3.3. Genes Related to Cell Cycle and Mitosis
Progression of the mitotic cycle is important for free nuclear mitosis in megagametogenesis. Many genes encoding components regulating the cell cycle showed different expression patterns during ovule development in FL compared with SL.
Cyclin-dependent kinases (CDKs) make a large contribution to the regulation of cell cycle progression and cell division. Among these proteins, CDKB members are plant-specific kinases and include the CDKB1 superfamily whose kinase activity reaches peak levels during mitosis [
17]. Deficiency of CDKB1 and CDKA1 kinases leads to the absence of the egg apparatus within the embryo sac [
18]. In the present study, one gene encoding CDKB was upregulated in FNM2-FL and FNM3-FL compared to FNM2-SL and FNM3-SL, respectively, suggesting the difference in gene expression is related to CDK between FL and SL. The CDK inhibitory protein (ICK) negatively regulates CDKs, and, therefore, degradation of ICK is also an essential component of cell cycle regulation [
19]. Accordingly, during megagametogenesis in
Arabidopsis thaliana, precise expression of the gene encoding ICK ensures mitotic cell cycle procession [
20]. The present study showed that one gene encoding ICK was downregulated in FNM2-FL and FNM3-FL compared to FNM2-SL and FNM3-SL, respectively. The downregulation of gene expression may lead to the reduction of inhibitors and upregulate the associated CDK protein in FL at the FNM2 and FNM3 stages, contributing to FNM during the developmental stages. It is suggested that the regulator related to CDK may responsible for the process of FNM in FG development. MYB proteins are important transcriptional factors in plants that are involved in various biological processes, including cell cycle, growth and developmental processes. Several members of the MYB family have been proven to be responsible for development of the female gametophyte in
Arabidopsis. FLP and
MYB88 regulate the FG development and their loss significantly increases the number of ovules [
21]. Attenuation of
MYB64 and
MYB119 expression, during transition to the FG5 phase, leads to failure of cellularization of the female gametophyte, resulting in supernumerary nuclei [
9], similar to the female gametophyte in gymnosperms. Similarly, in the present study, we found that the free nuclei were unable to conduct mitosis in SL ovules and the expression of 10
MYB-like sequences was downregulated in FL ovules compared with SL ovules, suggesting that the MYB family proteins play essential roles in the FNM process and putatively regulate cell cycle gene expression. The minichrome maintenance (MCM) protein family is indispensable in DNA replication and synthesis. The
PRL gene in
Arabidopsis encodes a homologue of MCM7, the subunit of a DNA replication licensing factor, which is required in all dividing cells, including FG [
22]. In our study, two genes encoding MCM10 and MCM3 proteins, were upregulated in comparison in FNM1-FL versus FNM1-SL and FNM2-FL versus FNM2-SL and the level of gene expression in FL ovules increased from the FNM1 to FNM2 stage, suggesting that MCM proteins function in developing ovules during the FNM process. The anaphase-promoting complex/cyclosome (APC/C) is an essential E3 ubiquitin protein ligase that regulates transition during the mitotic process and is responsible for the degradation of mitotic cyclins [
23]. Mutation in the
APC2 gene, which encodes one of APC/C’s subunits, impairs female megagametogenesis [
24]. Similarly, the
nomega mutant embryo sacs, which result in the arrest of mitosis at the two-nucleate stage, are unable to degrade Cyclin B and the gene product of
NOMEGA shows high homology to the APC6/cell division cycle (CDC) 16 subunit of APC/C [
25]. In the present research, two APC-like genes were downregulated in FL ovules compared with SL ovules at the FNM1 stage, implying that the APC/C ubiquitin-mediated proteolysis pathway may play an important role in the beginning of FNM.
