Obatoclax, a BH3 Mimetic, Enhances Cisplatin-Induced Apoptosis and Decreases the Clonogenicity of Muscle Invasive Bladder Cancer Cells via Mechanisms That Involve the Inhibition of Pro-Survival Molecules as Well as Cell Cycle Regulators

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
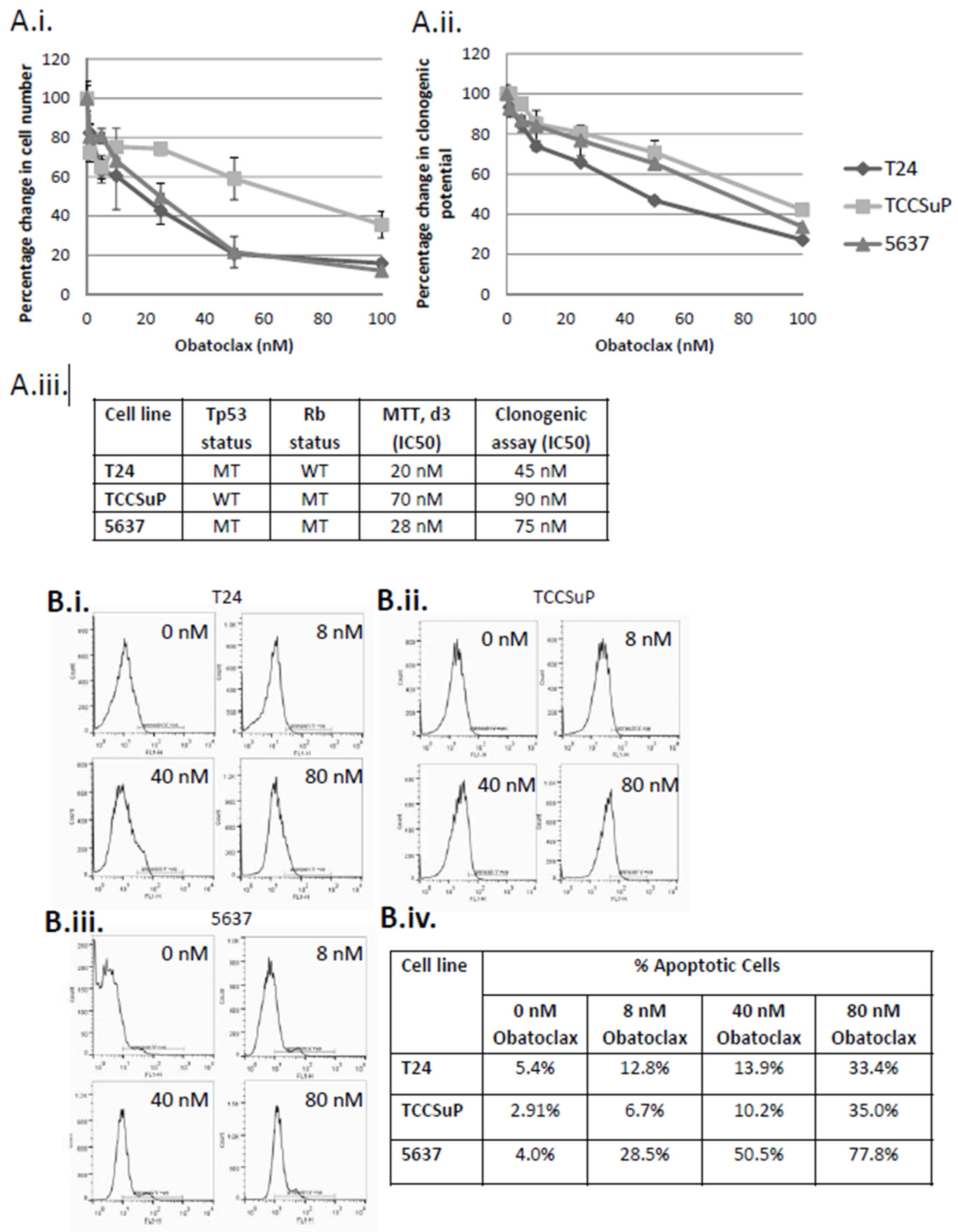
2.1. Treatment of Muscle Invasive Bladder Cancer Cell Lines with Obatoclax Inhibits Cell Proliferation and Clonogenicity and Promotes Apoptosis in a Dose-Dependent Manner
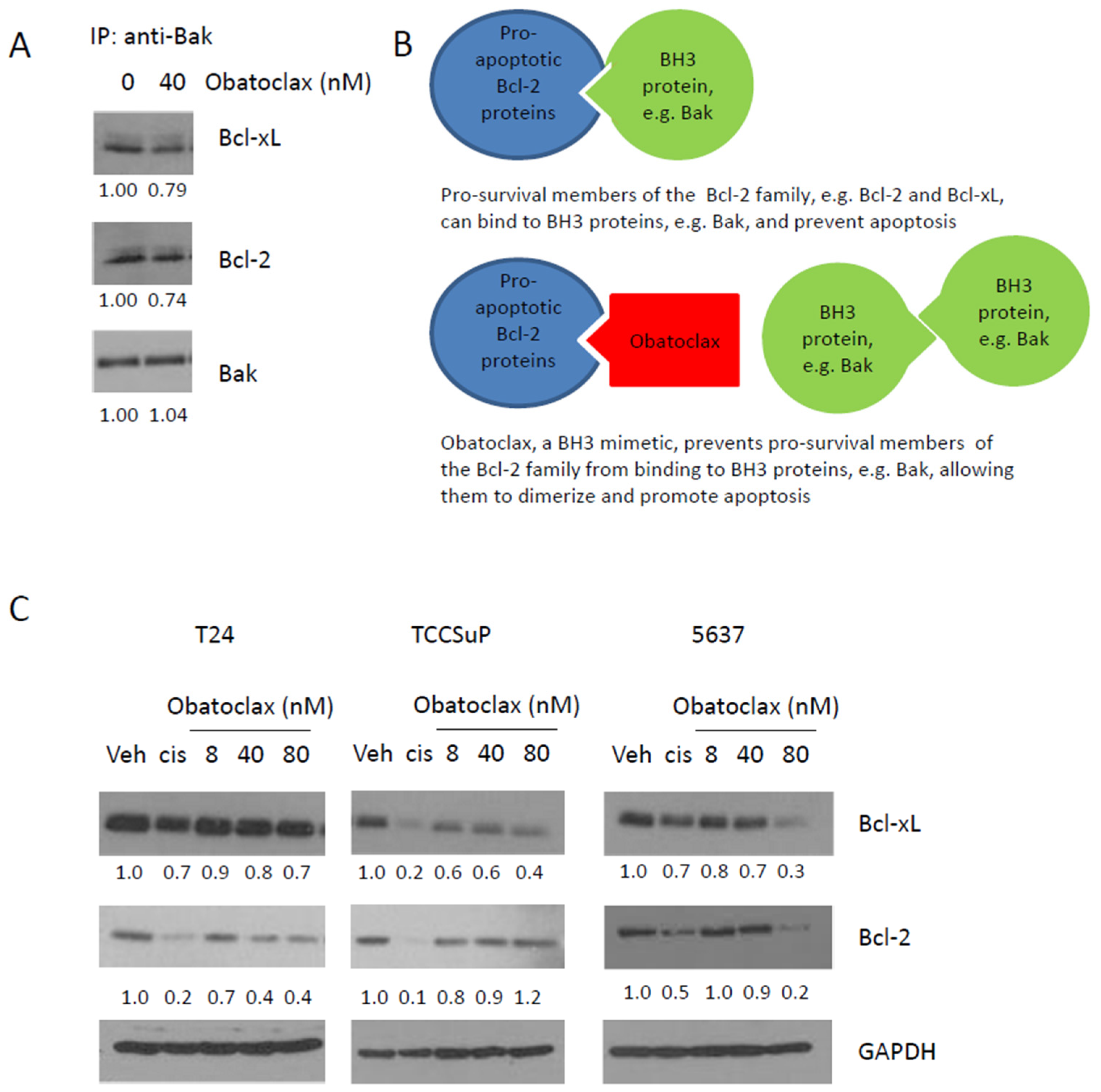
2.2. Obatoclax Can Inhibit the Binding of Bcl-2 and Bcl-xL to Bak, and Decreases Expression Levels of Bcl-2 and Bcl-xL
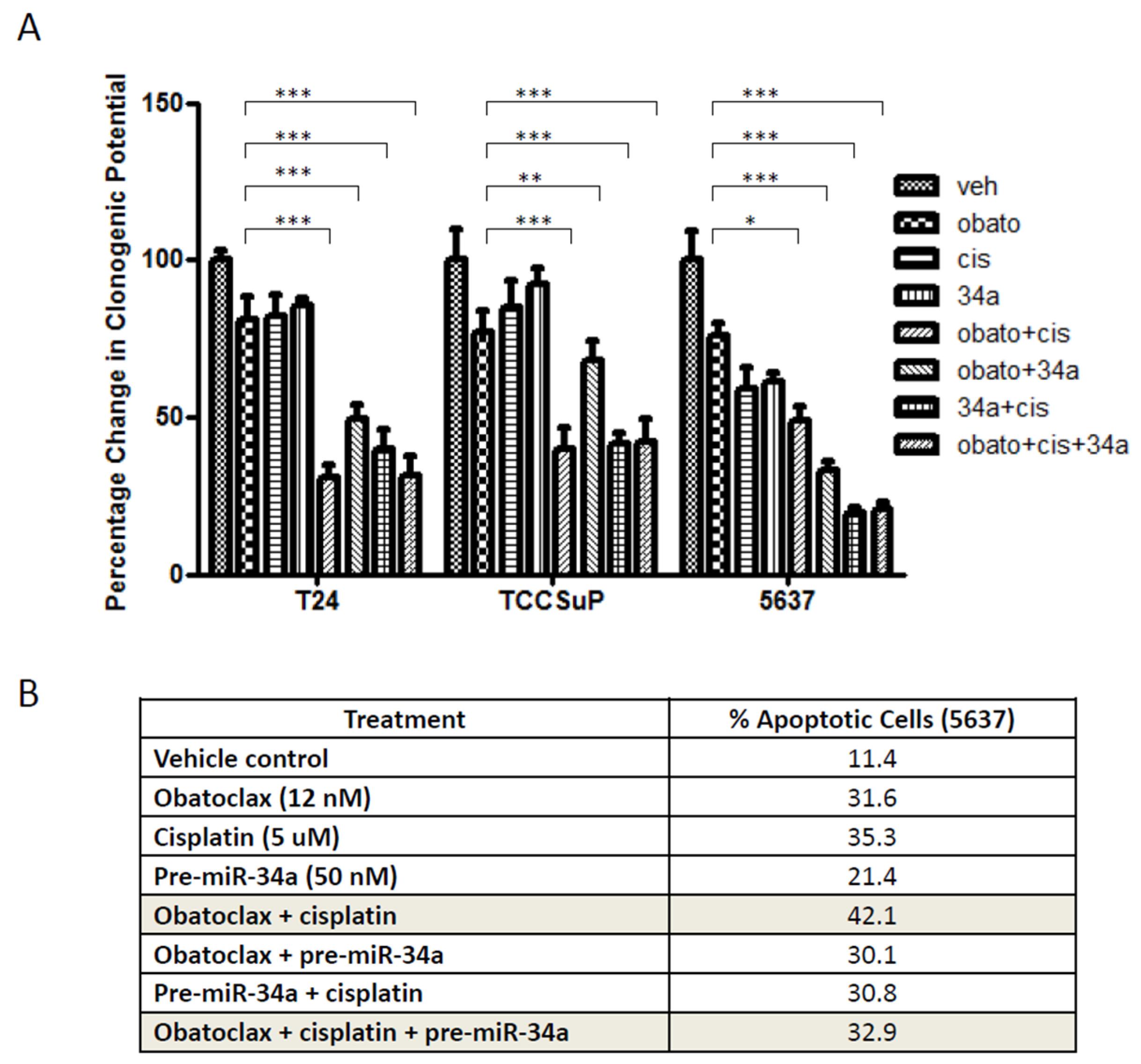
2.3. Combining Obatoclax with Cisplatin, the Standard of Care Treatment for Muscle Invasive Bladder Cancer, Can Further Decrease Cell Clonogenicity and Increase Apoptosis
2.4. Combining pre-miR-34a with Obatoclax and/or Cisplatin Can Decrease Cell Clonogenicity but Also Decreases Levels of Apoptosis
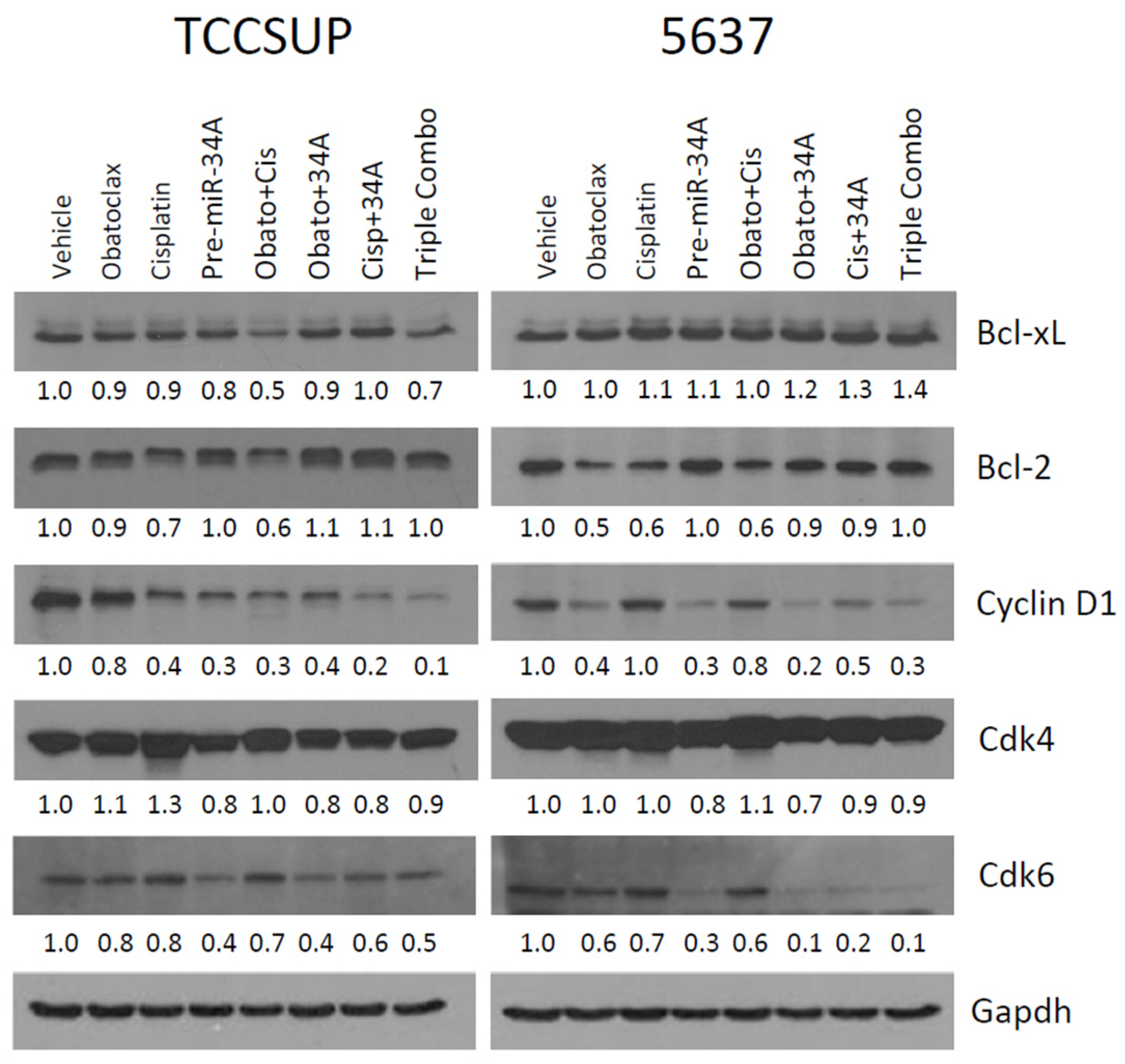
2.5. Treatment with Obatoclax and Cisplatin Can Further Reduce Bcl-2, Bcl-xL, and Cyclin D1 Expression Levels, While Treatment with pre-miR-34a Reduces Cdk6 and Cyclin D1 Expression Levels
2.6. Confirmation That Obatoclax Can Inhibit Cyclin D1 and Cdk6 Expression Levels, and Can Inhibit Cdk4 Expression Levels
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Line and Culture
4.3. Cell Proliferation Assays
4.4. Clonogenic Assay
4.5. Annexin V Flow Cytometry
4.6. Pre-miR-34 Transfection
4.7. Western Blot
4.8. Immunoprecipitation
4.9. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Cis | Cisplatin |
| CLL | Chronic lymphocytic leukemia |
| Cmax | Peak plasma concentration |
| IC30 | Thirty percent of maximal inhibitory concentration |
| IC50 | Half maximal inhibitory concentration |
| IP | Immunoprecipitation |
| MI-BC | Muscle invasive bladder cancer |
| NAC | Neoadjuvant chemotherapy |
| Veh | Vehicle |
References
