Unique Ca2+-Cycling Protein Abundance and Regulation Sustains Local Ca2+ Releases and Spontaneous Firing of Rabbit Sinoatrial Node Cells

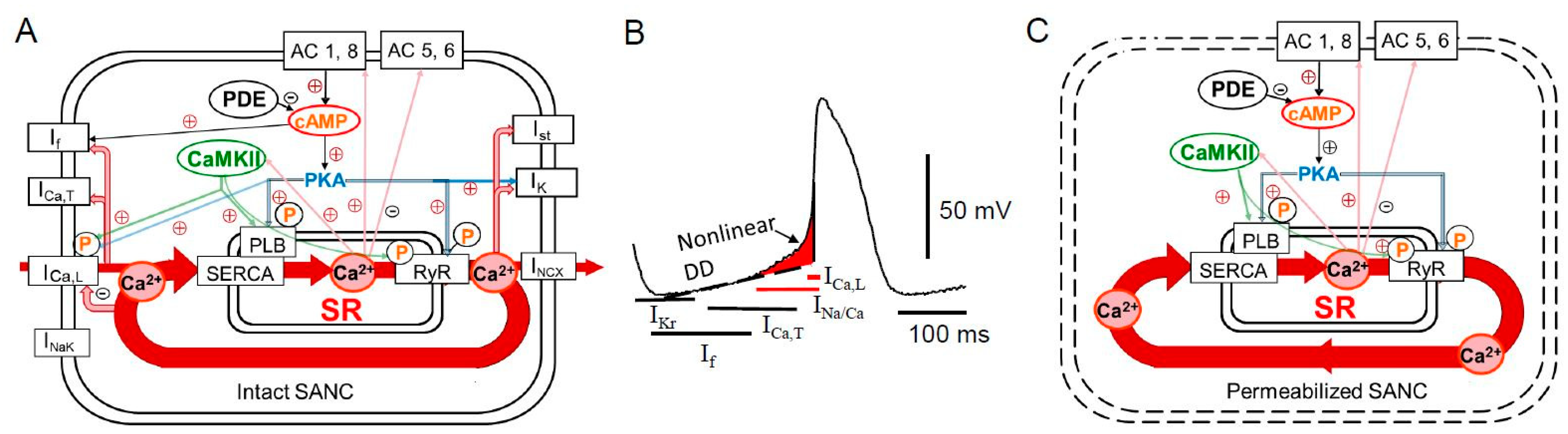{kind=link}
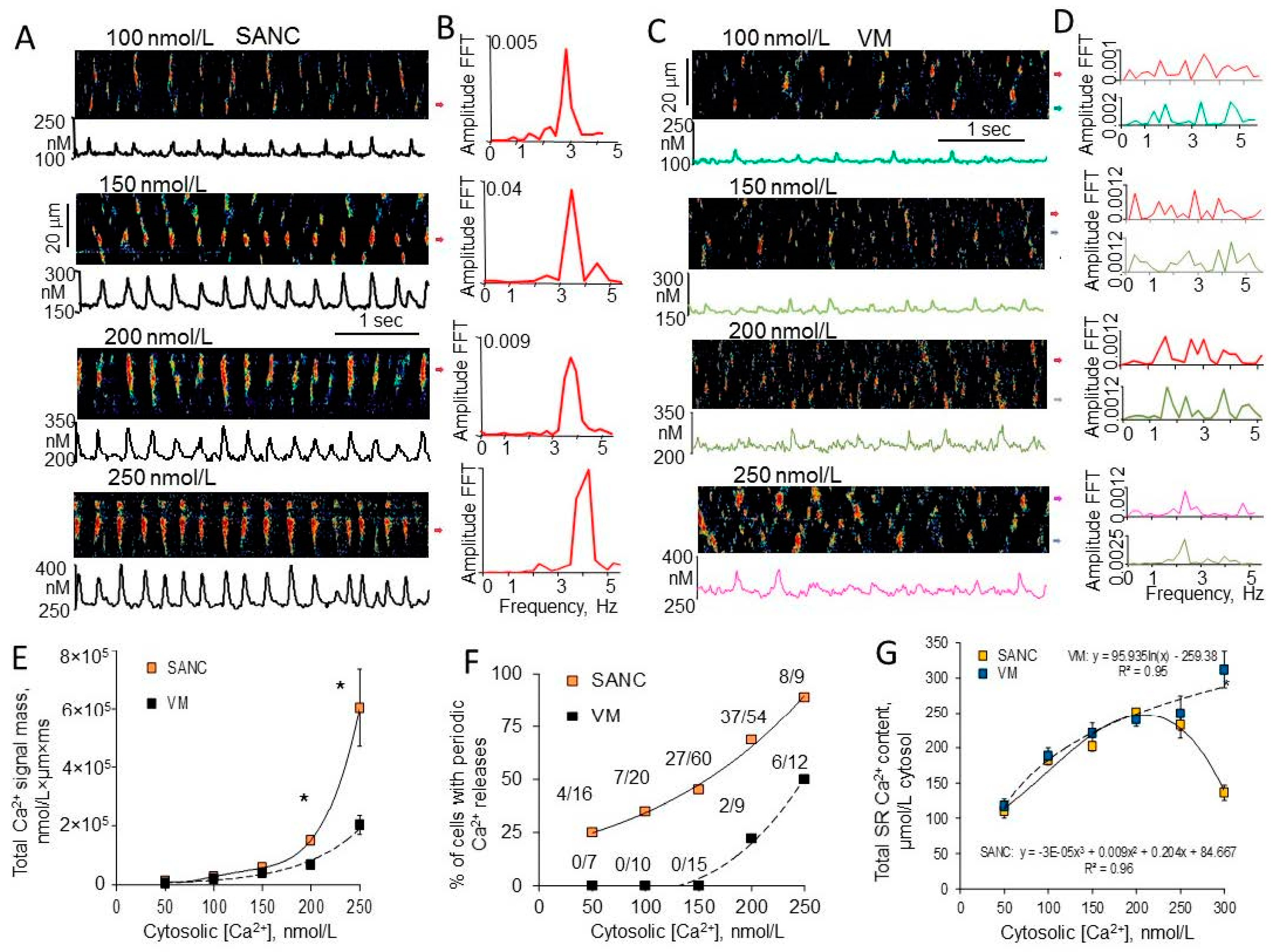{kind=link}
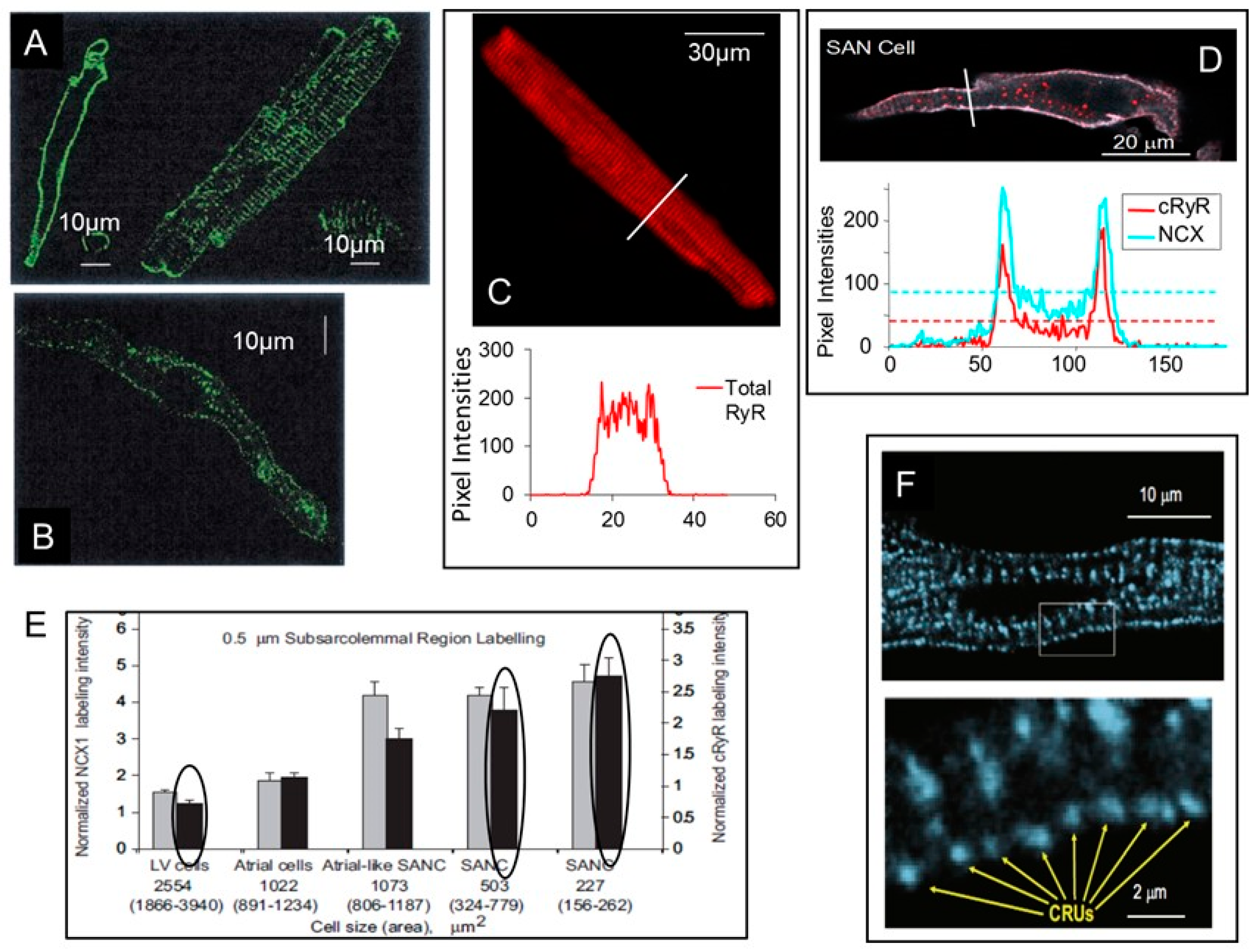{kind=link}
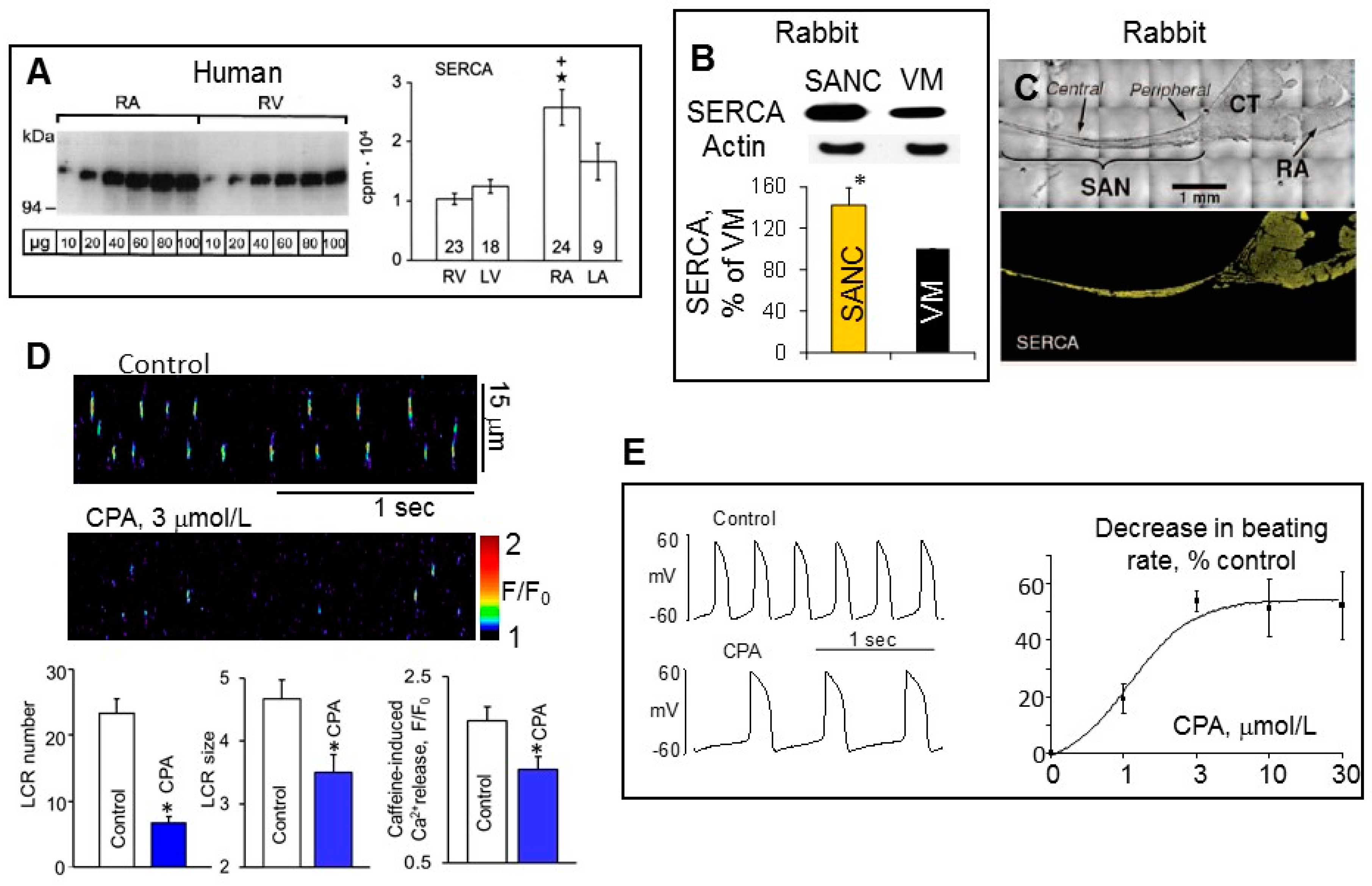{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Differences in Intrinsic SR Ca2+ Cycling between SANC and Ventricular Myocytes
3. Differences in RyR Abundance and Distribution in SANC and Ventricular Myocytes
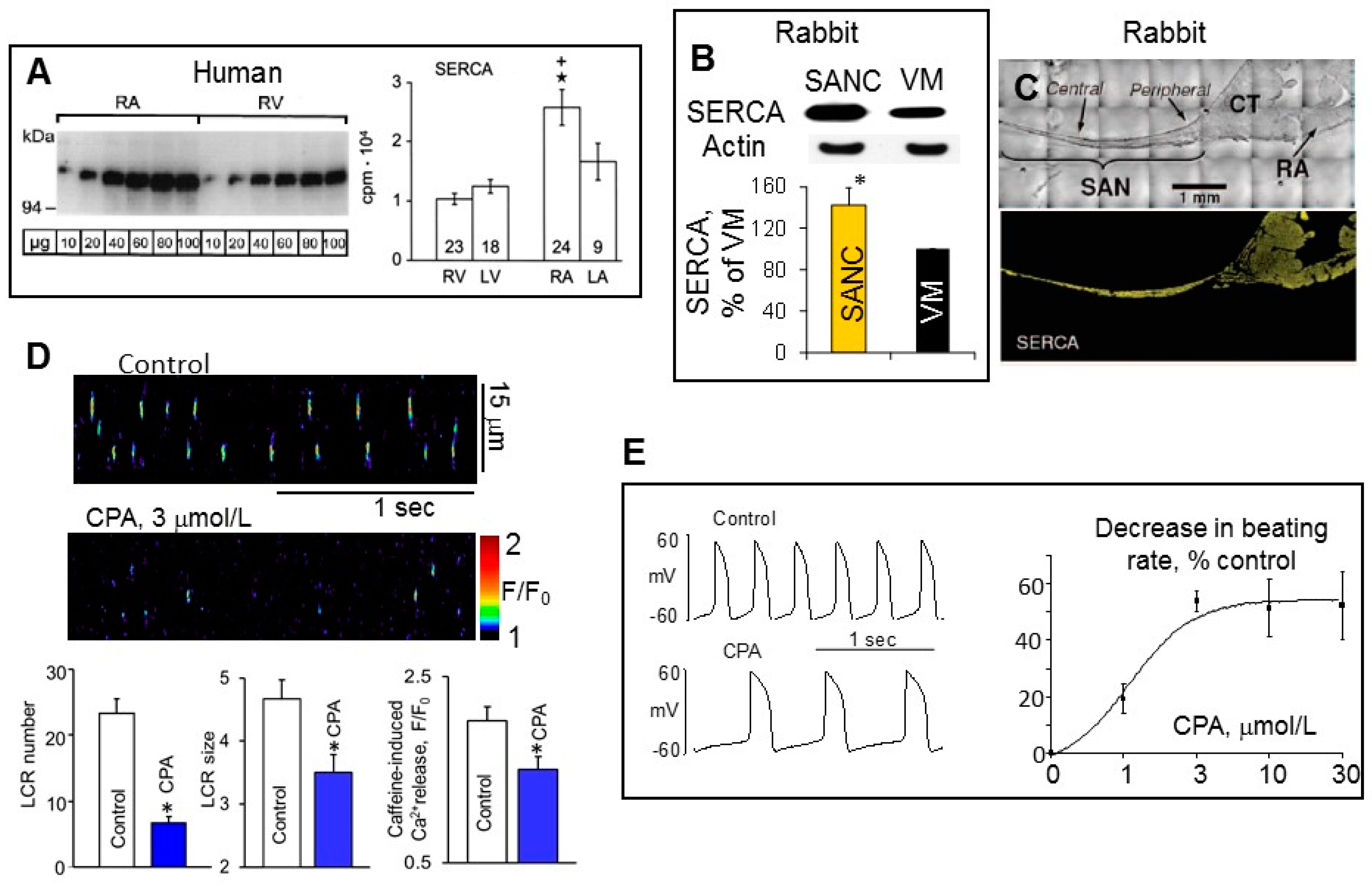
4. Differences in SERCA Abundance among SANC, Atrial and Ventricular Myocytes
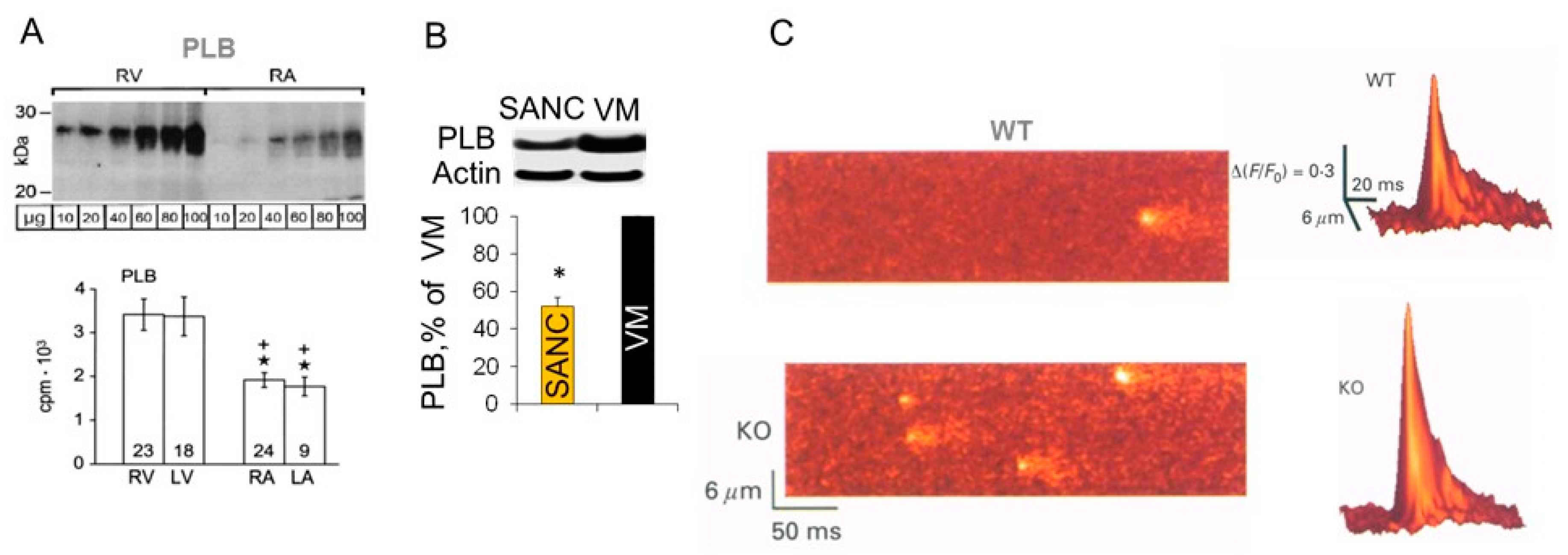
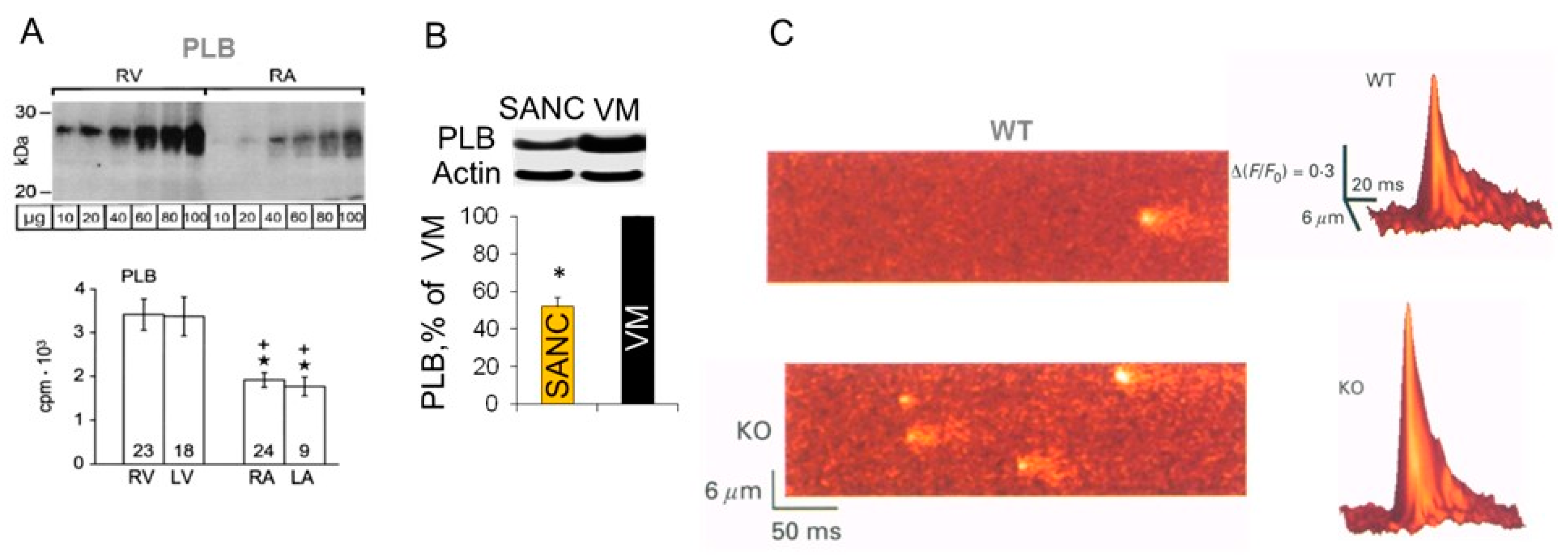
5. Differences in Phospholamban (PLB) Abundance in SANC and Ventricular Myocytes
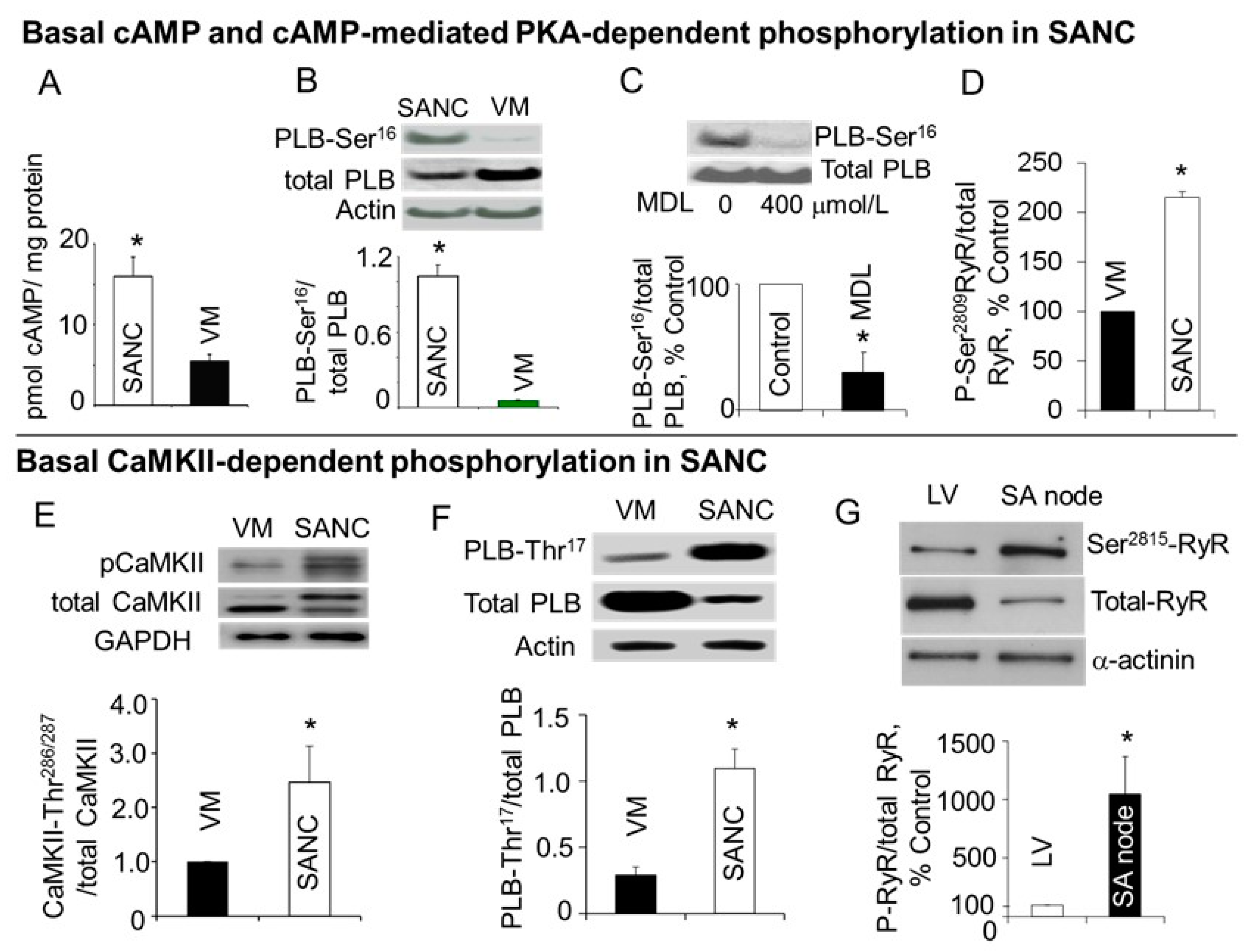
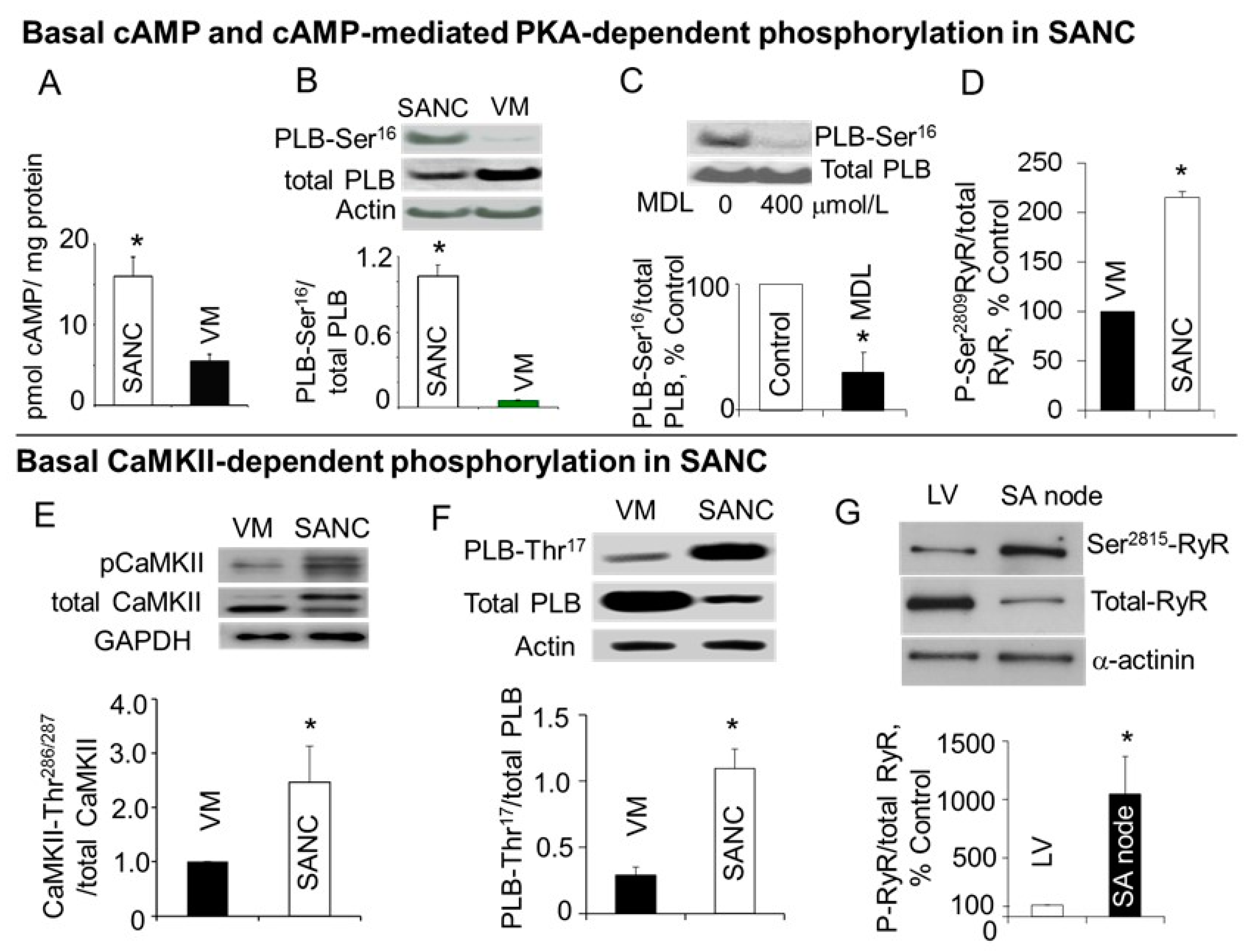
6. High Basal PKA- and CaMKII-Dependent Phosphorylation in Cardiac Pacemaker Cells
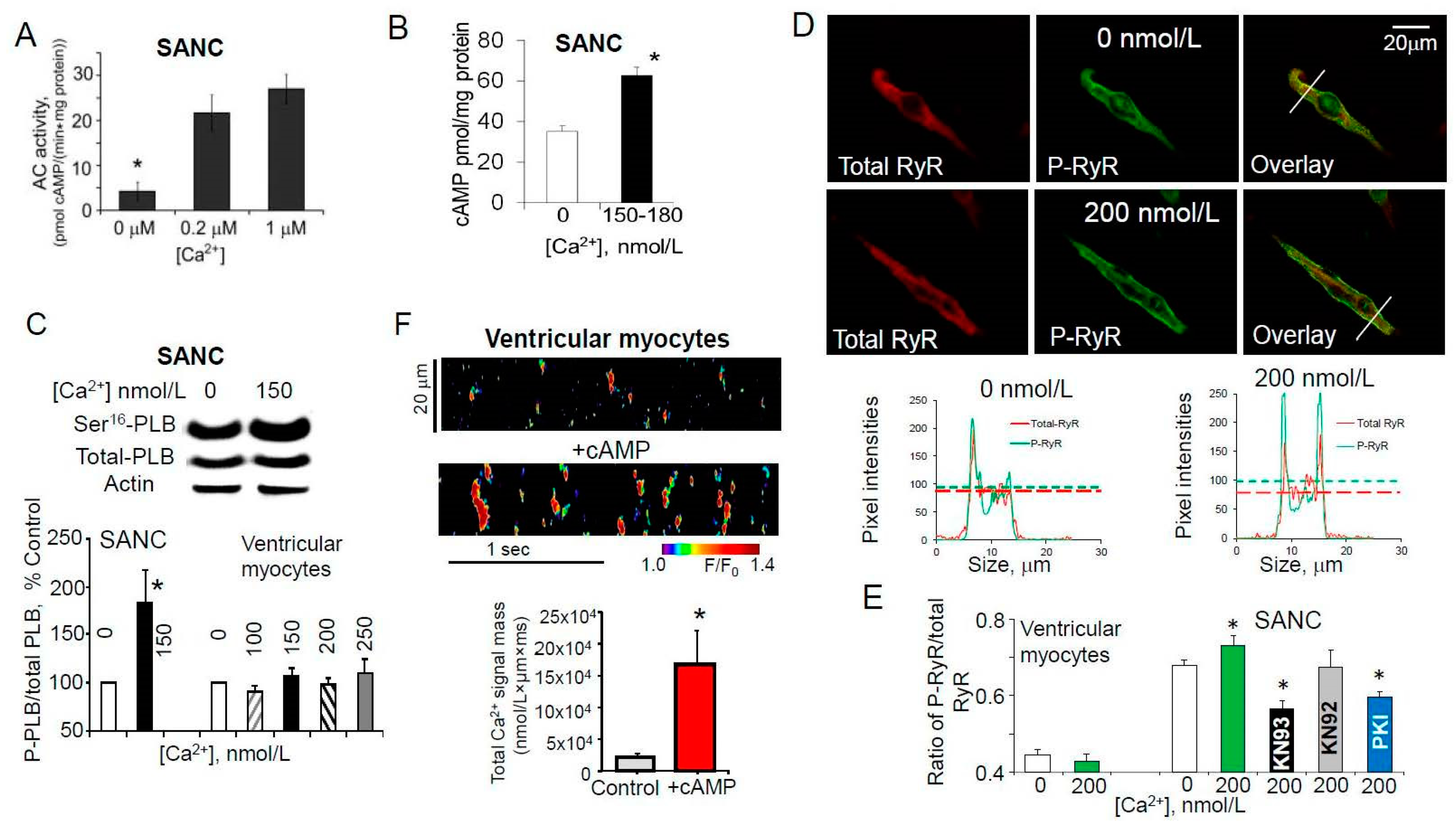
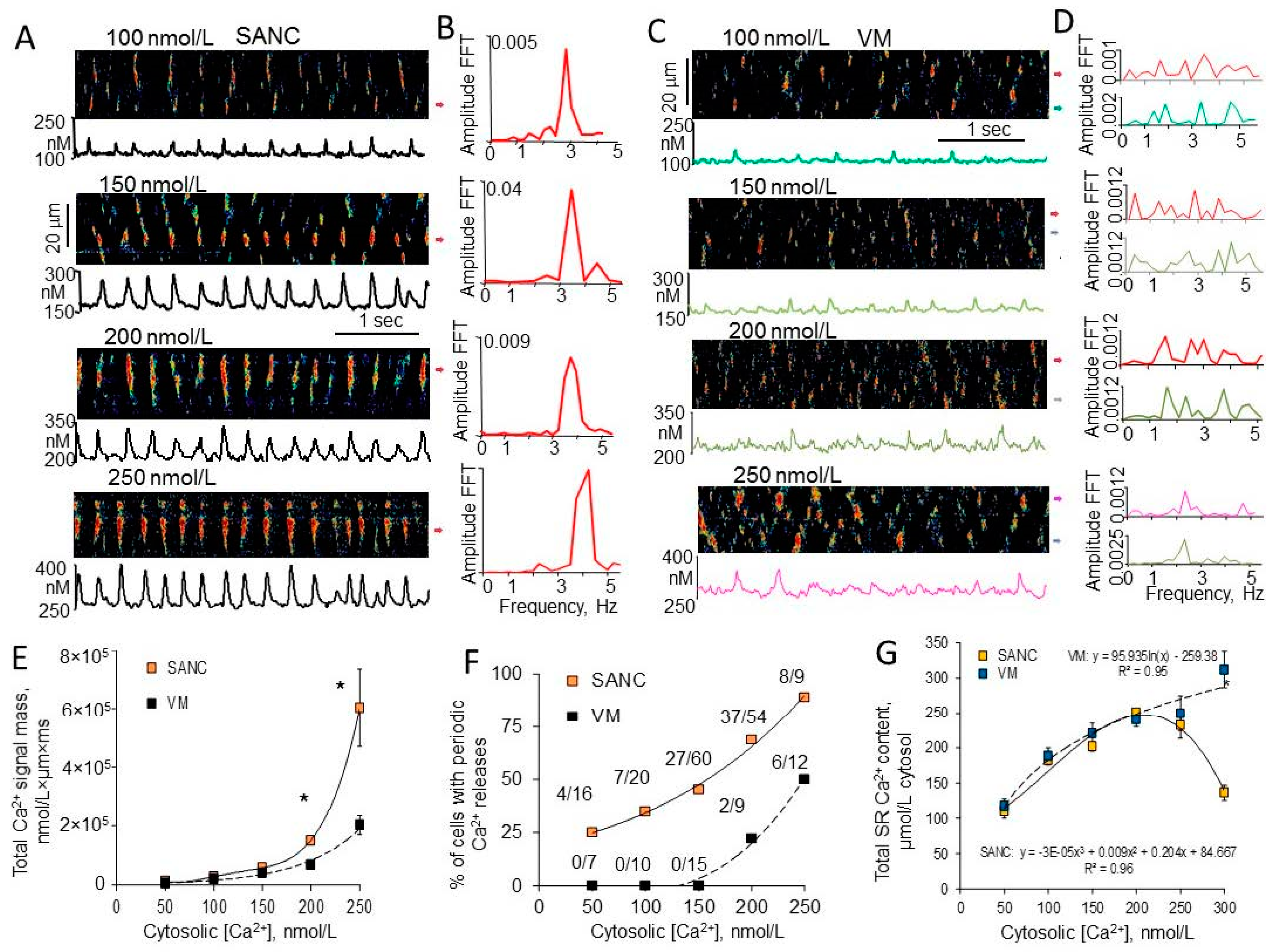
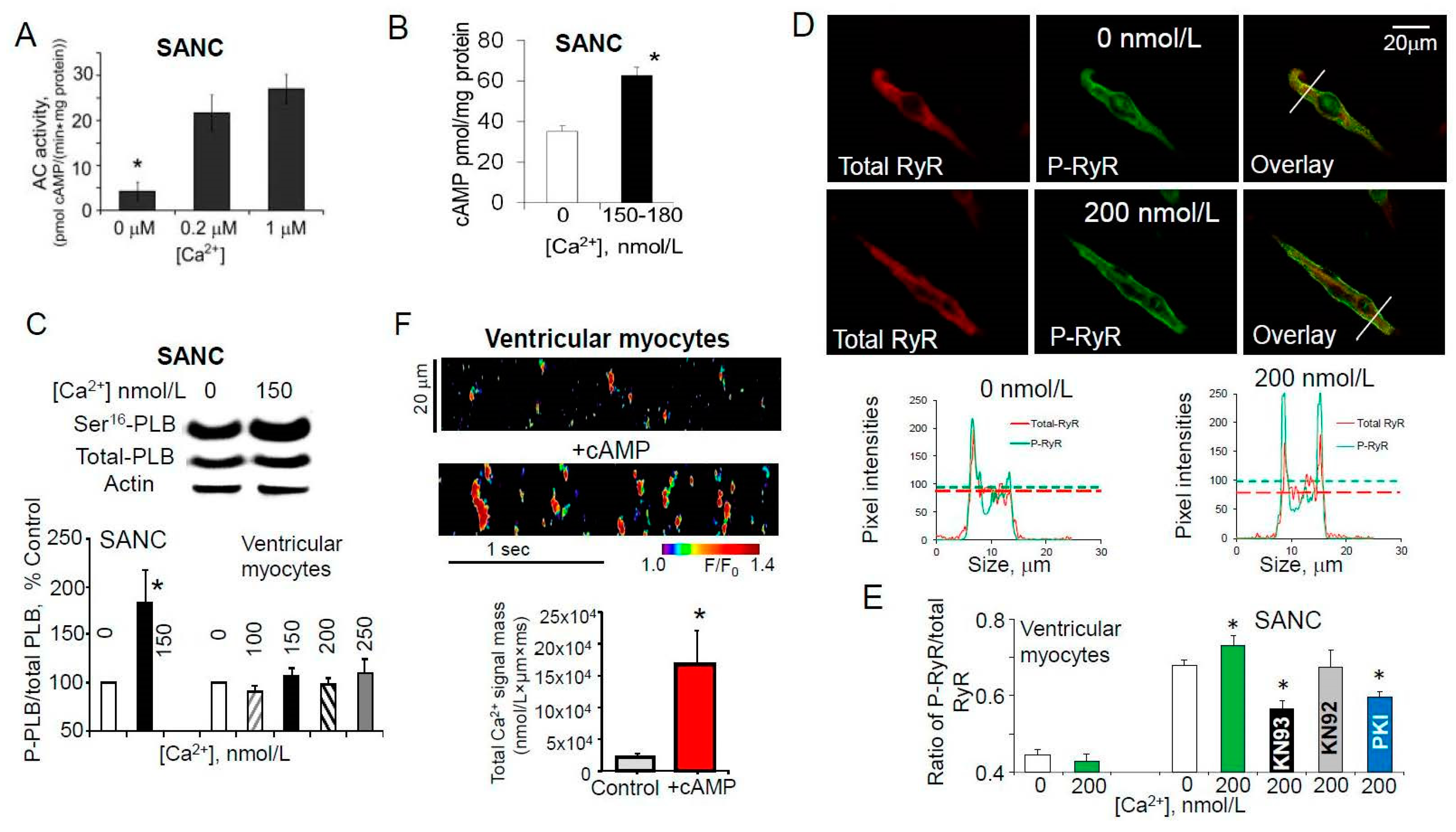
7. Ca2+-Dependent Phosphorylation of Both PLB and RyRs Was Associated with Amplified Ca2+ Release from SR
8. Conclusions
Funding
Conflicts of Interest
References
- Baruscotti, M.; Robinson, R.B. Electrophysiology and pacemaker function of the developing sinoatrial node. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2613–H2623. [Google Scholar] [CrossRef] [PubMed]
- Irisawa, H.; Brown, H.F.; Giles, W. Cardiac Pacemaking in the Sinoatrial Node. Physiol. Rev. 1993, 73, 197–227. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.E.; Nargeot, J. Genesis and regulation of the heart automaticity. Physiol. Rev. 2008, 88, 919–982. [Google Scholar] [CrossRef] [PubMed]
- Monfredi, O.; Dobrzynski, H.; Mondal, T.; Boyett, M.R.; Morris, G.M. The anatomy and physiology of the sinoatrial node--a contemporary review. Pacing Clin. Electrophysiol. 2010, 33, 1392–1406. [Google Scholar] [CrossRef] [PubMed]
- Cranefield, P. The Conduction of the Cardiac Impulse: The Slow Response and Cardiac Arrythmias, 1st ed.; Futura Comp: Mount Kisco, NY, USA, 1975. [Google Scholar]
- Seifen, E.; Schaer, H.; Marshall, J.M. Effect of Calcium on the Membrane Potentials of Single Pacemaker Fibres and Atrial Fibres in Isolated Rabbits Atria. Nature 1964, 202, 1223–1224. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, N.; Irisawa, H. Modulation by intracellular Ca2+ of the hyperpolarization-activated inward current in rabbit single sino-atrial node cells. J. Physiol. 1989, 409, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, N.; Irisawa, H.; Kameyama, M. Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J. Physiol. 1988, 395, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Heath, B.M.; Terrar, D.A. Protein kinase C enhances the rapidly activating delayed rectifier potassium current, IKr, through a reduction in C-type inactivation in guinea-pig ventricular myocytes. J. Physiol. 2000, 522, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Tohse, N. Calcium-sensitive delayed rectifier potassium current in guinea pig ventricular cells. Am. J. Physiol. 1990, 258, H1200–H1207. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Q.; Ono, K.I.; Noma, A.N. A Sustained Inward Current Activated at the Diastolic Potential Range in Rabbit Sinoatrial Node Cells. J. Physiol. 1995, 483, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Noda, T.; Nishimura, M.; Watanabe, Y. The role of Ca2+ release from sarcoplasmic reticulum in the regulation of sinoatrial node automaticity. Heart Vessel. 1996, 11, 234–241. [Google Scholar] [CrossRef]
- Huser, J.; Blatter, L.A.; Lipsius, S.L. Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J. Physiol. 2000, 524, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.K.; Allen, D.G. Intracellular calcium and Na+-Ca2+ exchange current in isolated toad pacemaker cells. J. Physiol. 1998, 508, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qu, J.; Nathan, R.D. Ionic basis of ryanodine’s negative chronotropic effect on pacemaker cells isolated from the sinoatrial node. Am. J. Physiol. 1997, 273, H2481–H2489. [Google Scholar] [CrossRef] [PubMed]
