Toll-Like Receptor Agonists Modulate Wound Regeneration in Airway Epithelial Cells


Abstract
:1. Introduction
2. Results
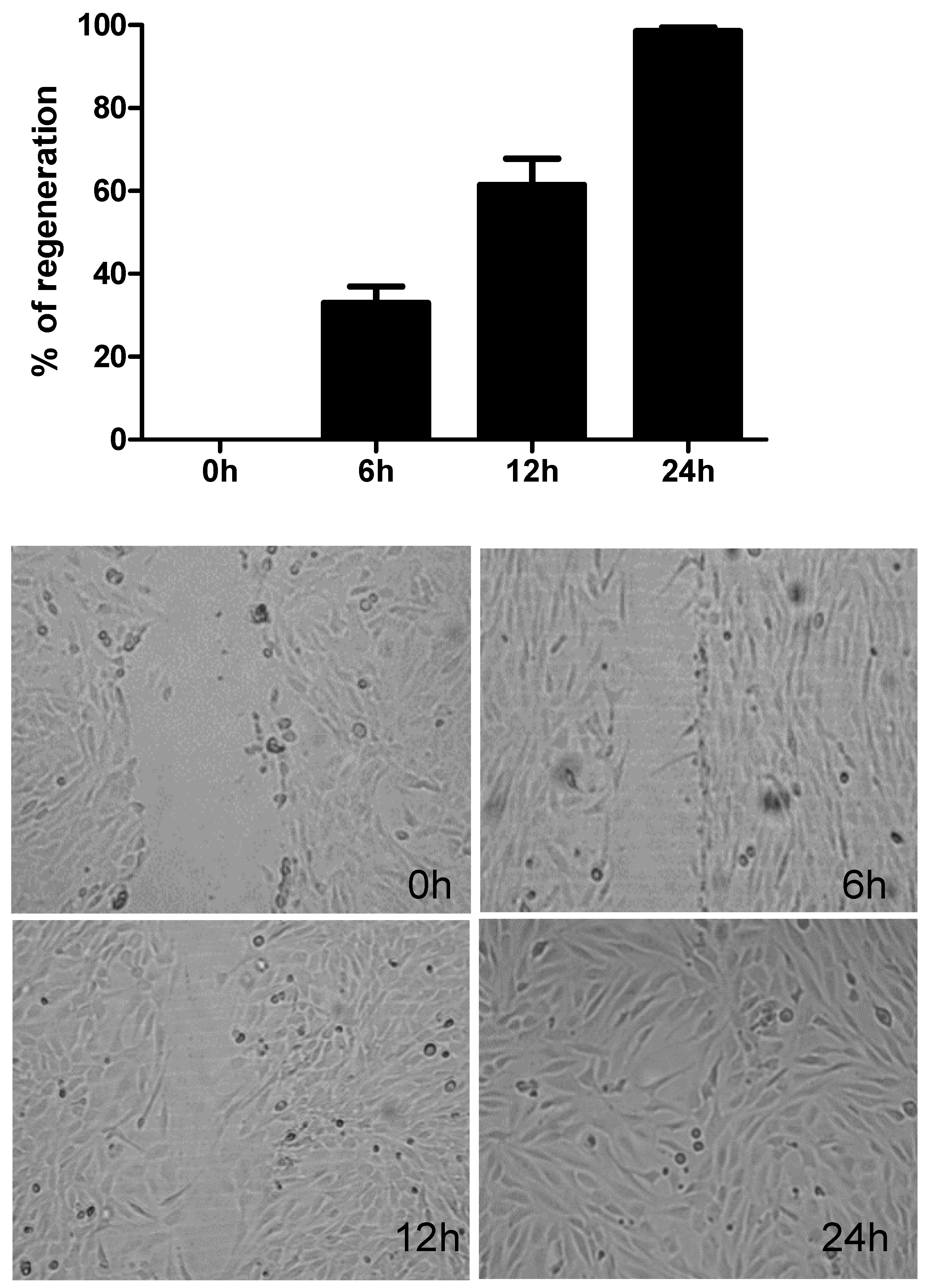
2.1. Repair Response in Human Bronchial Epithelial Cells
2.2. Viability of Injured Immortalized Human Bronchial Epithelial Cells-BEAS-2B
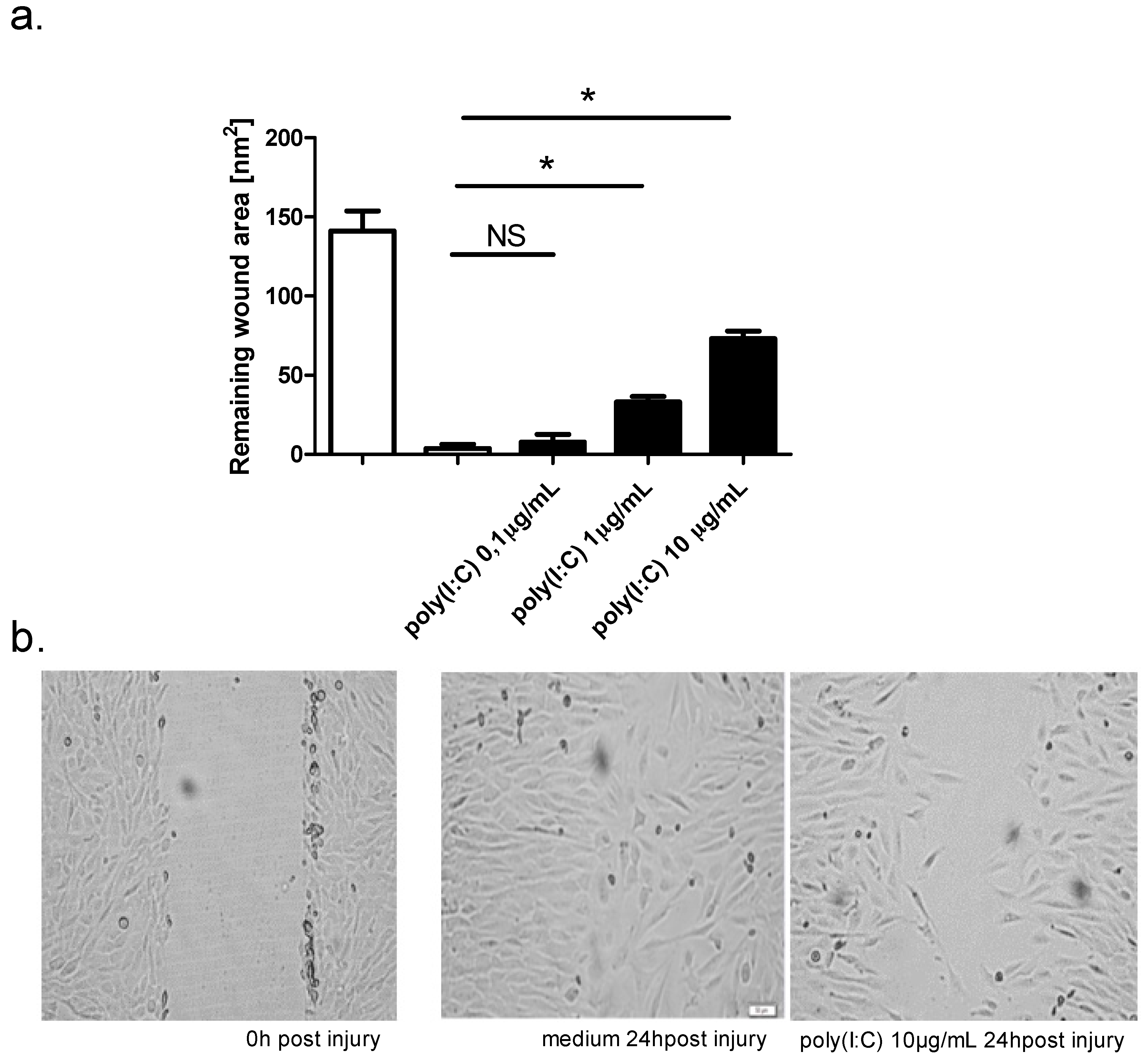
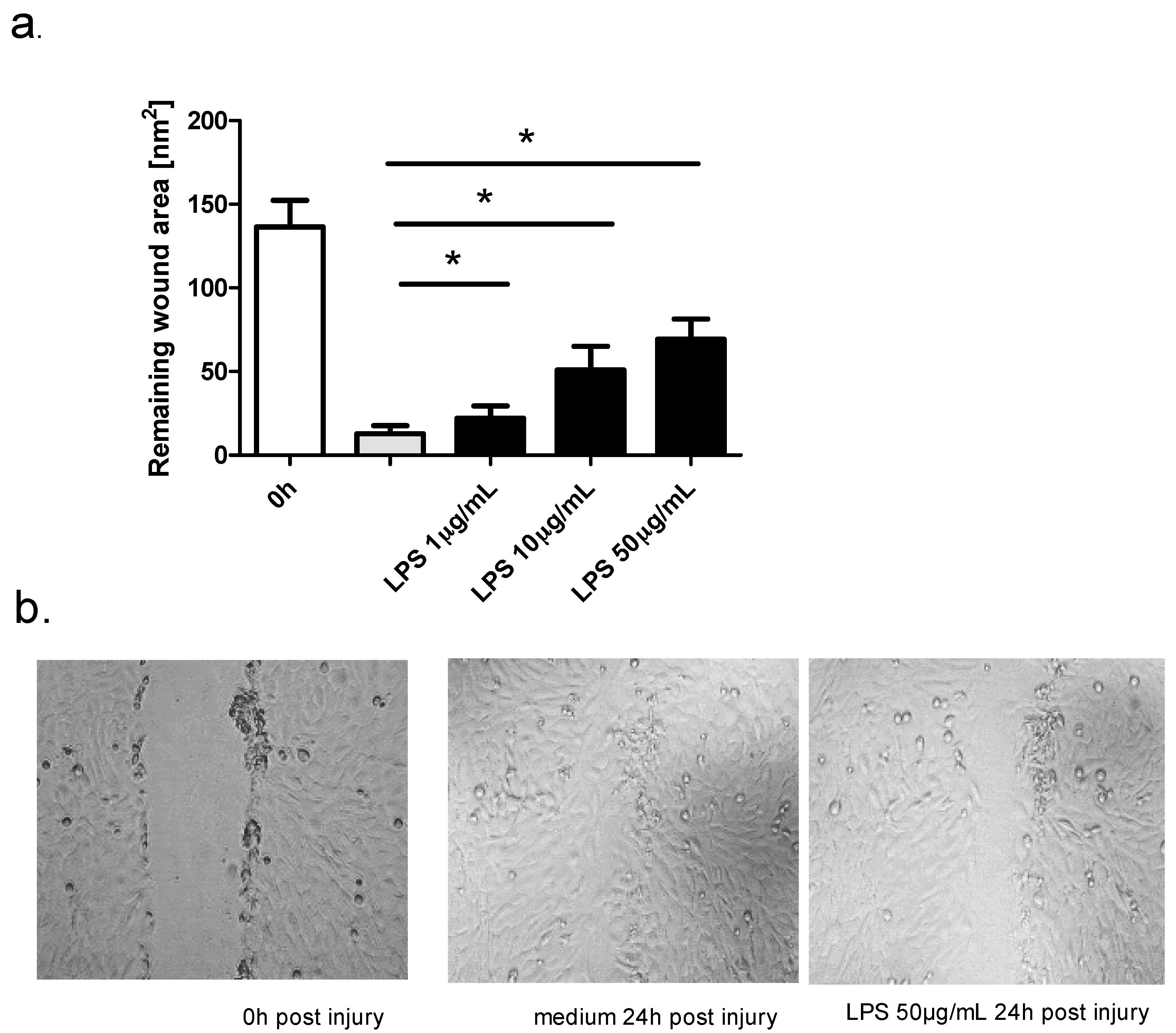
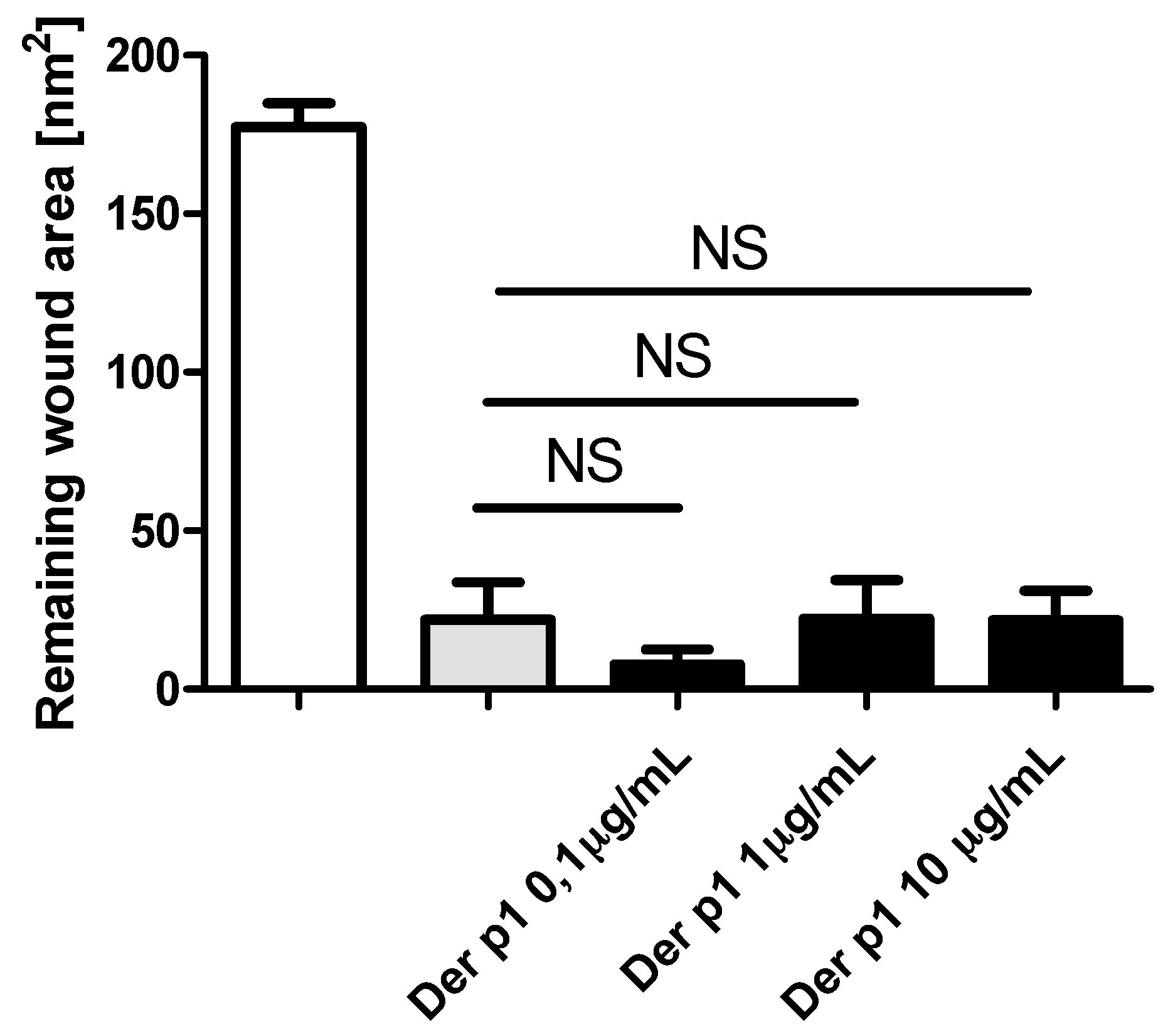
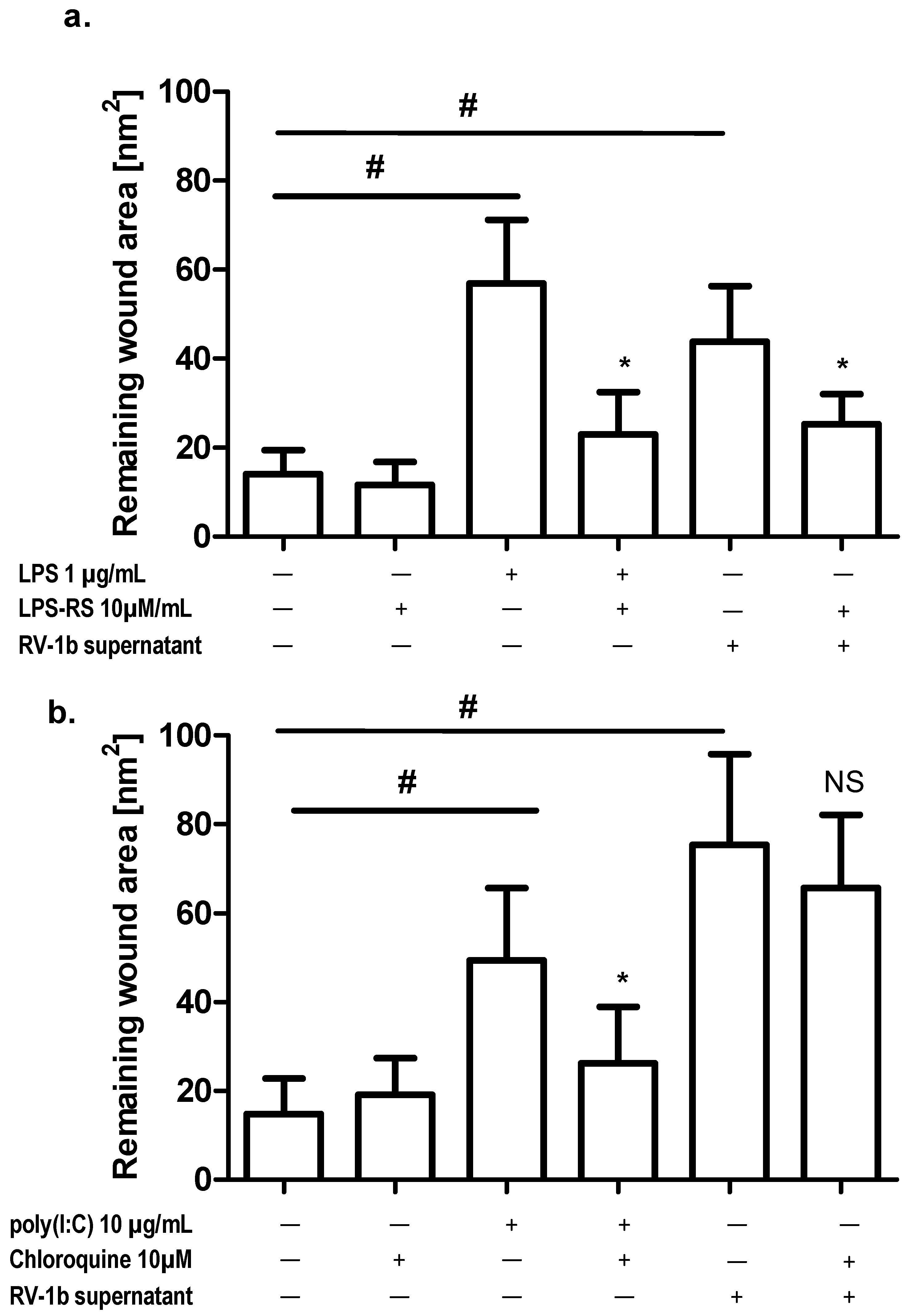
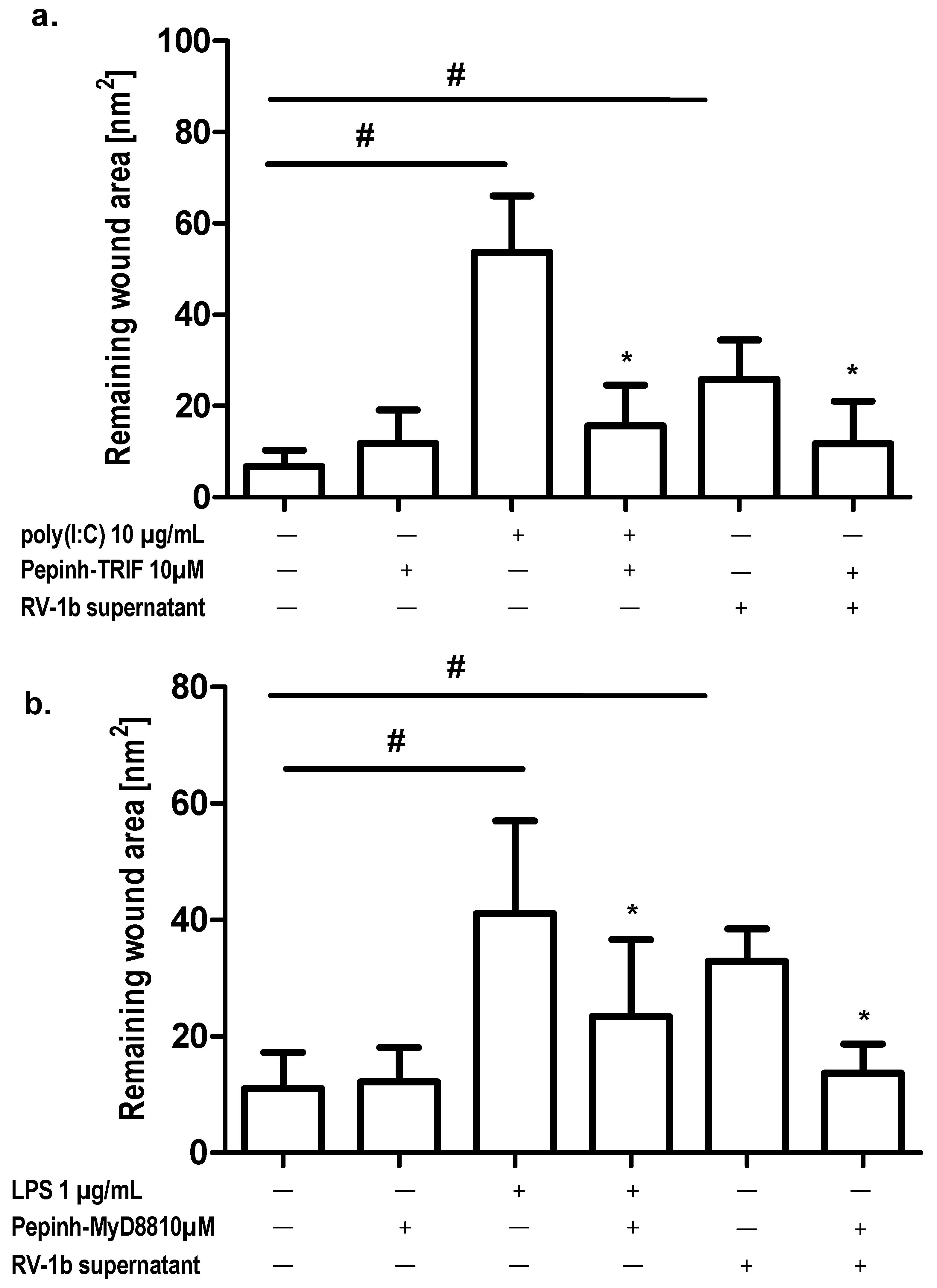
2.3. Effect of TLR Agonists and Allergen on Wound Repair in Bronchial Epithelial Cells
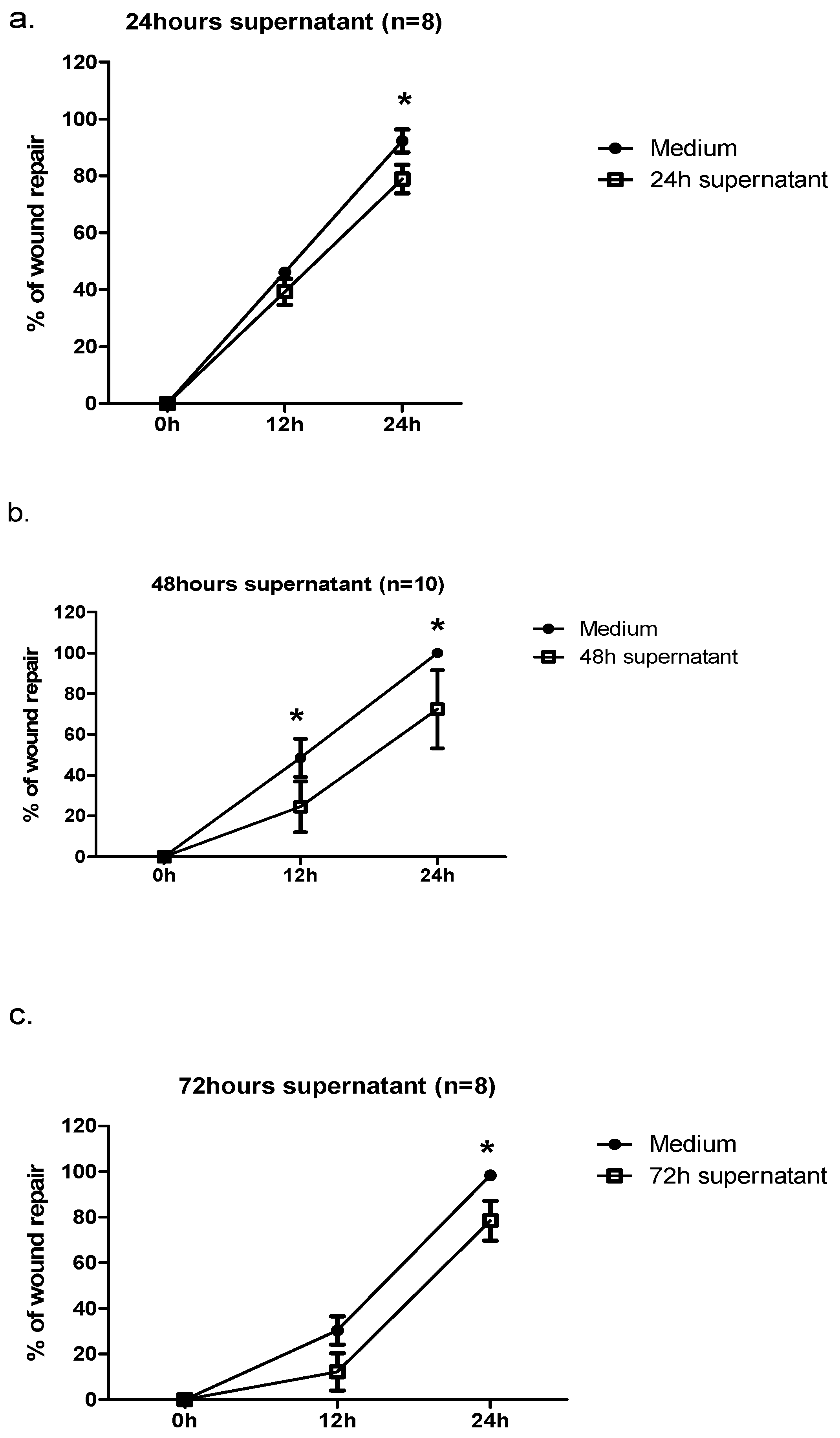
2.4. Preparation of Conditioned Media from RV1b- and PIV3-Infected Cells
2.5. Effect of Conditioned Media from RV1b-Infected Epithelial Cells (ECs) on Wound Repair
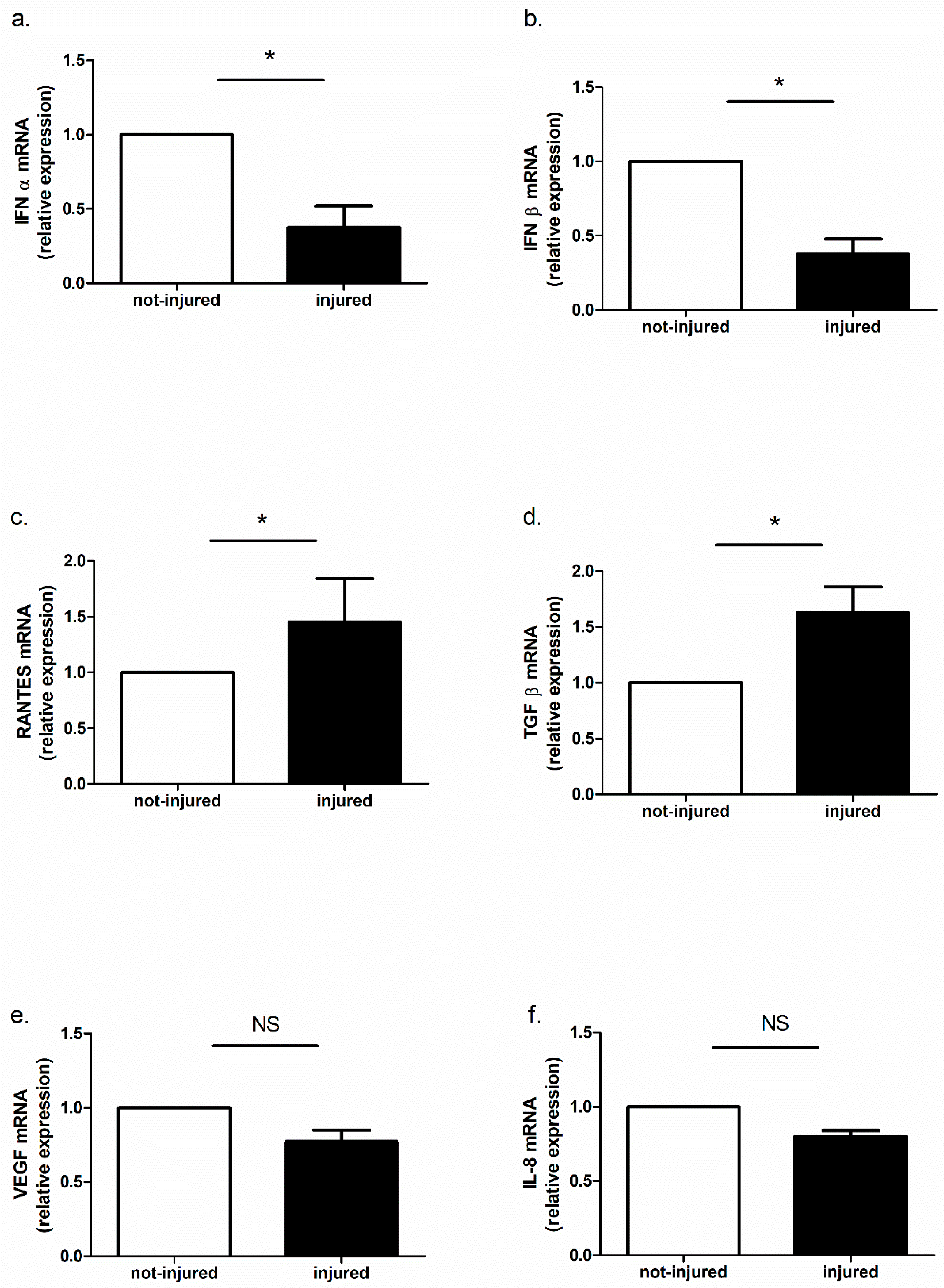
2.6. Immune Response in Injured and Not-Injured Airway Epithelial Cells
Cytokine mRNA Expression and Protein Release in Bronchial Epithelial Cells in Response to Injury
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Wound Repair Assay
4.3. Viability Assay
4.4. Cell Stimulation and Treatment of ECs with Conditioned Media from Infected Cells
4.5. Preparation of Conditioned Media from RV1b-Infected Cells
4.6. Measurement of Cytokines and Growth Factors
4.7. RNA Isolation and Quantitative Polymerase Chain Reaction (PCR)
4.8. Measurement of Mediators
4.9. Statistical Data Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ECs | epithelial cells |
| LPS | lipopolysaccharide |
| TLRs | toll-like receptors |
| RV1b | rhinovirus type 1B |
| PRRs | pathogen recognition receptors |
References