- Ebbing, J.; Heckmann, R.C.; Collins, J.W.; Miller, K.; Erber, B.; Friedersdorff, F.; Fuller, T.F.; Busch, J.; Seifert, H.H.; Ardelt, P.; et al. Oncological outcomes, quality of life outcomes and complications of partial cystectomy for selected cases of muscle-invasive bladder cancer. Sci. Rep. 2018, 8, 8360. [Google Scholar] [CrossRef]
- Lobo, N.; Mount, C.; Omar, K.; Nair, R.; Thurairaja, R.; Khan, M.S. Landmarks in the treatment of muscle-invasive bladder cancer. Nat. Rev. Urol. 2017, 14, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Dall’Era, M.A.; Cheng, L.; Pan, C.X. Contemporary management of muscle-invasive bladder cancer. Expert Rev. Anticancer Ther. 2012, 12, 941–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, H.B.; Natale, R.B.; Tangen, C.M.; Speights, V.O.; Vogelzang, N.J.; Trump, D.L.; deVere White, R.W.; Sarosdy, M.F.; Wood, D.P., Jr.; Raghavan, D.; et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N. Engl. J. Med. 2003, 349, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef]
- Martin-Doyle, W.; Kwiatkowski, D.J. Molecular biology of bladder cancer. Hematol./Oncol. Clin. N. Am. 2015, 29, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Mari, A.; D’Andrea, D.; Abufaraj, M.; Foerster, B.; Kimura, S.; Shariat, S.F. Genetic determinants for chemo- and radiotherapy resistance in bladder cancer. Transl. Androl. Urol. 2017, 6, 1081–1089. [Google Scholar] [CrossRef]
- Kirsh, E.J.; Baunoch, D.A.; Stadler, W.M. Expression of bcl-2 and bcl-X in bladder cancer. J. Urol. 1998, 159, 1348–1353. [Google Scholar] [CrossRef]
- Gazzaniga, P.; Gradilone, A.; Vercillo, R.; Gandini, O.; Silvestri, I.; Napolitano, M.; Albonici, L.; Vincenzoni, A.; Gallucci, M.; Frati, L.; et al. Bcl-2/bax mRNA expression ratio as prognostic factor in low-grade urinary bladder cancer. Int. J. Cancer 1996, 69, 100–104. [Google Scholar] [CrossRef]
- Wilson, T.R.; Longley, D.B.; Johnston, P.G. Chemoresistance in solid tumours. Ann. Oncol. 2006, 17 (Suppl. 10), x315–x324. [Google Scholar] [CrossRef] [Green Version]