- Rigg, L.; Terrar, D.A. Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp. Physiol. 1996, 81, 877–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubenstein, D.S.; Lipsius, S.L. Mechanisms of automaticity in subsidiary pacemakers from cat right atrium. Circ. Res. 1989, 64, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H. Electrophysiological actions of ryanodine on single rabbit sinoatrial nodal cells. Gen. Pharmacol. 1997, 28, 31–38. [Google Scholar] [CrossRef]
- Bogdanov, K.Y.; Vinogradova, T.M.; Lakatta, E.G. Sinoatrial nodal cell ryanodine receptor and Na(+)-Ca(2+) exchanger: Molecular partners in pacemaker regulation. Circ. Res. 2001, 88, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Lipsius, S.L.; Huser, J.; Blatter, L.A. Intracellular Ca2+ release sparks atrial pacemaker activity. Physiology 2001, 16, 101–106. [Google Scholar] [CrossRef]
- Rigg, L.; Heath, B.M.; Cui, Y.; Terrar, D.A. Localisation and functional significance of ryanodine receptors during beta-adrenoceptor stimulation in the guinea-pig sino-atrial node. Cardiovasc. Res. 2000, 48, 254–264. [Google Scholar] [CrossRef]
- Sanders, L.; Rakovic, S.; Lowe, M.; Mattick, P.A.; Terrar, D.A. Fundamental importance of Na+-Ca2+ exchange for the pacemaking mechanism in guinea-pig sino-atrial node. J. Physiol. 2006, 571, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, T.M.; Zhou, Y.Y.; Maltsev, V.; Lyashkov, A.; Stern, M.; Lakatta, E.G. Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ. Res. 2004, 94, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, V.A.; Vinogradova, T.M.; Lakatta, E.G. The emergence of a general theory of the initiation and strength of the heartbeat. J. Pharmacol. Sci. 2006, 100, 338–369. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Vinogradova, T.; Lyashkov, A.; Sirenko, S.; Zhu, W.; Ruknudin, A.; Maltsev, V.A. The integration of spontaneous intracellular Ca2+ cycling and surface membrane ion channel activation entrains normal automaticity in cells of the heart’s pacemaker. Ann. N. Y. Acad. Sci. 2006, 1080, 178–206. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, V.A.; Lakatta, E.G. Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H594–H615. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd ed.; Kluwer Academic: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Lakatta, E.G.; Maltsev, V.A.; Vinogradova, T.M. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 2010, 106, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Joung, B.; Tang, L.; Maruyama, M.; Han, S.; Chen, Z.; Stucky, M.; Jones, L.R.; Fishbein, M.C.; Weiss, J.N.; Chen, P.S.; et al. Intracellular calcium dynamics and acceleration of sinus rhythm by beta-adrenergic stimulation. Circulation 2009, 119, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Sirenko, S.G.; Yang, D.; Maltseva, L.A.; Kim, M.S.; Lakatta, E.G.; Maltsev, V.A. Spontaneous, local diastolic subsarcolemmal calcium releases in single, isolated guinea-pig sinoatrial nodal cells. PLoS ONE 2017, 12, e0185222. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Gao, Z.; Chen, B.Y.; Koval, O.M.; Singh, M.V.; Guan, X.Q.; Hund, T.J.; Kutschke, W.; Sarma, S.; Grumbach, I.M.; et al. Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc. Natl. Acad. Sci. USA 2009, 106, 5972–5977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsui, K.; Monfredi, O.J.; Sirenko-Tagirova, S.G.; Maltseva, L.A.; Bychkov, R.; Kim, M.S.; Ziman, B.D.; Tarasov, K.V.; Tarasova, Y.S.; Zhang, J.; et al. A coupled-clock system drives the automaticity of human sinoatrial nodal pacemaker cells. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Choate, J.K.; Feldman, R. Neuronal control of heart rate in isolated mouse atria. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1340–H1346. [Google Scholar] [CrossRef] [PubMed]
- Meissner, G. Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J. Biol. Chem. 1986, 261, 6300–6306. [Google Scholar] [PubMed]