3.4. Genes Related to Phytohormones
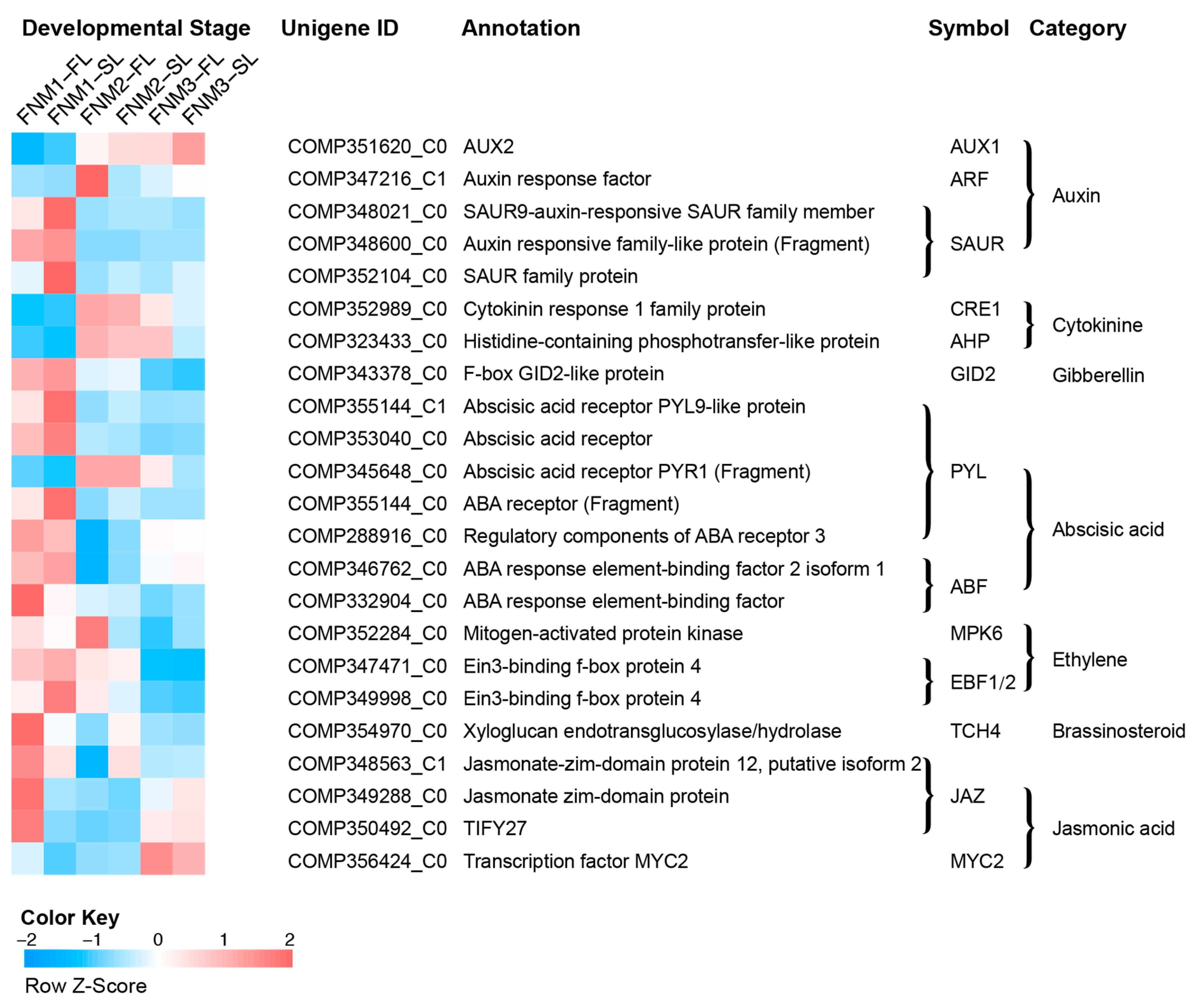
Plant hormones are generated within the plant and serve as signal functions, regulating a wide range of processes. The plant hormone signals are detected and transported to the nucleus by signal transduction to activate gene expression, leading to physiological processes. The DEGs were involved in all of the phytohormone signal transduction pathways. We mainly focused on the expression patterns of auxin- and cytokinin-related genes, which may be responsible for FNM in FG development.
Auxin, a mobile signaling molecule, functions as one of the most critical phytohormones, regulating various processes in plant development. Auxin has been shown as a key regulator in megagametogenesis. Genes related to auxin biosynthesis and auxin influx carriers are revealed, such as
YUCCA1 (
YUC1) and
AUX1 gene, implying a complicated mechanism of auxin involved in mitotic divisions, cell expansion and patterning during embryo sac development [
26].