- Kerbel, R.S.; Kobayashi, H.; Graham, C.H. Intrinsic or acquired drug resistance and metastasis: Are they linked phenotypes? J. Cell. Biochem. 1994, 56, 37–47. [Google Scholar] [CrossRef]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Montero, J.; Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef]
- Ittmann, M.; Huang, J.; Radaelli, E.; Martin, P.; Signoretti, S.; Sullivan, R.; Simons, B.W.; Ward, J.M.; Robinson, B.D.; Chu, G.C.; et al. Animal models of human prostate cancer: The consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013, 73, 2718–2736. [Google Scholar] [CrossRef]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Gerecitano, J.; Robak, T.; De la Serna, J.; et al. Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2018, 378, 1107–1120. [Google Scholar] [CrossRef]
- Herbst, R.S.; Frankel, S.R. Oblimersen sodium (Genasense bcl-2 antisense oligonucleotide): A rational therapeutic to enhance apoptosis in therapy of lung cancer. Clin. Cancer Res. 2004, 10, 4245s–4248s. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Dumez, H.; Van Poppel, H.; Skoneczna, I.; Sella, A.; Daugaard, G.; Gil, T.; Graham, J.; Carpentier, P.; Calabro, F.; et al. Docetaxel plus oblimersen sodium (Bcl-2 antisense oligonucleotide): An EORTC multicenter, randomized phase II study in patients with castration-resistant prostate cancer. Ann. Oncol. 2009, 20, 1264–1269. [Google Scholar] [CrossRef]
- Wang, J.L.; Zhang, Z.J.; Choksi, S.; Shan, S.; Lu, Z.; Croce, C.M.; Alnemri, E.S.; Korngold, R.; Huang, Z. Cell permeable Bcl-2 binding peptides: A chemical approach to apoptosis induction in tumor cells. Cancer Res. 2000, 60, 1498–1502. [Google Scholar]
- Pekarsky, Y.; Croce, C.M. Role of miR-15/16 in CLL. Cell Death Differ. 2015, 22, 6–11. [Google Scholar] [CrossRef]
- Friedrich, M.G.; Weisenberger, D.J.; Cheng, J.C.; Chandrasoma, S.; Siegmund, K.D.; Gonzalgo, M.L.; Toma, M.I.; Huland, H.; Yoo, C.; Tsai, Y.C.; et al. Detection of methylated apoptosis-associated genes in urine sediments of bladder cancer patients. Clin. Cancer Res. 2004, 10, 7457–7465. [Google Scholar] [CrossRef]