- Sirenko, S.; Yang, D.; Li, Y.; Lyashkov, A.E.; Lukyanenko, Y.O.; Lakatta, E.G.; Vinogradova, T.M. Ca2+-dependent phosphorylation of Ca2+ cycling proteins generates robust rhythmic local Ca2+ releases in cardiac pacemaker cells. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, T.M.; Brochet, D.X.; Sirenko, S.; Li, Y.; Spurgeon, H.; Lakatta, E.G. Sarcoplasmic reticulum Ca2+ pumping kinetics regulates timing of local Ca2+ releases and spontaneous beating rate of rabbit sinoatrial node pacemaker cells. Circ. Res. 2010, 107, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Lyashkov, A.E.; Beahr, J.; Lakatta, E.G.; Yaniv, Y.; Maltsev, V.A. Positive Feedback Mechanisms among Local Ca Releases, NCX, and I-CaL Ignite Pacemaker Action Potentials. Biophys. J. 2018, 114, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, Y.; Sirenko, S.; Ziman, B.D.; Spurgeon, H.A.; Maltsev, V.A.; Lakatta, E.G. New evidence for coupled clock regulation of the normal automaticity of sinoatrial nodal pacemaker cells: Bradycardic effects of ivabradine are linked to suppression of intracellular Ca2+ cycling. J. Mol. Cell. Cardiol. 2013, 62, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sirenko, S.; Riordon, D.R.; Yang, D.; Spurgeon, H.; Lakatta, E.G.; Vinogradova, T.M. CaMKII-dependent phosphorylation regulates basal cardiac pacemaker function via modulation of local Ca2+ releases. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H532–H544. [Google Scholar] [CrossRef] [PubMed]
- Yavari, A.; Bellahcene, M.; Bucchi, A.; Sirenko, S.; Pinter, K.; Herring, N.; Jung, J.J.; Tarasov, K.V.; Sharpe, E.J.; Wolfien, M.; et al. Mammalian gamma 2 AMPK regulates intrinsic heart rate. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Demion, M.; Bois, P.; Launay, P.; Guinamard, R. TRPM4, a Ca2+-activated nonselective cation channel in mouse sino-atrial node cells. Cardiovasc. Res. 2007, 73, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Hof, T.; Simard, C.; Rouet, R.; Salle, L.; Guinamard, R. Implication of the TRPM4 nonselective cation channel in mammalian sinus rhythm. Heart Rhythm 2013, 10, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, N.; Tran, A.; Kang, J.; Zhang, R.; Philipson, K.D.; Goldhaber, J.I. Regulation of calcium clock-mediated pacemaking by inositol-1,4,5-trisphosphate receptors in mouse sinoatrial nodal cells. J. Physiol. 2015, 593, 2649–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrente, A.G.; Zhang, R.; Wang, H.; Zaini, A.; Kim, B.; Yue, X.; Philipson, K.D.; Goldhaber, J.I. Contribution of small conductance K(+) channels to sinoatrial node pacemaker activity: Insights from atrial-specific Na(+)/Ca(2+) exchange knockout mice. J. Physiol. 2017, 595, 3847–3865. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, Y.; Spurgeon, H.A.; Lyashkov, A.E.; Yang, D.M.; Ziman, B.D.; Maltsev, V.A.; Lakatta, E.G. Crosstalk between Mitochondrial and Sarcoplasmic Reticulum Ca2+ Cycling Modulates Cardiac Pacemaker Cell Automaticity. PLoS ONE 2012, 7, e37582. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, Y.; Spurgeon, H.A.; Ziman, B.D.; Lyashkov, A.E.; Lakatta, E.G. Mechanisms that match ATP supply to demand in cardiac pacemaker cells during high ATP demand. Am. J. Physiol.-Heart Circ. Physiol. 2013, 304, H1428–H1438. [Google Scholar] [CrossRef] [PubMed]
- Allah, E.A.; Tellez, J.O.; Yanni, J.; Nelson, T.; Monfredi, O.; Boyett, M.R.; Dobrzynski, H. Changes in the expression of ion channels, connexins and Ca2+-handling proteins in the sino-atrial node during postnatal development. Exp. Physiol. 2011, 96, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Chandler, N.J.; Greener, I.D.; Tellez, J.O.; Inada, S.; Musa, H.; Molenaar, P.; DiFrancesco, D.; Baruscotti, M.; Longhi, R.; Anderson, R.H.; et al. Molecular Architecture of the Human Sinus Node Insights Into the Function of the Cardiac Pacemaker. Circulation 2009, 119, 1562–1575. [Google Scholar] [CrossRef] [PubMed]