Auxin biosynthesis plays an important role in ovule development. Data have been confirmed by Note-Wilson [
27], who associated ovule loss with a severe reduction in local auxin biosynthesis. Also, the disruption of auxin synthesis by the ectopic expression of the
YUC1 gene impairs cell specificity in the FG [
28]. Anthranilate synthase (ASA) is involved in the
l-tryptohphan synthesis pathway and is a precursor of IAA. The overexpression of the
α-SUBUNIT OF ANTHRANILATE SYNTHASE (
OASA1D) gene in a transgenic rice line increased the level of tryptophan and free IAA [
29]. In the present study, one gene encoding ASA was downregulated in FL compared with SL at the FNM3 stage and two
YUCCA genes were differentially expressed in FNM3-FL compared with FNM3-SL, implying a disorder of auxin biosynthesis in sterile FG.
Increasing evidence indicates that polar auxin transport (PAT) regulates significant processes of growth and development. An auxin gradient along the micropylar-chalazal axis can regulate the specification of cell fate in female gametophyte genesis [
28]. A transient application of N-1-naphthylphthalamic acid (an auxin efflux inhibitor) causes a significant loss of ovules [
30]. In
Arabidopsis, the AUX1/LAX auxin transporter family is an influx carrier, while the PIN-FORMED (PIN) auxin protein family is the main efflux carrier [
31]. In FG, downregulation of the
PIN1 gene induces mitosis arrest and subsequently, FG sterility [
32]. In our study, one gene encoding LAX4 was downregulated in FNM1-FL compared with FNM1-SL and two genes encoding PIN1 were differentially expressed in the FNM2 stage. It is indicated that PAT may be strengthened in SL FG.
Auxin primary response genes include
GH3, SAUR (
small auxin up RNA) and
AUX/IAA. During the process of FNM, genes related to the auxin response were preferentially expressed in different developmental stages. It is suggested that the correct expression of auxin response genes is necessary for the FG development. In our transcriptomic data, one
SAUR gene was downregulated in FL ovules at all three stages but was upregulated in SL ovules. One
IAA gene was downregulated in FL at the FNM1 stage. Auxin responsive factors (ARF) bind to the promoter region of the auxin-responsive element to stimulate or repress transcription. Overexpression of microRNA
miR167 leads to the downregulation of its target
ARF6 and
ARF8 genes, inducing the ensuing arrest of FG development [
33]. Several
ARF genes were preferentially expressed in FL and SL. It is hypothesized that the expression variations of auxin-responsive genes between FL and SL may affect the production and distribution of auxin during ovule development, resulting in FG sterility in
P. tabulaeformis.
However, evidence shows that the gradients in the FG cannot provide sufficient positional instruction for cell specification [
34]. This increases the complexity of auxin function in FG development. Although the function of auxin in female gametophyte development is not entirely clear, our study indicates the indispensable role of auxin in the FG formation.
Cytokinin is an essential hormone for cell proliferation and differentiation. Many studies have proven that cytokinin plays an important role during ovule development. Manipulation of cytokinin metabolism affects ovule fertility.
A reduction in the cytokinin gradient in plants induces a decrease in ovule number or female fertility [
35]. Cytokinin oxidases/dehydrogenases (CKX) are responsible for the irreversible degradation of cytokinin and the inhibition of hormone concentrations in tissues [
36]. Overexpression of cytokinin oxidase reduced the fertility rate [
37]. Also, alteration of
CKX gene expression influences the regulation of cytokinin levels and controls the developmental process of the ovule. The dual mutation of
CKX3 and
CKX5 genes in
Arabidopsis established supernumerary ovules, further confirming the relationship between cytokinin and ovule development [
38]. In the present study, two genes encoding CKX were downregulated in comparison between FL and SL at the FNM2 and FNM3 stages, resulting in less suppression of the cytokinin level and ensuring the FG development. Although the function of cytokinin in ovule development is still not clear, these findings confirm the necessity of cytokinin in ovule development.