- Bende, R.J.; Smit, L.A.; van Noesel, C.J. Molecular pathways in follicular lymphoma. Leukemia 2007, 21, 18–29. [Google Scholar] [CrossRef]
- Yamamoto, K.; Okamura, A.; Yakushijin, K.; Hayashi, Y.; Matsuoka, H.; Minami, H. Tandem triplication of the BCL2 gene in CD5-positive intravascular large B cell lymphoma with bone marrow involvement. Ann. Hematol. 2014, 93, 1791–1793. [Google Scholar] [CrossRef]
- Vinall, R.L.; Ripoll, A.Z.; Wang, S.; Pan, C.X.; deVere White, R.W. MiR-34a chemosensitizes bladder cancer cells to cisplatin treatment regardless of p53-Rb pathway status. Int. J. Cancer 2012, 130, 2526–2538. [Google Scholar] [CrossRef]
- Li, X.J.; Ren, Z.J.; Tang, J.H. MicroRNA-34a: A potential therapeutic target in human cancer. Cell Death Dis. 2014, 5, e1327. [Google Scholar] [CrossRef]
- Saito, Y.; Nakaoka, T.; Saito, H. microRNA-34a as a Therapeutic Agent against Human Cancer. J. Clin. Med. 2015, 4, 1951–1959. [Google Scholar] [CrossRef]
- McConkey, D.J.; Choi, W.; Dinney, C.P. Genetic subtypes of invasive bladder cancer. Curr. Opin. Urol. 2015, 25, 449–458. [Google Scholar] [CrossRef]
- Xiao, Z.; Chen, Y. Small molecule targeting miR-34a for cancer therapy. Mol. Cell. Oncol. 2015, 2, e977160. [Google Scholar] [CrossRef]
- Li, H.; Yu, G.; Shi, R.; Lang, B.; Chen, X.; Xia, D.; Xiao, H.; Guo, X.; Guan, W.; Ye, Z.; et al. Cisplatin-induced epigenetic activation of miR-34a sensitizes bladder cancer cells to chemotherapy. Mol. Cancer 2014, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Tian, J.; Xian, W.; Xie, T.; Yang, X. miR-34a inhibits proliferation and invasion of bladder cancer cells by targeting orphan nuclear receptor HNF4G. Dis. Mark. 2015, 2015, 879254. [Google Scholar] [CrossRef]
- Yu, G.; Yao, W.; Xiao, W.; Li, H.; Xu, H.; Lang, B. MicroRNA-34a functions as an anti-metastatic microRNA and suppresses angiogenesis in bladder cancer by directly targeting CD44. J. Exp. Clin. Cancer Res. 2014, 33, 779. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, V.; Winkler, J. miRNA-based therapies: Strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Futur. Med. Chem. 2014, 6, 1967–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.Y.; Ferrajoli, A.; Sood, A.K.; Lopez-Berestein, G.; Calin, G.A. microRNA Therapeutics in Cancer—An Emerging Concept. EBioMedicine 2016, 12, 34–42. [Google Scholar] [CrossRef]
- Goard, C.A.; Schimmer, A.D. An evidence-based review of obatoclax mesylate in the treatment of hematological malignancies. Core Evid. 2013, 8, 15–26. [Google Scholar] [CrossRef]
- Li, J.; Viallet, J.; Haura, E.B. A small molecule pan-Bcl-2 family inhibitor, GX15-070, induces apoptosis and enhances cisplatin-induced apoptosis in non-small cell lung cancer cells. Cancer Chemother. Pharmacol. 2008, 61, 525–534. [Google Scholar] [CrossRef]
- Arisan, E.D.; Kutuk, O.; Tezil, T.; Bodur, C.; Telci, D.; Basaga, H. Small inhibitor of Bcl-2, HA14-1, selectively enhanced the apoptotic effect of cisplatin by modulating Bcl-2 family members in MDA-MB-231 breast cancer cells. Breast Cancer Res. Treat. 2010, 119, 271–281. [Google Scholar] [CrossRef]
- Yu, L.; Wu, W.K.; Gu, C.; Zhong, D.; Zhao, X.; Kong, Y.; Lin, Q.; Chan, M.T.; Zhou, Z.; Liu, S. Obatoclax impairs lysosomal function to block autophagy in cisplatin-sensitive and -resistant esophageal cancer cells. Oncotarget 2016, 7, 14693–14707. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Guerrero, R.; Gasca, J.; Flores, M.L.; Perez-Valderrama, B.; Tejera-Parrado, C.; Medina, R.; Tortolero, M.; Romero, F.; Japon, M.A.; Saez, C. Obatoclax and Paclitaxel Synergistically Induce Apoptosis and Overcome Paclitaxel Resistance in Urothelial Cancer Cells. Cancers 2018, 10, 490. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Meeks, J.J.; Kuzel, T.M.; Scaranti, M.; Abdulkadir, S.A.; Giles, F.J. Emerging therapeutic targets in bladder cancer. Cancer Treat. Rev. 2015, 41, 170–178. [Google Scholar] [CrossRef]
- Or, C.R.; Chang, Y.; Lin, W.C.; Lee, W.C.; Su, H.L.; Cheung, M.W.; Huang, C.P.; Ho, C.; Chang, C.C. Obatoclax, a Pan-BCL-2 Inhibitor, Targets Cyclin D1 for Degradation to Induce Antiproliferation in Human Colorectal Carcinoma Cells. Int. J. Mol. Sci. 2016, 18, 44. [Google Scholar] [CrossRef]
