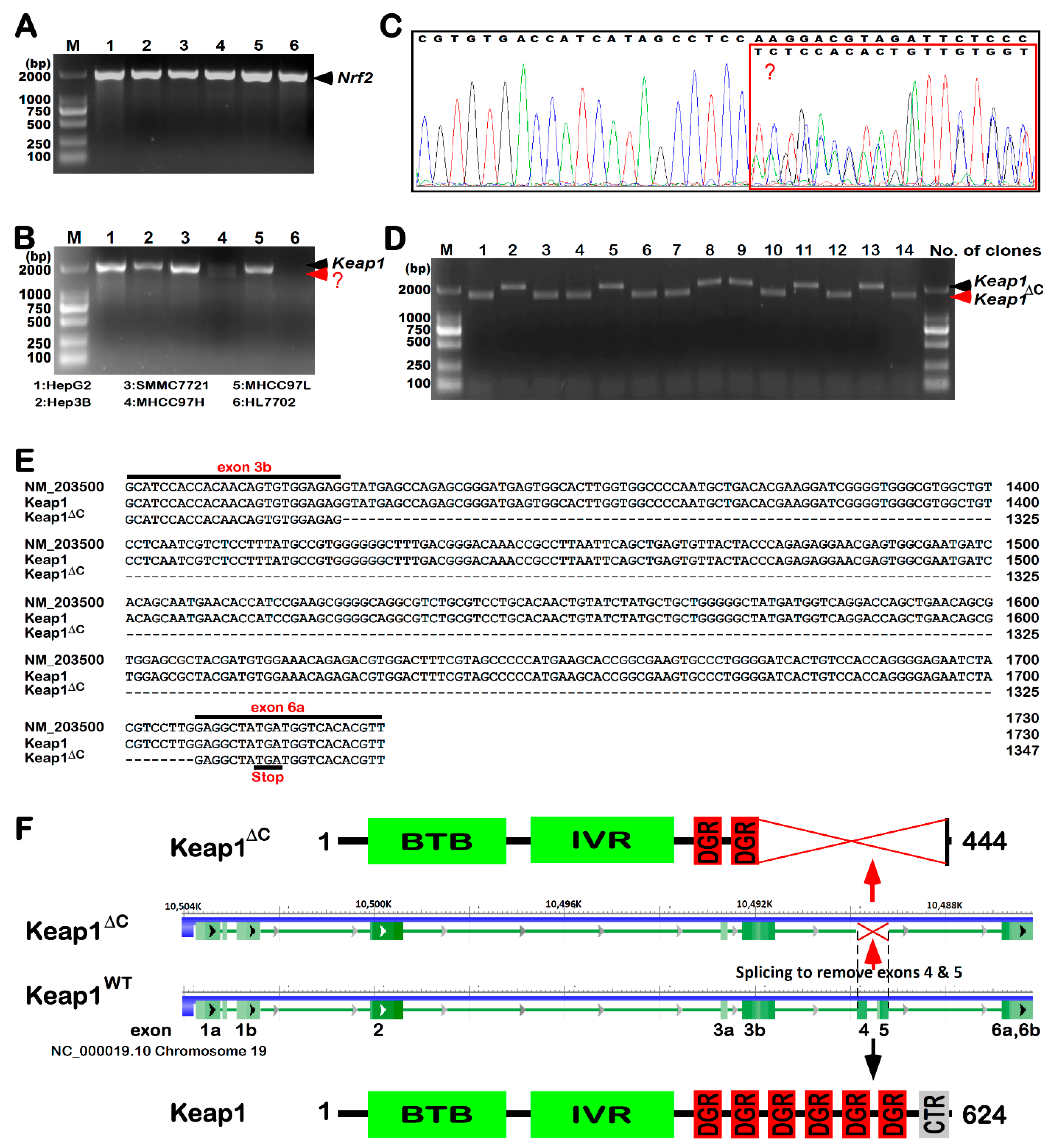
2.1. Discovery of KEAP1ΔC as an Alternatively-Spliced Variant
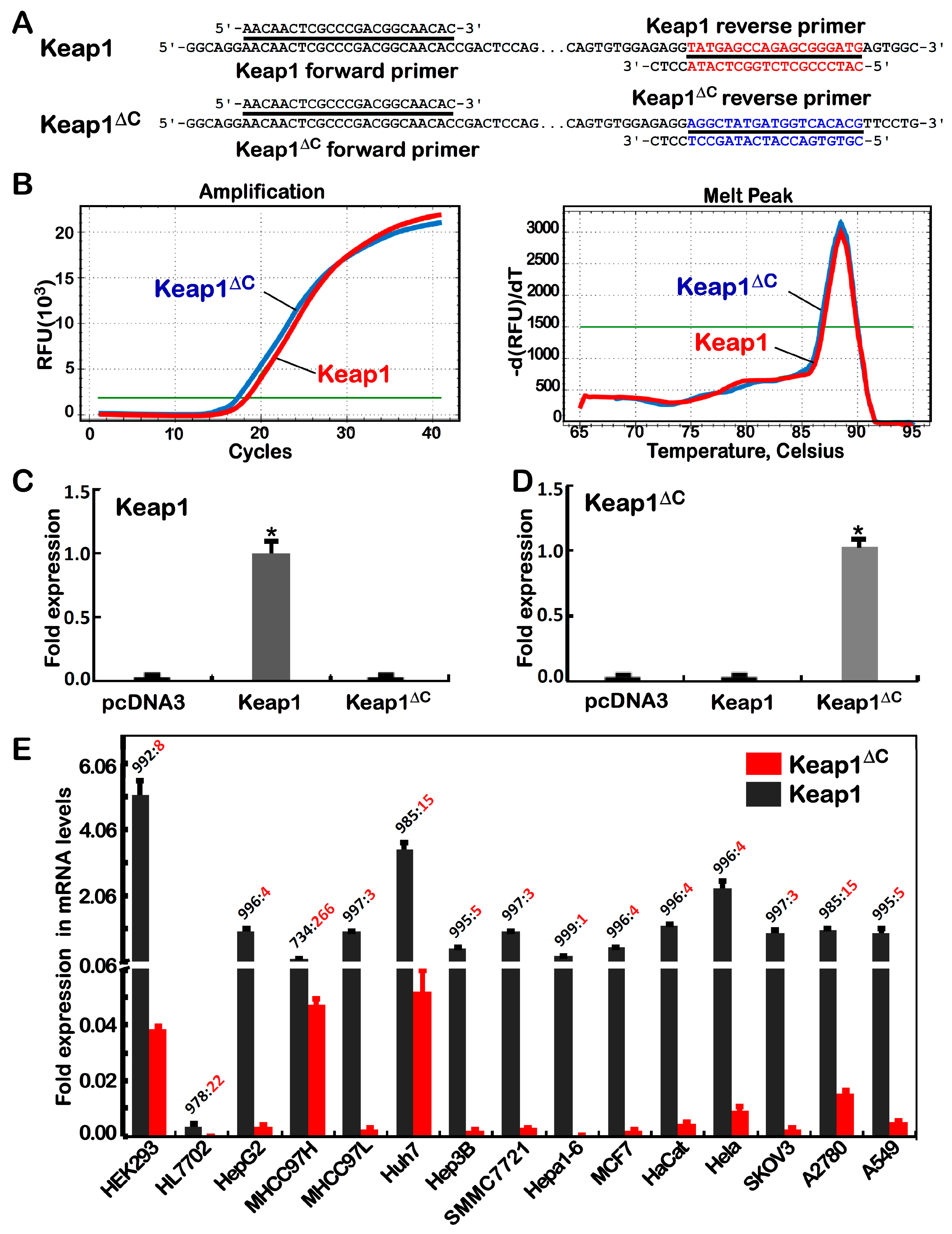
2.2. Differential Expression of Keap1ΔC in Distinct Cell Lines
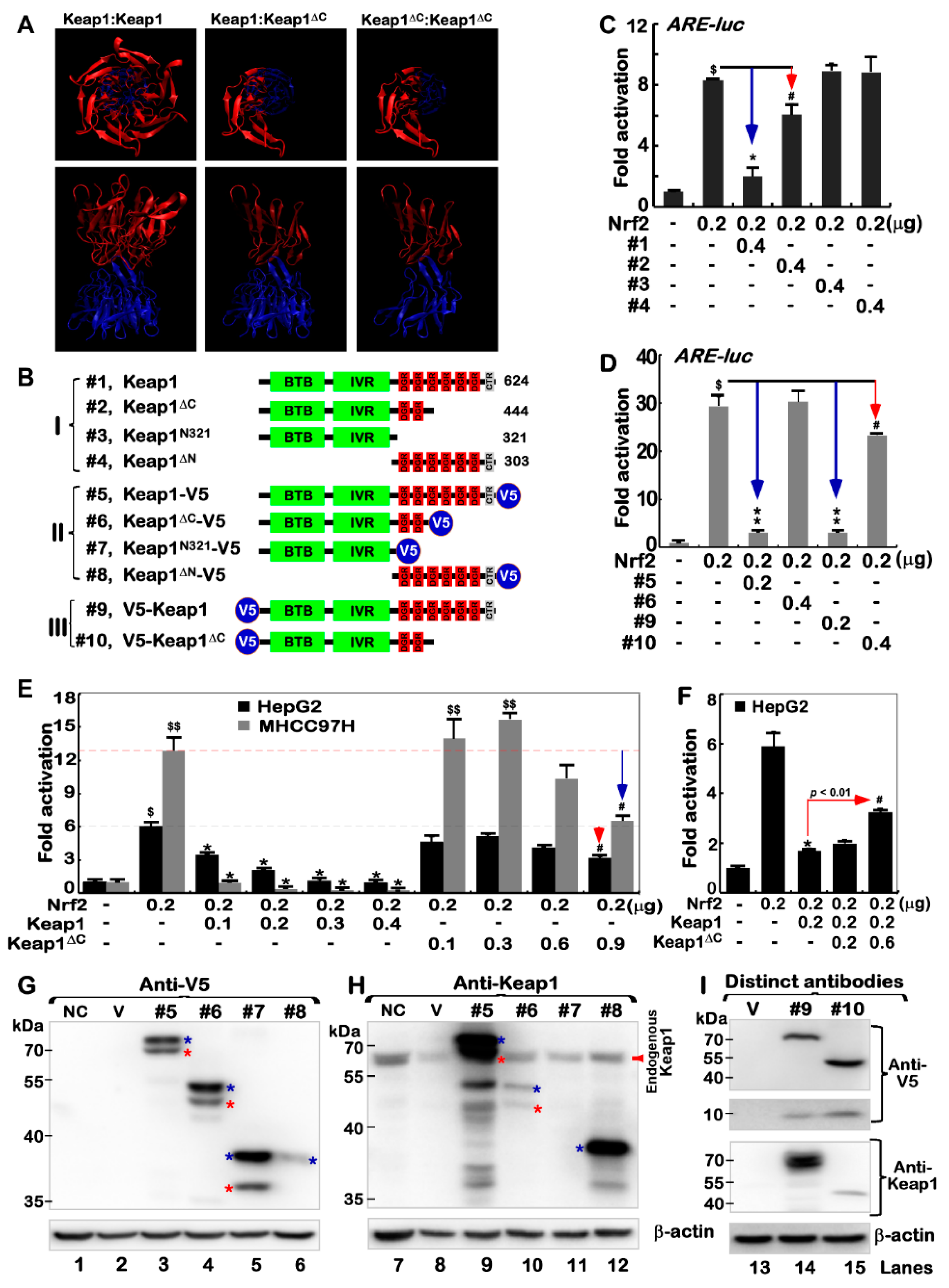
2.3. Nrf2 Could Be Partially Inhibited by Keap1ΔC
2.4. Keap1ΔC Acts as a Dominant-Negative Competitor Partially against Intact Keap1
2.5. Putative Processing of Keap1 and Keap1ΔC to Yield Distinct Lengths of Isoforms
2.6. An Antagonist Effect of Keap1ΔC on Keap1-Mediated Turnover of Nrf2
3. Materials and Methods
3.1. Chemicals, Antibodies and Other Reagents
3.2. Cell Lines
3.3. Expression Constructs and Transfection
3.4. Luciferase Reporter Assay
3.5. Real-Time Quantitative PCR
3.6. Western Blotting
3.7. Pulse-Chase Experiments
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Gene Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural basis for defects of keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Maher, J.; Yamamoto, M. Select heterozygous Keap1 mutations have a dominant-negative effect on wild-type Keap1 in vivo. Cancer Res. 2011, 71, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Yoo, N.J.; Kim, H.R.; Kim, Y.R.; An, C.H.; Lee, S.H. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology 2012, 60, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Hast, B.E.; Cloer, E.W.; Goldfarb, D.; Li, H.; Siesser, P.F.; Yan, F.; Walter, V.; Zheng, N.; Hayes, D.N.; Major, M.B. Cancer-derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res. 