- Grainge, C.L.; Davies, D.E. Epithelial injury and repair in airways diseases. Chest 2013, 144, 1906–1912. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.; Wadsworth, S.; Dorscheid, D.; Man, S.F.; Sin, D.D. The airway epithelium: More than just a structural barrier. Ther. Adv. Respir. Dis. 2011, 5, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Li, D.X.; Liu, M. Knowledge translation: Airway epithelial cell migration and respiratory diseases. Cell. Mol. Life Sci. 2012, 69, 4149–4162. [Google Scholar] [CrossRef] [PubMed]
- Loxham, M.; Davies, D.E.; Blume, C. Epithelial function and dysfunction in asthma. Clin. Exp. Allergy 2014, 44, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Georas, S.N.; Rezaee, F. Epithelial barrier function: At the front line of asthma immunology and allergic airway inflammation. J. Allergy Clin. Immune 2014, 134, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahy, J.V. Type 2 inflammation in asthma—Present in most, absent in many. Nat. Rev. Immun. 2015, 15, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.R.; Knight, D.A. Structural changes in the airways in asthma: Observations and consequences. Clin. Sci. 2005, 108, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T.; Wenzel, S.; Postma, D.S.; Weiss, S.T.; Renz, H.; Sly, P.D. Asthma. Nat. Rev. Dis. Primers 2015, 1, 15025. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol. Rev. 2011, 242, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Sajjan, U.; Wang, Q.; Zhao, Y.; Gruenert, D.C.; Hershenson, M.B. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am. J. Respir. Crit. Care Med. 2008, 178, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T.; Roberts, G.; Arshad, H.S.; Howarth, P.H.; Davies, D.E. The role of the airway epithelium and its interaction with environmental factors in asthma pathogenesis. Proc. Am. Thorac. Soc. 2009, 6, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Comstock, A.T.; Ganesan, S.; Chattoraj, A.; Faris, A.N.; Margolis, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J. Virol. 2011, 85, 6795–6808. [Google Scholar] [CrossRef] [PubMed]