- Marionneau, C.; Couette, B.; Liu, J.; Li, H.; Mangoni, M.E.; Nargeot, J.; Lei, M.; Escande, D.; Demolombe, S. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J. Physiol. 2005, 562, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Musa, H.; Lei, M.; Honjo, H.; Jones, S.A.; Dobrzynski, H.; Lancaster, M.K.; Takagishi, Y.; Henderson, Z.; Kodama, I.; Boyett, M.R. Heterogeneous expression of Ca(2+) handling proteins in rabbit sinoatrial node. J. Histochem. Cytochem. 2002, 50, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H. Sino-atrial nodal cells of mammalian hearts: Ionic currents and gene expression of pacemaker ionic channels. J. Smooth Muscle Res. 2003, 39, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Tellez, J.O.; Dobrzynski, H.; Greener, I.D.; Graham, G.M.; Laing, E.; Honjo, H.; Hubbard, S.J.; Boyett, M.R.; Billeter, R. Differential expression of ion channel transcripts in atrial muscle and sinoatrial node in rabbit. Circ. Res. 2006, 99, 1384–1393. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Fermini, B.; Nattel, S. Potassium channel blocking properties of propafenone in rabbit atrial myocytes. J. Pharmacol. Exp. Ther. 1993, 264, 1113–1123. [Google Scholar] [PubMed]
- Klimek-Piotrowska, W.; Krawczyk-Ozog, A.; Suski, M.; Kapusta, P.; Wolkow, P.P.; Holda, M.K. Comparative iTRAQ analysis of protein abundance in the human sinoatrial node and working cardiomyocytes. J. Anat. 2018, 232, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Christoffels, V.M.; Smits, G.J.; Kispert, A.; Moorman, A.F. Development of the pacemaker tissues of the heart. Circ. Res. 2010, 106, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Gourdie, R.G.; Harris, B.S.; Bond, J.; Justus, C.; Hewett, K.W.; O’Brien, T.X.; Thompson, R.P.; Sedmera, D. Development of the cardiac pacemaking and conduction system. Birth Defects Res. C Embryo Today 2003, 69, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Mommersteeg, M.T.; Hoogaars, W.M.; Prall, O.W.; de Gier-de Vries, C.; Wiese, C.; Clout, D.E.; Papaioannou, V.E.; Brown, N.A.; Harvey, R.P.; Moorman, A.F.; et al. Molecular pathway for the localized formation of the sinoatrial node. Circ. Res. 2007, 100, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Wiese, C.; Grieskamp, T.; Airik, R.; Mommersteeg, M.T.; Gardiwal, A.; de Gier-de Vries, C.; Schuster-Gossler, K.; Moorman, A.F.; Kispert, A.; Christoffels, V.M. Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ. Res. 2009, 104, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Lederer, W.J.; Cannell, M.B. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; van Borren, M.M.; Wilders, R. Calcium transient and sodium-calcium exchange current in human versus rabbit sinoatrial node pacemaker cells. ScientificWorldJournal 2013, 2013, 507872. [Google Scholar] [CrossRef] [PubMed]
- Lyashkov, A.E.; Juhaszova, M.; Dobrzynski, H.; Vinogradova, T.M.; Maltsev, V.A.; Juhasz, O.; Spurgeon, H.A.; Sollott, S.J.; Lakatta, E.G. Calcium cycling protein density and functional importance to automaticity of isolated sinoatrial nodal cells are independent of cell size. Circ. Res. 2007, 100, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.D.; Maltseva, L.A.; Juhaszova, M.; Sollott, S.J.; Lakatta, E.G.; Maltsev, V.A. Hierarchical clustering of ryanodine receptors enables emergence of a calcium clock in sinoatrial node cells. J. Gen. Physiol. 2014, 143, 577–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukyanenko, V.; Gyorke, I.; Subramanian, S.; Smirnov, A.; Wiesner, T.F.; Gyorke, S. Inhibition of Ca2+ sparks by ruthenium red in permeabilized rat ventricular myocytes. Biophys. J. 2000, 79, 1273–1284. [Google Scholar] [CrossRef]
- Lukyanenko, V.; Gyorke, S. Ca2+ sparks and Ca2+ waves in saponin-permeabilized rat ventricular myocytes. J. Physiol. 1999, 521, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.V.; Bovo, E.; Bers, D.M.; Blatter, L.A. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J. Physiol. 2010, 588, 4743–4757. [Google Scholar] [CrossRef] [PubMed]
- Sirenko, S.; Maltsev, V.A.; Maltseva, L.A.; Yang, D.; Lukyanenko, Y.; Vinogradova, T.M.; Jones, L.R.; Lakatta, E.G. Sarcoplasmic reticulum Ca cycling protein phosphorylation in a physiologic Ca milieu unleashes a high-power, rhythmic Ca clock in ventricular myocytes: Relevance to arrhythmias and bio-pacemaker design. J. Mol. Cell. Cardiol. 2014, 66C, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Fabiato, A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 1985, 85, 247–289. [Google Scholar] [CrossRef] [PubMed]