2014, 74, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of Nf-E2-Related Factor-2 (Nrf2), a Nf-E2-like basic leucine-zipper transcriptional activator that binds to the tandem Nf-E2/Ap1 repeat of the beta-globin locus-control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived Cnc family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell. Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Lu, F.; Stewart, D.; Zhang, Y. Mechanisms underlying chemopreventive effects of flavonoids via multiple signaling nodes within Nrf2-ARE and AhR-XRE gene regulatory networks. Curr. Chem. Biol. 2013, 7, 151–176. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate for proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.Y.; Cooley, L. Kelch encodes a component of intercellular bridges in drosophila egg chambers. Cell 1993, 72, 681–693. [Google Scholar] [CrossRef]
- Zipper, L.M.; Mulcahy, R.T. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; de Galarreta, C.M.R.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, K.R.; Spitz, D.R.; Harris, S.; Nguyen, T.T.; Meredith, M.J.; Holt, J.T.; Gius, D.; Marnett, L.J.; Summar, M.L.; Freeman, M.L. Redox-sensitive interaction between KIAA0132 and Nrf2 mediates indomethacin-induced expression of gamma-glutamylcysteine synthetase. Free Radic. Biol. Med. 2002, 33, 149. [Google Scholar]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase(1) gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef] [PubMed]