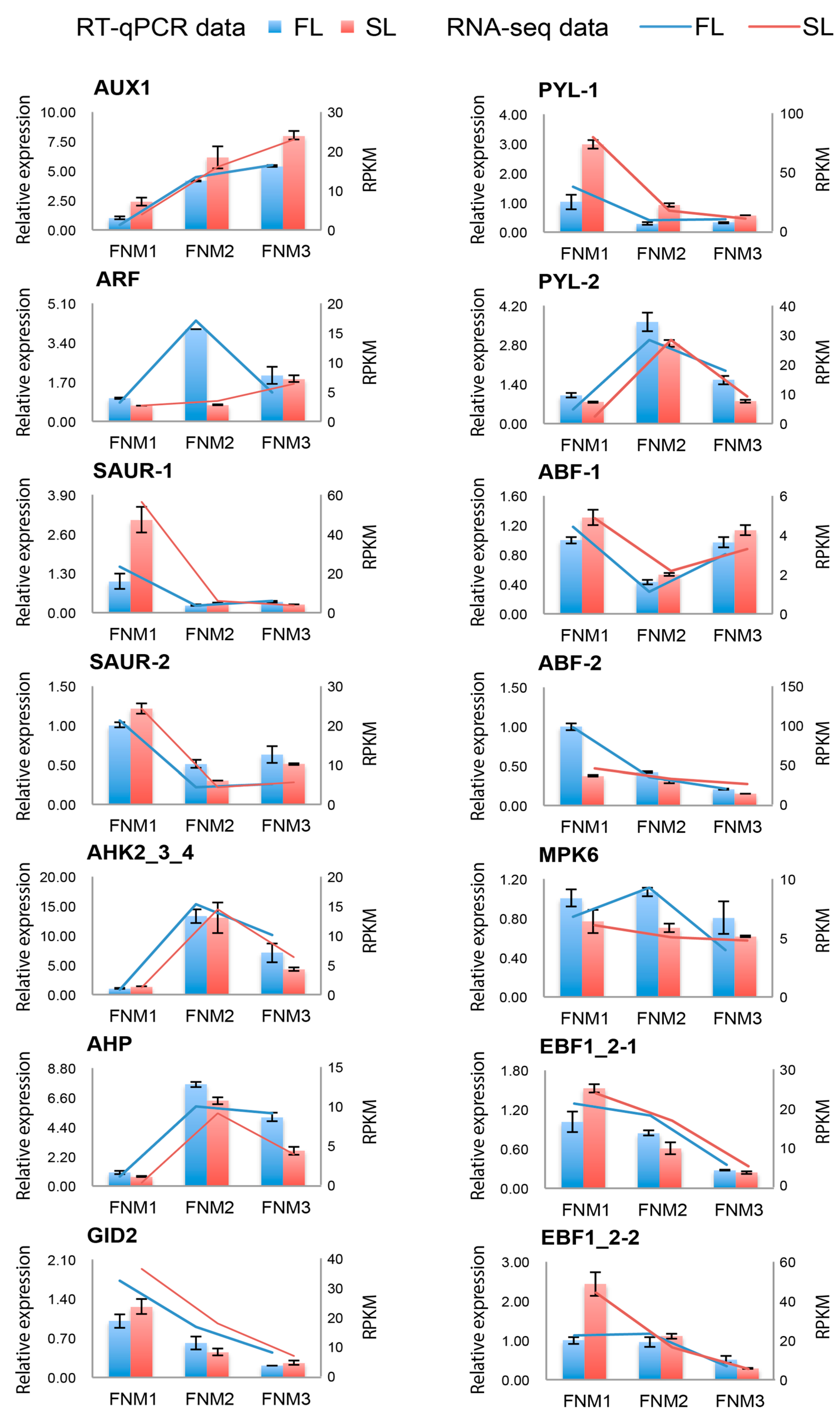
In cytokinin signal transduction, the interference of three genes, encoding Arabidopsis histidine kinases (AHK2, AHK3 and AHK4/CRE1), induced a reduction in fertility, phenocopying of the quintuple mutant
ahp1,
2,
3,
4,
5 and impairment of the cytokinin signaling pathway [
39]. On the basis of these studies, we focused on gene expression in cytokinin signal transduction in our research, especially at the FNM1 and FNM2 stages. In the present study, no DEG in a comparison between FL and SL ovules was found in the cytokinin signal transduction pathway but some genes were specifically expressed at different developmental stages. The expression level of genes encoding AHK2/3/4 and AHP was upregulated from the FNM1 to FNM2 stage in FL ovules, suggesting the cytokinin response is enhanced at FNM2 stage. During the period of functional megaspore mitosis, genes related to the cytokinin signaling pathway (
CRE1 and
AHP) and degradation (
CKX) were significantly differentially expressed between FNM1 and FNM2 in different lines.