- Looi, K.; Troy, N.M.; Garratt, L.W.; Iosifidis, T.; Bosco, A.; Buckley, A.G.; Ling, K.M.; Martinovich, K.M.; Kicic-Starcevich, E.; Shaw, N.C.; et al. Effect of human rhinovirus infection on airway epithelium tight junction protein disassembly and transepithelial permeability. Exp. Lung Res. 2016, 42, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Bossios, A.; Psarras, S.; Gourgiotis, D.; Skevaki, C.L.; Constantopoulos, A.G.; Saxoni-Papageorgiou, P.; Papadopoulos, N.G. Rhinovirus infection induces cytotoxicity and delays wound healing in bronchial epithelial cells. Respir. Res. 2005, 6, 114. [Google Scholar] [CrossRef] [PubMed]
- Faris, A.N.; Ganesan, S.; Chattoraj, A.; Chattoraj, S.S.; Comstock, A.T.; Unger, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus delays cell repolarization in a model of injured/regenerating human airway epithelium. Am. J. Respir. Cell Mol. Biol. 2016, 55, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Medzhitov, R. Role of toll-like receptors in tissue repair and tumorigenesis. Biochemistry 2008, 73, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Pull, S.L.; Doherty, J.M.; Mills, J.C.; Gordon, J.I.; Stappenbeck, T.S. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc. Nat. Acad. Natl. Sci. USA 2005, 102, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.L.; Riehl, T.E.; Walker, M.R.; Geske, M.J.; Doherty, J.M.; Stenson, W.F.; Stappenbeck, T.S. Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury. J. Clin. Investig. 