- Koehler, B.C.; Scherr, A.L.; Lorenz, S.; Elssner, C.; Kautz, N.; Welte, S.; Jaeger, D.; Urbanik, T.; Schulze-Bergkamen, H. Pan-Bcl-2 inhibitor obatoclax delays cell cycle progression and blocks migration of colorectal cancer cells. PLoS ONE 2014, 9, e106571. [Google Scholar] [CrossRef]
- Schettini, F.; De Santo, I.; Rea, C.G.; De Placido, P.; Formisano, L.; Giuliano, M.; Arpino, G.; De Laurentiis, M.; Puglisi, F.; De Placido, S.; et al. CDK 4/6 Inhibitors as Single Agent in Advanced Solid Tumors. Front. Oncol. 2018, 8, 608. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2018, 139, 471–488. [Google Scholar] [CrossRef]
- Sathe, A.; Koshy, N.; Schmid, S.C.; Thalgott, M.; Schwarzenbock, S.M.; Krause, B.J.; Holm, P.S.; Gschwend, J.E.; Retz, M.; Nawroth, R. CDK4/6 Inhibition Controls Proliferation of Bladder Cancer and Transcription of RB1. J. Urol. 2016, 195, 771–779. [Google Scholar] [CrossRef]
- Shariat, S.F.; Tokunaga, H.; Zhou, J.; Kim, J.; Ayala, G.E.; Benedict, W.F.; Lerner, S.P. p53, p21, pRB, and p16 expression predict clinical outcome in cystectomy with bladder cancer. J. Clin. Oncol. 2004, 22, 1014–1024. [Google Scholar] [CrossRef]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef]
- Hong, J.H.; Lee, E.; Hong, J.; Shin, Y.J.; Ahn, H. Antisense Bcl2 oligonucleotide in cisplatin-resistant bladder cancer cell lines. BJU Int. 2002, 90, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Ho, J.N.; Jin, H.; Lee, S.C.; Lee, S.E.; Hong, S.K.; Lee, J.W.; Lee, E.S.; Byun, S.S. Upregulated expression of BCL2, MCM7, and CCNE1 indicate cisplatin-resistance in the set of two human bladder cancer cell lines: T24 cisplatin sensitive and T24R2 cisplatin resistant bladder cancer cell lines. Investig. Clin. Urol. 2016, 57, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.J.; Kim, J.K.; Kim, K.D.; Yoon, H.K.; Cho, M.Y.; Park, Y.P.; Jeon, J.H.; Lee, E.S.; Byun, S.S.; Lim, H.M.; et