- Nioi, P.; Hayes, J.D. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat. Res. 2004, 555, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Chew, E.H.; Go, M.L. Functionalized aurones as inducers of NAD(P)H:quinone oxidoreductase 1 that activate AhR/XRE and Nrf2/ARE signaling pathways: Synthesis, evaluation and SAR. Eur. J. Med. Chem. 2010, 45, 2957–2971. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, Thermodynamic, and structural characterizations of the association between Nrf2-DLGex eegron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Kombairaju, P.; Bodas, M.; Wu, H.; Bova, S.G.; Biswal, S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol. Cancer Ther. 2010, 9, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Boyden, L.M.; Choi, M.; Choate, K.A.; Nelson-Williams, C.J.; Farhi, A.; Toka, H.R.; Tikhonova, I.R.; Bjornson, R.; Mane, S.M.; Colussi, G.; et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012, 482, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, E.; Susa, K.; Mori, T.; Isobe, K.; Araki, Y.; Inoue, Y.; Yoshizaki, Y.; Ando, F.; Mori, Y.; Mandai, S.; et al. KLHL3 knockout mice reveal the physiological role of KLHL3 and the pathophysiology of pseudohypoaldosteronism type II caused by mutant KLHL3. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Poetsch, T.; Ju, J.H.; Eyler, D.E.; Dang, Y.J.; Bhat, S.; Merrick, W.C.; Green, R.; Shen, B.; Liu, J.O. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 2010, 6, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lucocq, J.M.; Yamamoto, M.; Hayes, J.D. The NHB1 (N-terminal homology box 1) sequence in transcription factor Nrf1 is required to anchor it to the endoplasmic reticulum and also to enable its asparagine-glycosylation. Biochem. J. 2007, 408, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Sihvola, V.; Levonen, A.L. Keap1 as the redox sensor of the antioxidant response. Arch. Biochem. Biophys. 2017, 617, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 system in cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| 1, The following primers used for expression plasmids | ||
| Keap1 | GCTTATCTTCTGGTACCCCATGCA | CAAGAAACTCGAGTTAACAGGTACAGTTCTGCTG |
| Keap1ΔC | GCTTATCTTCTGGTACCCCATGCA | ACTCATCCTCGAGTTATAGCCTCCTCTCCACACTG |
| Keap1N321 | GCTTATCTTCTGGTACCCCATGCA | GCGGCCCTCGAGTTACGCCCGGCAGGGCA |
| Keap1ΔN | GATGCCCGGTACCATGCCCAAGGTGGGCCG | CAAGAAACTCGAGTTAACAGGTACAGTTCTGCTG |
| Keap1:V5 | GCTTATCTTCTGGTACCCCATGCA | CAAGAAACTCGAGCGACAGGTACAGTTCTGCTG |
| Keap1ΔC:V5 | GCTTATCTTCTGGTACCCCATGCA | ACTCATCCTCGAGCGTAGCCTCCTCTCCACACTG |
| Keap1N321:V5 | GCTTATCTTCTGGTACCCCATGCA | GCGGCCCTCGAGCGCGCCCGGCAGGGCA |
| Keap1ΔN:V5 | GATGCCCGGTACCATGCCCAAGGTGGGCCG | CAAGAAACTCGAGCGACAGGTACAGTTCTGCTG |
| Nrf2 | GAGCCCGAATTCACGGTCCACAGCTC | AAAACTAGCTCGAGAAAGGTCAAATCCTCCT |