- Bassani, J.W.; Bassani, R.A.; Bers, D.M. Calibration of indo-1 and resting intracellular [Ca]i in intact rabbit cardiac myocytes. Biophys. J. 1995, 68, 1453–1460. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, A.V.; Maltsev, V.A.; Mikheev, M.; Maltseva, L.A.; Sirenko, S.G.; Lakatta, E.G.; Stern, M.D. Synchronization of stochastic Ca2+ release units creates a rhythmic Ca2+ clock in cardiac pacemaker cells. Biophys. J. 2011, 100, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.T.; Blatter, L.A. A novel mechanism of tandem activation of ryanodine receptors by cytosolic and SR luminal Ca(2+) during excitation-contraction coupling in atrial myocytes. J. Physiol. 2017, 595, 3835–3845. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Bhupathy, P.; Babu, G.J. Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc. Res. 2008, 77, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.F.; Bolck, B.; Erdmann, E.; Schwinger, R.H. Sarcoplasmic reticulum Ca2+-ATPase modulates cardiac contraction and relaxation. Cardiovasc. Res. 2003, 57, 20–27. [Google Scholar] [CrossRef]
- Luss, I.; Boknik, P.; Jones, L.R.; Kirchhefer, U.; Knapp, J.; Linck, B.; Luss, H.; Meissner, A.; Muller, F.U.; Schmitz, W.; et al. Expression of cardiac calcium regulatory proteins in atrium v ventricle in different species. J. Mol. Cell. Cardiol. 1999, 31, 1299–1314. [Google Scholar] [CrossRef] [PubMed]
- Minajeva, A.; Kaasik, A.; Paju, K.; Seppet, E.; Lompre, A.M.; Veksler, V.; Ventura-Clapier, R. Sarcoplasmic reticulum function in determining atrioventricular contractile differences in rat heart. Am. J. Physiol. 1997, 273, H2498–H2507. [Google Scholar] [CrossRef] [PubMed]
- Boknik, P.; Unkel, C.; Kirchhefer, U.; Kleideiter, U.; Klein-Wiele, O.; Knapp, J.; Linck, B.; Luss, H.; Muller, F.U.; Schmitz, W.; et al. Regional expression of phospholamban in the human heart. Cardiovasc. Res. 1999, 43, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Eisner, D.; Bode, E.; Venetucci, L.; Trafford, A. Calcium flux balance in the heart. J. Mol. Cell. Cardiol. 2013, 58, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Goeger, D.E.; Riley, R.T.; Dorner, J.W.; Cole, R.J. Cyclopiazonic acid inhibition of the Ca2+-transport ATPase in rat skeletal muscle sarcoplasmic reticulum vesicles. Biochem. Pharmacol. 1988, 37, 978–981. [Google Scholar] [CrossRef]
- Kranias, E.G.; Hajjar, R.J. Modulation of Cardiac Contractility by the Phopholamban/SERCA2a Regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.L.; Hashimoto, K.; Grupp, I.L.; Ji, Y.; Reed, T.; Loukianov, E.; Grupp, G.; Bhagwhat, A.; Hoit, B.; Walsh, R.; et al. Targeted overexpression of the sarcoplasmic reticulum Ca2+-ATPase increases cardiac contractility in transgenic mouse hearts. Circ. Res. 1998, 83, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.B.; Birkeland, J.A.; Finsen, A.V.; Louch, W.E.; Sjaastad, I.; Wang, Y.; Chen, J.; Molkentin, J.D.; Chien, K.R.; Sejersted, O.M.; et al. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J. Mol. Cell. Cardiol. 2009, 47, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Louch, W.E.; Hougen, K.; Mork, H.K.; Swift, F.; Aronsen, J.M.; Sjaastad, I.; Reims, H.M.; Roald, B.; Andersson, K.B.; Christensen, G.; et al. Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J. Physiol. 2010, 588, 465–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, B.S.; Meldrum, D.R.; Joo, K.S.; Wang, J.F.; Meng, X.; Cleveland, J.C., Jr.; Banerjee, A.; Harken, A.H. Human SERCA2a levels correlate inversely with age in senescent human myocardium. J. Am. Coll. Cardiol. 1998, 32, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Lakatta, E.G. Myocardial adaptations in advanced age. Basic Res. Cardiol. 1993, 88, 125–133. [Google Scholar] [PubMed]
- Lompre, A.M.; Lambert, F.; Lakatta, E.G.; Schwartz, K. Expression of sarcoplasmic reticulum Ca(2+)-ATPase and calsequestrin genes in rat heart during ontogenic development and aging. Circ. Res. 1991, 69, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sirenko, S.; Juhaszova, M.; Sollott, S.J.; Shukla, S.; Yaniv, Y.; Lakatta, E.G. Age-associated abnormalities of intrinsic automaticity of sinoatrial nodal cells are linked to deficient cAMP-PKA-Ca(2+) signaling. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1385–H1397. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Alpert, N.R.; Maclennan, D.H.; Barton, P.; Periasamy, M. Alterations in Sarcoplasmic-Reticulum Gene-Expression in Human Heart-Failure—A Possible Mechanism for Alterations in Systolic and Diastolic Properties of the Failing Myocardium. Circ. Res. 1993, 72, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Matsui, H.; Periasamy, M. Sarcoplasmic reticulum gene expression in cardiac hypertrophy and heart failure. Circ. Res. 1994, 74, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Hasenfuss, G.; Meyer, M.; Schillinger, W.; Preuss, M.; Pieske, B.; Just, H. Calcium handling proteins in the failing human heart. Basic Res. Cardiol. 1997, 92, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Hasenfuss, G. Alterations of calcium-regulatory proteins in heart failure. Cardiovasc. Res. 1998, 37, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Winslow, R.L.; Rice, J.; Jafri, S.; Marban, E.; O’Rourke, B. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: Model studies. Circ. Res. 1999, 84, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Huke, S. SERCA pump level is a critical determinant of Ca2+ homeostasis and cardiac contractility. J. Mol. Cell. Cardiol. 2001, 33, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Sakata, S.; Lebeche, D.; Sakata, N.; Sakata, Y.; Chemaly, E.R.; Liang, L.F.; Tsuji, T.; Takewa, Y.; del Monte, F.; Peluso, R.; et al. Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J. Mol. Cell. Cardiol. 2007, 42, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Beeri, R.; Chaput, M.; Guerrero, J.L.; Kawase, Y.; Yosefy, C.; Abedat, S.; Karakikes, I.; Morel, C.; Tisosky, A.; Sullivan, S.; et al. Gene delivery of sarcoplasmic reticulum calcium ATPase inhibits ventricular remodeling in ischemic mitral regurgitation. Circ. Heart Fail. 2010, 3, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.A.; Smolic, A.; Preovolos, A.; Byrne, M.J.; Power, J.M.; Kaye, D.M. Augmentation of left ventricular mechanics by recirculation-mediated AAV2/1-SERCA2a gene delivery in experimental heart failure. Eur. J. Heart Fail. 2011, 13, 247–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papolos, A.; Frishman, W.H. Sarcoendoplasmic reticulum calcium transport ATPase 2a: A potential gene therapy target in heart failure. Cardiol. Rev. 2013, 21, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Laraia, P.J.; Morkin, E. Adenosine 3′,5′-Monophosphate-Dependent Membrane Phosphorylation—Possible Mechanism for Control of Microsomal Calcium-Transport in Heart-Muscle. Circ. Res. 1974, 35, 298–306. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell. Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Wolska, B.M.; Grupp, I.L.; Harrer, J.M.; Haghighi, K.; Ferguson, D.G.; Slack, J.P.; Grupp, G.; Doetschman, T.; Solaro, R.J.; et al. Phospholamban gene dosage effects in the mammalian heart. Circ. Res. 1996, 78, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Santana, L.F.; Kranias, E.G.; Lederer, W.J. Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. J. Physiol. 1997, 503, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Koss, K.L.; Grupp, I.L.; Kranias, E.G. The relative phospholamban and SERCA2 ratio: A critical determinant of myocardial contractility. Basic Res. Cardiol. 1997, 92, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Walden, A.P.; Dibb, K.M.; Trafford, A.W. Differences in intracellular calcium homeostasis between atrial and ventricular myocytes. J. Mol. Cell. Cardiol. 2009, 46, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Wolska, B.M.; Stojanovic, M.O.; Luo, W.; Kranias, E.G.; Solaro, R.J. Effect of ablation of phospholamban on dynamics of cardiac myocyte contraction and intracellular Ca2+. Am. J. Physiol. 1996, 271, C391–C397. [Google Scholar] [CrossRef] [PubMed]
- Del Monte, F.; Harding, S.E.; Dec, G.W.; Gwathmey, J.K.; Hajjar, R.J. Targeting phospholamban by gene transfer in human heart failure. Circulation 2002, 105, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W.; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghighi, K.; Kolokathis, F.; Pater, L.; Lynch, R.A.; Asahi, M.; Gramolini, A.O.; Fan, G.C.; Tsiapras, D.; Hahn, H.S.; Adamopoulos, S.; et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J. Clin. Investig. 2003, 111, 869–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradova, T.M.; Lyashkov, A.E.; Zhu, W.; Ruknudin, A.M.; Sirenko, S.; Yang, D.; Deo, S.; Barlow, M.; Johnson, S.; Caffrey, J.L.; et al. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ. Res. 2006, 98, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Katsushika, S.; Chen, L.; Kawabe, J.; Nilakantan, R.; Halnon, N.J.; Homcy, C.J.; Ishikawa, Y. Cloning and characterization of a sixth adenylyl cyclase isoform: Types V and VI constitute a subgroup within the mammalian adenylyl cyclase family. Proc. Natl. Acad. Sci. USA 1992, 89, 8774–8778. [Google Scholar] [CrossRef] [PubMed]