2007, 117, 258–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaykhiev, R.; Behr, J.; Bals, R. Microbial patterns signaling via Toll-like receptors 2 and 5 contribute to epithelial repair, growth and survival. PLoS ONE 2008, 3, e1393. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Liang, J.; Fan, J.; Yu, S.; Chen, S.; Luo, Y.; Prestwich, G.D.; Mascarenhas, M.M.; Garg, H.G.; Quinn, D.A.; et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 2005, 11, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska-Polak, A.; Brauncajs, M.; Paradowska, E.; Jarzebska, M.; Kurowski, M.; Moskwa, S.; Lesnikowski, Z.J.; Kowalski, M.L. Human parainfluenza virus type 3 (HPIV3) induces production of IFNgamma and RANTES in human nasal epithelial cells (HNECs). J. Inflamm. 2015, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Globinska, A.; Pawelczyk, M.; Piechota-Polanczyk, A.; Olszewska-Ziaber, A.; Moskwa, S.; Mikolajczyk, A.; Jablonska, A.; Zakrzewski, P.K.; Brauncajs, M.; Jarzebska, M.; et al. Impaired virus replication and decreased innate immune responses to viral infections in nasal epithelial cells from patients with allergic rhinitis. Clin. Exp. Immunol. 2017, 187, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Moskwa, S.; Piotrowski, W.; Marczak, J.; Pawelczyk, M.; Lewandowska-Polak, A.; Jarzebska, M.; Brauncajs, M.; Globinska, A.; Gorski, P.; Papadopoulos, N.G.; et al. Innate immune response to viral infections in primary bronchial epithelial cells is modified by the atopic status of asthmatic patients. Allergy Asthma Immunol. Res. 2018, 10, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska-Polak, A.; Jarzębska, M.; Brauncajs, M.; Grzegorczyk, J.; Kowalski, M.L. Wound repair and regeneration of upper and lower airway epithelium-validation of experimental methodology. Alergy Astma Immunol. 2016, 21, 162–168. [Google Scholar]