| V5 tag | CTAGCATGGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACGGA | AGCTTCCGTAGAATCGAGACCGAGGAGAGGGTTAGGGATAGGCTTACCCATG |
| 2, The following primers used for RT-qPCR analysis | ||
| HO1 | CAGAGCCTGGAAGACACCCTAA | AAACCACCCCAACCCTGCTAT |
| GCLM | GTGTGATGCCACCAGATTTGAC | CACAATGACCGAATACCGCAGT |
| NQO1 | AAGAAGAAAGGATGGGAGGTGG | GAACAGACTCGGCAGGATACTGA |
| Nrf2 | TCAGCGACGGAAAGAGTATGA | CCACTGGTTTCTGACTGGATGT |
| Keap1 | AACAACTCGCCCGACGGCAACAC | CATCCCGCTCTGGCTCATACCTC |
| Keap1ΔC | AACAACTCGCCCGACGGCAACAC | CGTGTGACCATCATAGCCTCCTC |
| β-actin | CATGTACGTTGCTATCCAGGC | CTCCTTAATGTCACGCACGAT |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, L.; Wang, M.; Zhu, Y.; Xiang, Y.; Zhang, Y. A Naturally-Occurring Dominant-Negative Inhibitor of Keap1 Competitively against Its Negative Regulation of Nrf2. Int. J. Mol. Sci. 2018, 19, 2150. https://doi.org/10.3390/ijms19082150
Qiu L, Wang M, Zhu Y, Xiang Y, Zhang Y. A Naturally-Occurring Dominant-Negative Inhibitor of Keap1 Competitively against Its Negative Regulation of Nrf2. International Journal of Molecular Sciences. 2018; 19(8):2150. https://doi.org/10.3390/ijms19082150
Chicago/Turabian StyleQiu, Lu, Meng Wang, Yuping Zhu, Yuancai Xiang, and Yiguo Zhang. 2018. "A Naturally-Occurring Dominant-Negative Inhibitor of Keap1 Competitively against Its Negative Regulation of Nrf2" International Journal of Molecular Sciences 19, no. 8: 2150. https://doi.org/10.3390/ijms19082150