al. Upregulation of Bcl-2 is associated with cisplatin-resistance via inhibition of Bax translocation in human bladder cancer cells. Cancer Lett. 2006, 237, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Cooke, P.W.; James, N.D.; Ganesan, R.; Burton, A.; Young, L.S.; Wallace, D.M. Bcl-2 expression identifies patients with advanced bladder cancer treated by radiotherapy who benefit from neoadjuvant chemotherapy. BJU Int. 2000, 85, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Yoshimine, S.; Kikuchi, E.; Kosaka, T.; Mikami, S.; Miyajima, A.; Okada, Y.; Oya, M. Prognostic significance of Bcl-xL expression and efficacy of Bcl-xL targeting therapy in urothelial carcinoma. Br. J. Cancer 2013, 108, 2312–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karam, J.A.; Lotan, Y.; Karakiewicz, P.I.; Ashfaq, R.; Sagalowsky, A.I.; Roehrborn, C.G.; Shariat, S.F. Use of combined apoptosis biomarkers for prediction of bladder cancer recurrence and mortality after radical cystectomy. Lancet Oncol. 2007, 8, 128–136. [Google Scholar] [CrossRef]
- Kong, G.; Shin, K.Y.; Oh, Y.H.; Lee, J.J.; Park, H.Y.; Woo, Y.N.; Lee, J.D. Bcl-2 and p53 expressions in invasive bladder cancers. Acta Oncol. 1998, 37, 715–720. [Google Scholar] [CrossRef]
- Liang, L.Z.; Ma, B.; Liang, Y.J.; Liu, H.C.; Zhang, T.H.; Zheng, G.S.; Su, Y.X.; Liao, G.Q. Obatoclax induces Beclin 1- and ATG5-dependent apoptosis and autophagy in adenoid cystic carcinoma cells. Oral Dis. 2015, 21, 470–477. [Google Scholar] [CrossRef]
- Pan, J.; Cheng, C.; Verstovsek, S.; Chen, Q.; Jin, Y.; Cao, Q. The BH3-mimetic GX15-070 induces autophagy, potentiates the cytotoxicity of carboplatin and 5-fluorouracil in esophageal carcinoma cells. Cancer Lett. 2010, 293, 167–174. [Google Scholar] [CrossRef]
- Champa, D.; Orlacchio, A.; Patel, B.; Ranieri, M.; Shemetov, A.A.; Verkhusha, V.V.; Cuervo, A.M.; Di Cristofano, A. Obatoclax kills anaplastic thyroid cancer cells by inducing lysosome neutralization and necrosis. Oncotarget 2016, 7, 34453–34471. [Google Scholar] [CrossRef] [Green Version]