- Liu, X.; Meng, J. Tanshinone IIA ameliorates lipopolysaccharide-induced inflammatory response in bronchial epithelium cell line BEAS-2B by down-regulating miR-27a. Biomed. Pharmacother. 2018, 104, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Encarnacion-Medina, J.; Rodriguez-Cotto, R.I.; Bloom-Oquendo, J.; Ortiz-Martinez, M.G.; Duconge, J.; Jimenez-Velez, B. Selective ATP-binding cassette subfamily C gene expression and proinflammatory mediators released by BEAS-2B after PM2.5, budesonide, and cotreated exposures. Mediat. Inflamm. 2017, 2017, 6827194. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mychajlowycz, M.; Lau, C.; Gutierrez, C.; Scott, J.A.; Chow, C.W. Spleen tyrosine kinase mediates BEAS-2B cell migration and proliferation and human rhinovirus-induced expression of vascular endothelial growth factor and interleukin-8. J. Pharmacol. Exp. Ther. 2012, 340, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Xagorari, A.; Chlichlia, K. Toll-like receptors and viruses: Induction of innate antiviral immune responses. Open Microbiol. J. 2008, 2, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Kuznik, A.; Bencina, M.; Svajger, U.; Jeras, M.; Rozman, B.; Jerala, R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J. Immunol. 2011, 186, 4794–4804. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors-redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivera, D.S.; Boggs, S.E.; Beenhouwer, C.; Aden, J.; Knall, C. Cellular mechanisms of mainstream cigarette smoke-induced lung epithelial tight junction permeability changes in vitro. Inhal. Toxicol. 