- Premont, R.T.; Chen, J.; Ma, H.W.; Ponnapalli, M.; Iyengar, R. Two members of a widely expressed subfamily of hormone-stimulated adenylyl cyclases. Proc. Natl. Acad. Sci. USA 1992, 89, 9809–9813. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, M.; Cooper, D.M.F. Cloning and Expression of a Ca2+-Inhibitable Adenylyl Cyclase from Ncb-20 Cells. Proc. Natl. Acad. Sci. USA 1992, 89, 6716–6720. [Google Scholar] [CrossRef] [PubMed]
- Mattick, P.; Parrington, J.; Odia, E.; Simpson, A.; Collins, T.; Terrar, D. Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the If pacemaker current. J. Physiol. 2007, 582, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Lyashkov, A.E.; Graham, D.; Sheydina, A.; Volkova, M.V.; Mitsak, M.; Vinogradova, T.M.; Lukyanenko, Y.O.; Li, Y.; Ruknudin, A.M.; et al. Ca2+ -stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J. Biol. Chem. 2008, 283, 14461–14468. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, T.M.; Zhou, Y.Y.; Bogdanov, K.Y.; Yang, D.; Kuschel, M.; Cheng, H.; Xiao, R.P. Sinoatrial node pacemaker activity requires Ca(2+)/calmodulin-dependent protein kinase II activation. Circ. Res. 2000, 87, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, T.M.; Sirenko, S.; Lyashkov, A.E.; Younes, A.; Li, Y.; Zhu, W.; Yang, D.; Ruknudin, A.M.; Spurgeon, H.; Lakatta, E.G. Constitutive phosphodiesterase activity restricts spontaneous beating rate of cardiac pacemaker cells by suppressing local Ca2+ releases. Circ. Res. 2008, 102, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Kranias, E.G.; Solaro, R.J. Phosphorylation of troponin I and phospholamban during catecholamine stimulation of rabbit heart. Nature 1982, 298, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, J.P.; Jones, L.R.; Hathaway, D.R.; Henry, B.G.; Watanabe, A.M. beta-Adrenergic stimulation of phospholamban phosphorylation and Ca2+-ATPase activity in guinea pig ventricles. J. Biol. Chem. 1983, 258, 464–471. [Google Scholar] [PubMed]
- Garvey, J.L.; Kranias, E.G.; Solaro, R.J. Phosphorylation of C-protein, troponin I and phospholamban in isolated rabbit hearts. Biochem. J. 1988, 249, 709–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegener, A.D.; Simmerman, H.K.; Lindemann, J.P.; Jones, L.R. Phospholamban phosphorylation in intact ventricles. Phosphorylation of serine 16 and threonine 17 in response to beta-adrenergic stimulation. J. Biol. Chem. 1989, 264, 11468–11474. [Google Scholar] [PubMed]
- Talosi, L.; Edes, I.; Kranias, E.G. Intracellular mechanisms mediating reversal of beta-adrenergic stimulation in intact beating hearts. Am. J. Physiol. 1993, 264, H791–H797. [Google Scholar] [CrossRef] [PubMed]
- MundinaWeilenmann, C.; Vittone, L.; Ortale, M.; deCingolani, G.C.; Mattiazzi, A. Immunodetection of phosphorylation sites gives new insights into the mechanisms underlying phospholamban phosphorylation in the intact heart. J. Biol. Chem. 1996, 271, 33561–33567. [Google Scholar] [CrossRef]
- Gomez, A.M.; Cheng, H.P.; Lederer, W.J.; Bers, D.M. Ca2+ diffusion and sarcoplasmic reticulum transport both contribute to [Ca2+](i) decline during Ca2+ sparks in rat ventricular myocytes. J. Physiol. 1996, 496, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zhang, T.; Ginsburg, K.S.; Mishra, S.; Brown, J.H.; Bers, D.M. CaMKII delta(C) Slows [Ca](i) Decline in Cardiac Myocytes by Promoting Ca Sparks. Biophys. J. 2012, 102, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Currie, S.; Loughrey, C.M.; Craig, M.A.; Smith, G.L. Calcium/calmodulin-dependent protein kinase II delta associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochem. J. 2004, 377, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.S.; Bers, D.M. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 2007, 73, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, X.H.; Lehnart, S.E.; Reiken, S.R.; Marks, A.R. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 2004, 94, e61–e70. [Google Scholar] [CrossRef] [PubMed]