- Stamelos, V.A.; Fisher, N.; Bamrah, H.; Voisey, C.; Price, J.C.; Farrell, W.E.; Redman, C.W.; Richardson, A. The BH3 Mimetic Obatoclax Accumulates in Lysosomes and Causes Their Alkalinization. PLoS ONE 2016, 11, e0150696. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Nomura, M.; Shuck, R.; Yustein, J. Cancer’s Achilles’ Heel: Apoptosis and Necroptosis to the Rescue. Int. J. Mol. Sci. 2016, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Vinall, R.L.; Tepper, C.G.; Shi, X.B.; Xue, L.A.; Gandour-Edwards, R.; de Vere White, R.W. The R273H p53 mutation can facilitate the androgen-independent growth of LNCaP by a mechanism that involves H2 relaxin and its cognate receptor LGR7. Oncogene 2006, 25, 2082–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steele, T.M.; Talbott, G.C.; Sam, A.; Tepper, C.G.; Ghosh, P.M.; Vinall, R.L. Obatoclax, a BH3 Mimetic, Enhances Cisplatin-Induced Apoptosis and Decreases the Clonogenicity of Muscle Invasive Bladder Cancer Cells via Mechanisms That Involve the Inhibition of Pro-Survival Molecules as Well as Cell Cycle Regulators. Int. J. Mol. Sci. 2019, 20, 1285. https://doi.org/10.3390/ijms20061285
Steele TM, Talbott GC, Sam A, Tepper CG, Ghosh PM, Vinall RL. Obatoclax, a BH3 Mimetic, Enhances Cisplatin-Induced Apoptosis and Decreases the Clonogenicity of Muscle Invasive Bladder Cancer Cells via Mechanisms That Involve the Inhibition of Pro-Survival Molecules as Well as Cell Cycle Regulators. International Journal of Molecular Sciences. 2019; 20(6):1285. https://doi.org/10.3390/ijms20061285
Chicago/Turabian StyleSteele, Thomas M., George C. Talbott, Anhao Sam, Clifford G. Tepper, Paramita M. Ghosh, and Ruth L. Vinall. 2019. "Obatoclax, a BH3 Mimetic, Enhances Cisplatin-Induced Apoptosis and Decreases the Clonogenicity of Muscle Invasive Bladder Cancer Cells via Mechanisms That Involve the Inhibition of Pro-Survival Molecules as Well as Cell Cycle Regulators" International Journal of Molecular Sciences 20, no. 6: 1285. https://doi.org/10.3390/ijms20061285