2007, 19, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Petecchia, L.; Sabatini, F.; Varesio, L.; Camoirano, A.; Usai, C.; Pezzolo, A.; Rossi, G.A. Bronchial airway epithelial cell damage following exposure to cigarette smoke includes disassembly of tight junction components mediated by the extracellular signal-regulated kinase 1/2 pathway. Chest 2009, 135, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Winton, H.L.; Soeller, C.; Taylor, G.W.; Gruenert, D.C.; Thompson, P.J.; Cannell, M.B.; Stewart, G.A.; Garrod, D.R.; Robinson, C. The transmembrane protein occludin of epithelial tight junctions is a functional target for serine peptidases from faecal pellets of Dermatophagoides pteronyssinus. Clin. Exp. Allergy 2001, 31, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Winton, H.L.; Soeller, C.; Tovey, E.R.; Gruenert, D.C.; Thompson, P.J.; Stewart, G.A.; Taylor, G.W.; Garrod, D.R.; Cannell, M.B.; et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J. Clin. Investig. 1999, 104, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellson, C.D.; Dunmore, R.; Hogaboam, C.M.; Sleeman, M.A.; Murray, L.A. Danger-associated molecular patterns and danger signals in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 51, 163–168. [Google Scholar] [PubMed]
- Kang, J.H.; Hwang, S.M.; Chung, I.Y. S100A8, S100A9 and S100A12 activate airway epithelial cells to produce MUC5AC via extracellular signal-regulated kinase and nuclear factor-kappaB pathways. Immunology 2015, 144, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, S.D.; Heijink, I.H.; ten Hacken, N.H.; Vandenabeele, P.; Krysko, D.V.; Nawijn, M.C.; van Oosterhout, A.J. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 2014, 7, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Sumi, Y.; Prefontaine, D.; Al Abri, J.; Al Heialy, N.; Al-Ramli, W.; Michoud, M.C.; Martin, J.G.; Hamid, Q. Epithelium-derived chemokines induce airway smooth muscle cell migration. Clin. Exp. Allergy 2009, 39, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Puddicombe, S.M.; Polosa, R.; Richter, A.; Krishna, M.T.; Howarth, P.H.; Holgate, S.T.; Davies, D.E. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 2000, 14, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Guillot, L.; Le Goffic, R.; Bloch, S.; Escriou, N.; Akira, S.; Chignard, M.; Si-Tahar, M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 2005, 280, 5571–5580. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
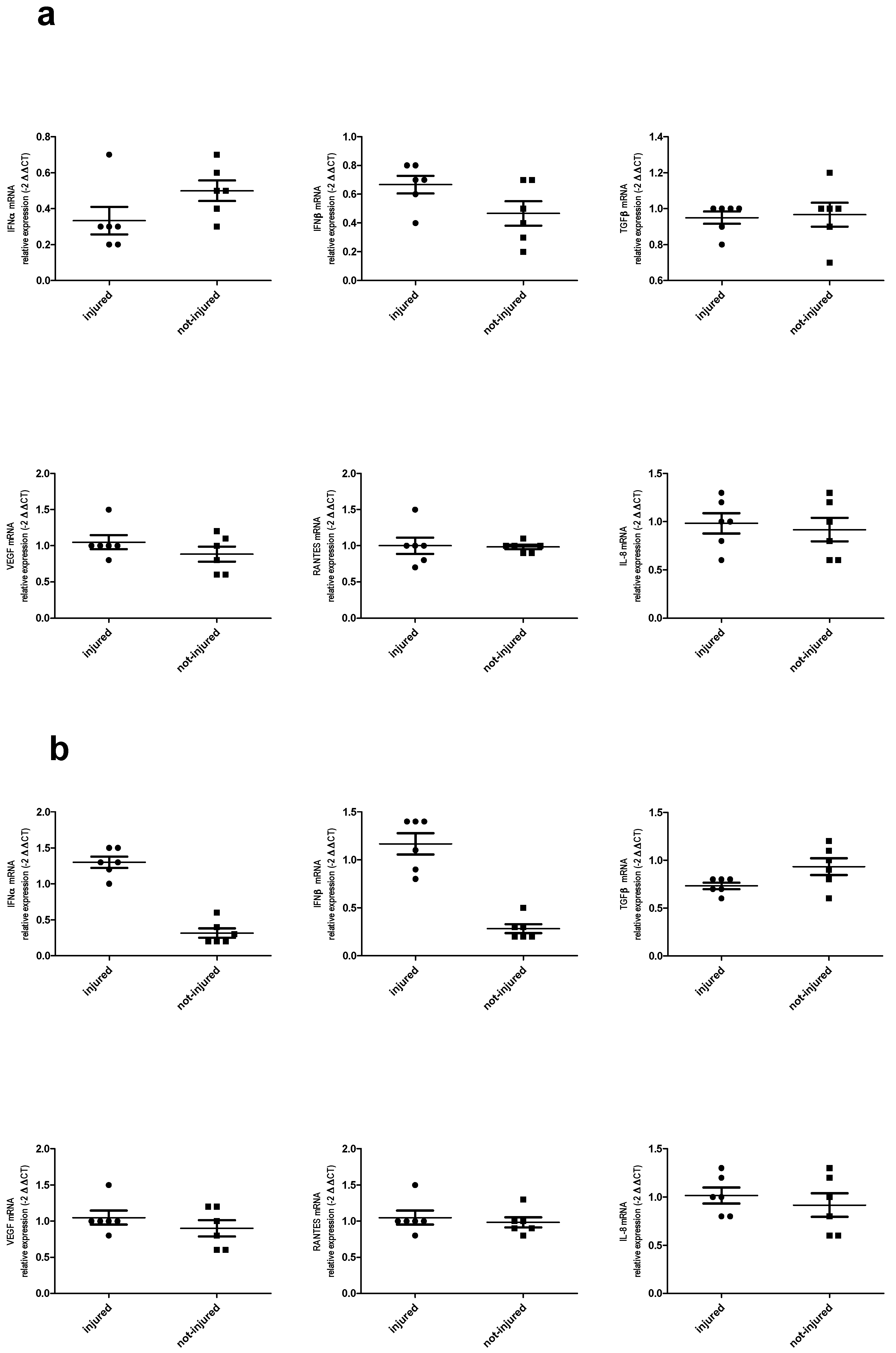{kind=link}
{kind=link}
| (a) | ||||
| Time Post Injury | 0 h | 24 h | ||
| Mean wound area (nm2) | control | LPS 1 μg/mL | control | LPS 1 μg/mL |
| 143.2 ± 13.8 | 151.8 ± 9.9 | 22.1 ± 7.3 | ||
| LPS 10 μg/mL | LPS 10 μg/mL | |||
| 150.2 ± 14.9 | 14.1 ± 4.7 | 56.8 ± 14.3 | ||
| LPS 50 μg/mL | LPS 50 μg/mL | |||
| 137.2 ± 16.1 | 69.2 ± 12.2 | |||
| Time Post Injury | 0 h | 24 h | ||
| Mean wound area (nm2) | control | poly(I:C) 0.1 μg/mL | control | poly(I:C) 0.1 μg/mL |
| 133.6 ± 13.1 | 151.8 ± 9.9 | 7.7 ± 4.9 | ||
| poly(I:C) 1 μg/mL | 3.7 ± 2.6 | poly(I:C) 1 μg/ml | ||
| 150.2 ± 14.9 | 33.1 ± 3.5 | |||
| poly(I:C) 10 μg/mL | poly(I:C) 10 μg/ml | |||
| 137.2 ± 16.1 | 73.1 ± 4.8 | |||
| (b) | ||||
| Time Post Injury | 0 h | 24 h | ||
| Mean wound area (nm2) | control | RV1b 24 h supernatant | control | RV1b 24 h supernatant |
| 163.4 ± 14.5 | 143.7 ± 12.7 | 17.4 ± 6.7 | 33.8 ± 18.1 | |
| control | RV1b 48 h supernatant | control | RV1b 48 h supernatant | |
| 167.9 ± 13.6 | 150.2 ± 14.9 | 1.1 ± 0.8 | 44.6 ± 23.9 | |
| control | RV1b 72 h supernatant | control | RV1b 72 h supernatant | |