ABA-mediated signaling is believed to be involved in stress response, seed germination and the developmental process of plants. In the ABA biosynthesis pathway, the rate-limiting step is the decomposition of 9-
cis-epoxycarotenoids and the generation of the ABA precursor xanthoxin and this key process is catalyzed by 9-
cis-epoxycarotenoid dioxygenase (NCED). In our study, two genes encoding NCED were upregulated in SL ovules compared with FL ovules at the FNM2 and FNM3 stages, suggesting the acceleration of ABA biosynthesis in SL ovules. In the FNM process, the ABA content is higher in SL than in FL at all of the developmental stages [
12], which is in agreement with gene expression result in the present study. In the ABA signal transduction pathway, PYL, the receptor for ABA, suppresses protein phosphatase (PP2C) and is the origin of ABA negative regulation [
40]. In our study, two genes encoding PYL/PYR were upregulated in FNM1-SL compared with FNM1-FL. Taken together, these data indicate that ABA biosynthesis and signal transduction might be more active in SL ovules.
In addition, an alteration of the phytohormone level not only regulates FG development directly but also controls the cell cycle process and affects FG development indirectly. Auxin induces mitosis cyclin (CYCA, CYCB) in
Arabidopsis roots and mRNA accumulation of CYCD in the G1 phase. As for the G2/M phase transition, auxin is necessary and regulates the cell cycle process through cyclin degradation by ubiquitin-related protein [
41]. Cytokinin induces the synthesis of CYCD3, promoting G1/S phase transition. From the G0 to S phase, exogenous cytokinin activates potential DNA replication and reduces the time span of DNA replication in the S phase. In tobacco, cytokinin activates CDKA kinase in G2/M phase transition [
42]. ABA induces ICK and regulates the activity of CDKA in the G1 and G2 phase. It is implied that genes involved in the cell cycle process and phytohormone biosynthesis and signal transduction pathway constitute a complex regulation network to guarantee the FG development; these genes require further exploration.
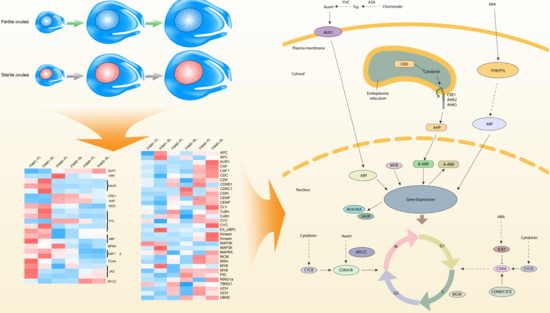
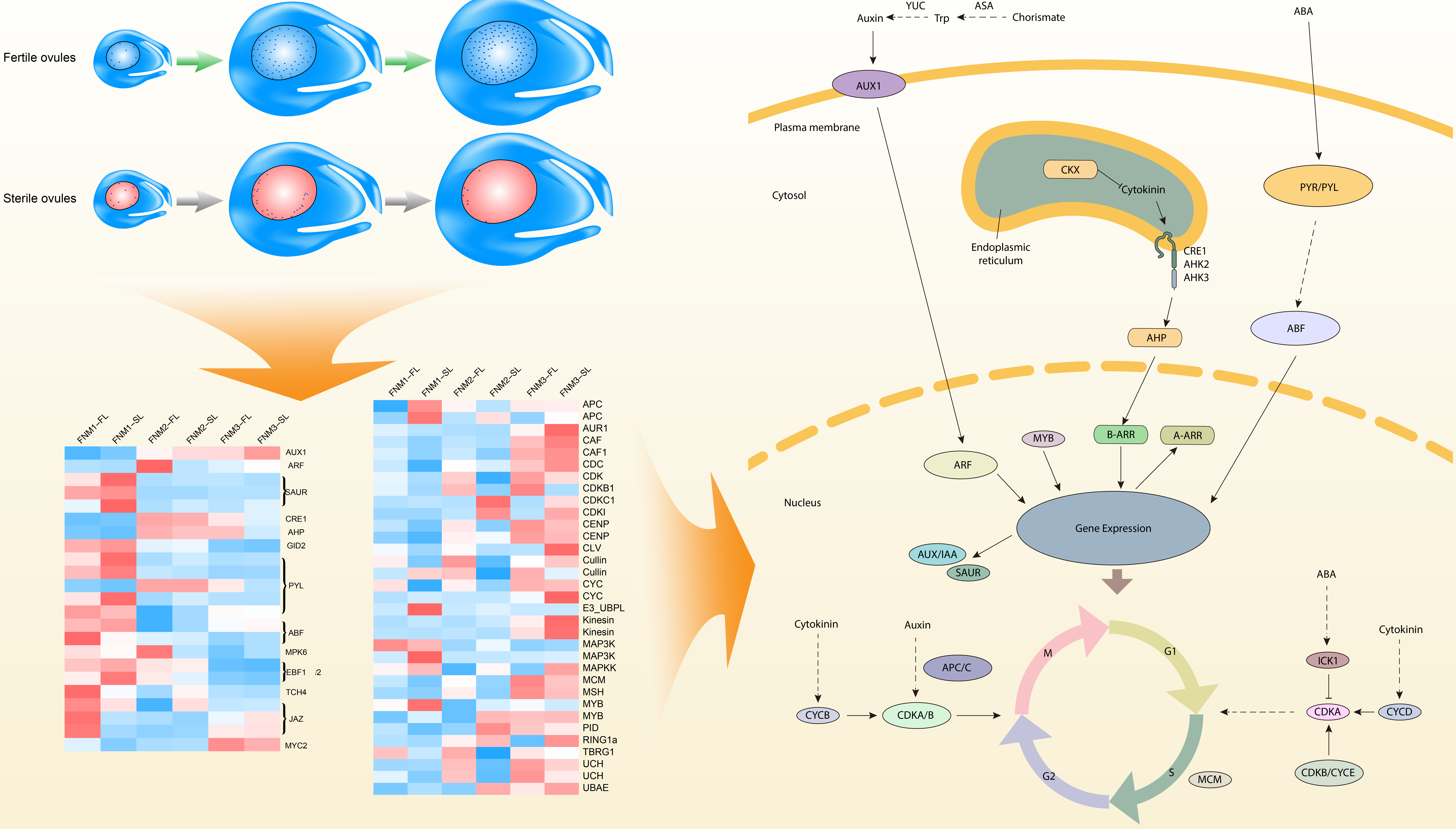
In summary, we reported a comprehensive
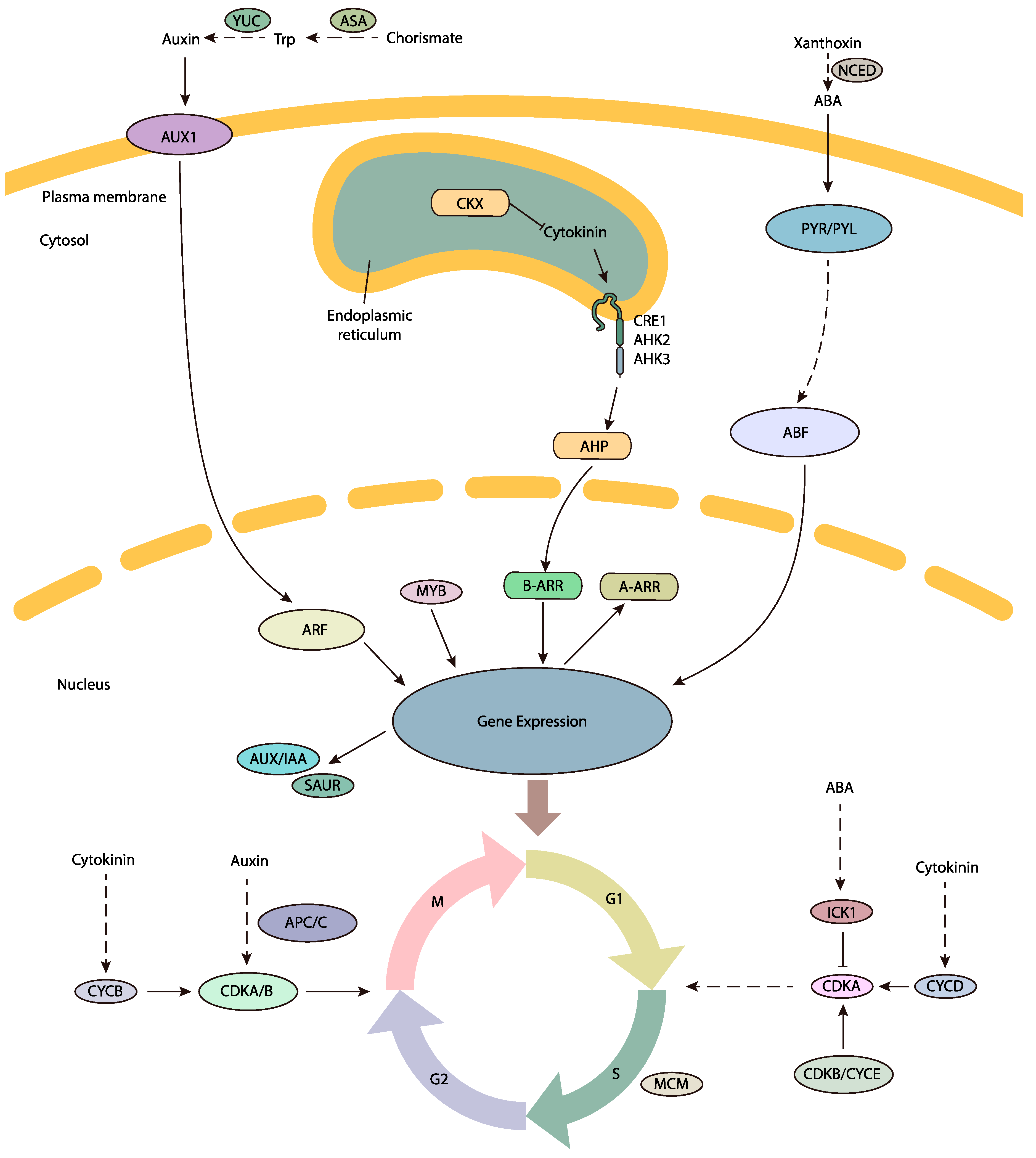
P. tabulaeformis ovule transcriptome dataset generated by de novo assembly of next-generation sequencing data, allowing gene expression changes between developing and abortive ovules to be compared. The DEG analysis revealed that two groups of genes were putatively involved in the FNM process, including cell cycle regulation and phytohormone pathways (shown in
Figure 9). DEGs encoding CDKB, ICK, MCM, MYB and the APC/C protein were preferentially expressed in developing ovules, securing the cell cycle process. DEGs encoding ASA, YUC, AUX1, ARF, AUX/IAA and SAUR in the auxin pathway, CKX, CRE/AHK and AHP in the cytokinin pathway and NCED and PYR/PYL in the ABA pathway may potentially induce the phytohormone regulation and trigger the FNM process. In addition, phytohormones might regulate cell cycle process and indirectly regulate the FNM, constructing a comprehensive regulation network for the FNM process. The
P. tabulaeformis transcriptome data is a valuable resource for future genomic studies and will be helpful for researchers working on other closely related species. The differentially expressed genes dataset will also provide useful candidate genes for functional analysis of ovule development in woody gymnosperms.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}