- Curran, J.; Hinton, M.J.; Rios, E.; Bers, D.M.; Shannon, T.R. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ. Res. 2007, 100, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Kushnir, A.; Betzenhauser, M.J.; Reiken, S.; Li, J.; Lehnart, S.E.; Lindegger, N.; Mongillo, M.; Mohler, P.J.; Marks, A.R. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J. Clin. Investig. 2010, 120, 4388–4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit-Jacques, J.; Bois, P.; Bescond, J.; Lenfant, J. Mechanism of muscarinic control of the high-threshold calcium current in rabbit sino-atrial node myocytes. Pflugers Arch. 1993, 423, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Zhao, Y.T.; Guo, Y.B.; Xu, S.M.; Bai, S.H.; Lakatta, E.G.; Cheng, H.; Hao, X.M.; Wang, S.Q. Beta-adrenergic signaling accelerates and synchronizes cardiac ryanodine receptor response to a single L-type Ca2+ channel. Proc. Natl. Acad. Sci. USA 2009, 106, 18028–18033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Khoo, M.S.; Wu, Y.; Yang, Y.; Grueter, C.E.; Ni, G.; Price, E.E., Jr.; Thiel, W.; Guatimosim, S.; Song, L.S.; et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat. Med. 2005, 11, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lai, D.; Cheng, J.; Lim, H.J.; Keskanokwong, T.; Backs, J.; Olson, E.N.; Wang, Y. Alterations of L-type calcium current and cardiac function in CaMKII{delta} knockout mice. Circ. Res. 2010, 107, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Singh, M.V.; Hall, D.D.; Koval, O.M.; Luczak, E.D.; Joiner, M.L.; Chen, B.; Wu, Y.; Chaudhary, A.K.; Martins, J.B.; et al. Catecholamine-independent heart rate increases require Ca2+/calmodulin-dependent protein kinase II. Circ. Arrhythm. Electrophysiol. 2011, 4, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.P.; Terrar, D.A. Ca(2+)-stimulated adenylyl cyclases regulate the L-type Ca(2+) current in guinea-pig atrial myocytes. J. Physiol. 2012, 590, 1881–1893. [Google Scholar] [CrossRef] [PubMed]
- De Koninck, P.; Schulman, H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 1998, 279, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Rich, R.C.; Schulman, H. Substrate-directed function of calmodulin in autophosphorylation of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1998, 273, 28424–28429. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, K.S.; Weber, C.R.; Bers, D.M. Control of maximum sarcoplasmic reticulum Ca load in intact ferret ventricular myocytes. Effects Of thapsigargin and isoproterenol. J. Gen. Physiol. 1998, 111, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Akin, B.L.; Chen, Z.; Jones, L.R. Superinhibitory phospholamban mutants compete with Ca2+ for binding to SERCA2a by stabilizing a unique nucleotide-dependent conformational state. J. Biol. Chem. 2010, 285, 28540–28552. [Google Scholar] [CrossRef] [PubMed]
- Akin, B.L.; Jones, L.R. Characterizing phospholamban to sarco(endo)plasmic reticulum Ca2+-ATPase 2a (SERCA2a) protein binding interactions in human cardiac sarcoplasmic reticulum vesicles using chemical cross-linking. J. Biol. Chem. 2012, 287, 7582–7593. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinogradova, T.M.; Tagirova, S.; Lakatta, E.G. Unique Ca2+-Cycling Protein Abundance and Regulation Sustains Local Ca2+ Releases and Spontaneous Firing of Rabbit Sinoatrial Node Cells. Int. J. Mol. Sci. 2018, 19, 2173. https://doi.org/10.3390/ijms19082173
Vinogradova TM, Tagirova S, Lakatta EG. Unique Ca2+-Cycling Protein Abundance and Regulation Sustains Local Ca2+ Releases and Spontaneous Firing of Rabbit Sinoatrial Node Cells. International Journal of Molecular Sciences. 2018; 19(8):2173. https://doi.org/10.3390/ijms19082173
Chicago/Turabian StyleVinogradova, Tatiana M., Syevda Tagirova (Sirenko), and Edward G. Lakatta. 2018. "Unique Ca2+-Cycling Protein Abundance and Regulation Sustains Local Ca2+ Releases and Spontaneous Firing of Rabbit Sinoatrial Node Cells" International Journal of Molecular Sciences 19, no. 8: 2173. https://doi.org/10.3390/ijms19082173