| 146.9 ± 19.1 | 137.2 ± 16.1 | 1.1 ± 0.7 | 23.1 ± 11.6 | |
| Mean wound area (nm2) | control | PIV3 24 h supernatant | control | PIV3 24 h supernatant |
| 168.0 ± 18.6 | 151.0 ± 18.5 | 0 | 66.6 ± 36.8 | |
| control | PIV3 48 h supernatant | control | PIV3 48 h supernatant | |
| 120.2 ± 12.7 | 147.7 ± 13.3 | 11.6 ± 8.7 | 73.9 ± 32.1 | |
| control | PIV3 72 h supernatant | control | PIV3 72 h supernatant | |
| 120.8 ± 13.7 | 137.0 ± 15.5 | 0 | 90.2 ± 35.3 | |
| Injured versus Not-Injured | LPS | poly(I:C) | |||
|---|---|---|---|---|---|
| Not-Injured | Injured | Not-Injured | Injured | ||
| IFN-α | ↓ | ↓ | ↓ | ↓ | ↑ |
| IFN-β | ↓ | ↓ | ↓ | ↓ | ↑ |
| TGF-β | ↑ | NS | NS | NS | ↑ |
| VEGF | ↓ | NS | NS | NS | NS |
| RANTES | ↑ | NS | NS | NS | NS |
| IL-8 | ↓ | NS | NS | NS | NS |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewandowska-Polak, A.; Brauncajs, M.; Jarzębska, M.; Pawełczyk, M.; Kurowski, M.; Chałubiński, M.; Makowska, J.; Kowalski, M.L. Toll-Like Receptor Agonists Modulate Wound Regeneration in Airway Epithelial Cells. Int. J. Mol. Sci. 2018, 19, 2456. https://doi.org/10.3390/ijms19082456
Lewandowska-Polak A, Brauncajs M, Jarzębska M, Pawełczyk M, Kurowski M, Chałubiński M, Makowska J, Kowalski ML. Toll-Like Receptor Agonists Modulate Wound Regeneration in Airway Epithelial Cells. International Journal of Molecular Sciences. 2018; 19(8):2456. https://doi.org/10.3390/ijms19082456
Chicago/Turabian StyleLewandowska-Polak, Anna, Małgorzata Brauncajs, Marzanna Jarzębska, Małgorzata Pawełczyk, Marcin Kurowski, Maciej Chałubiński, Joanna Makowska, and Marek L. Kowalski. 2018. "Toll-Like Receptor Agonists Modulate Wound Regeneration in Airway Epithelial Cells" International Journal of Molecular Sciences 19, no. 8: 2456. https://doi.org/10.3390/ijms19082456