
Discovery of New 3,3-Diethylazetidine-2,4-dione Based Thiazoles as Nanomolar Human Neutrophil Elastase Inhibitors with Broad-Spectrum Antiproliferative Activity

 , , and
, , and
Abstract
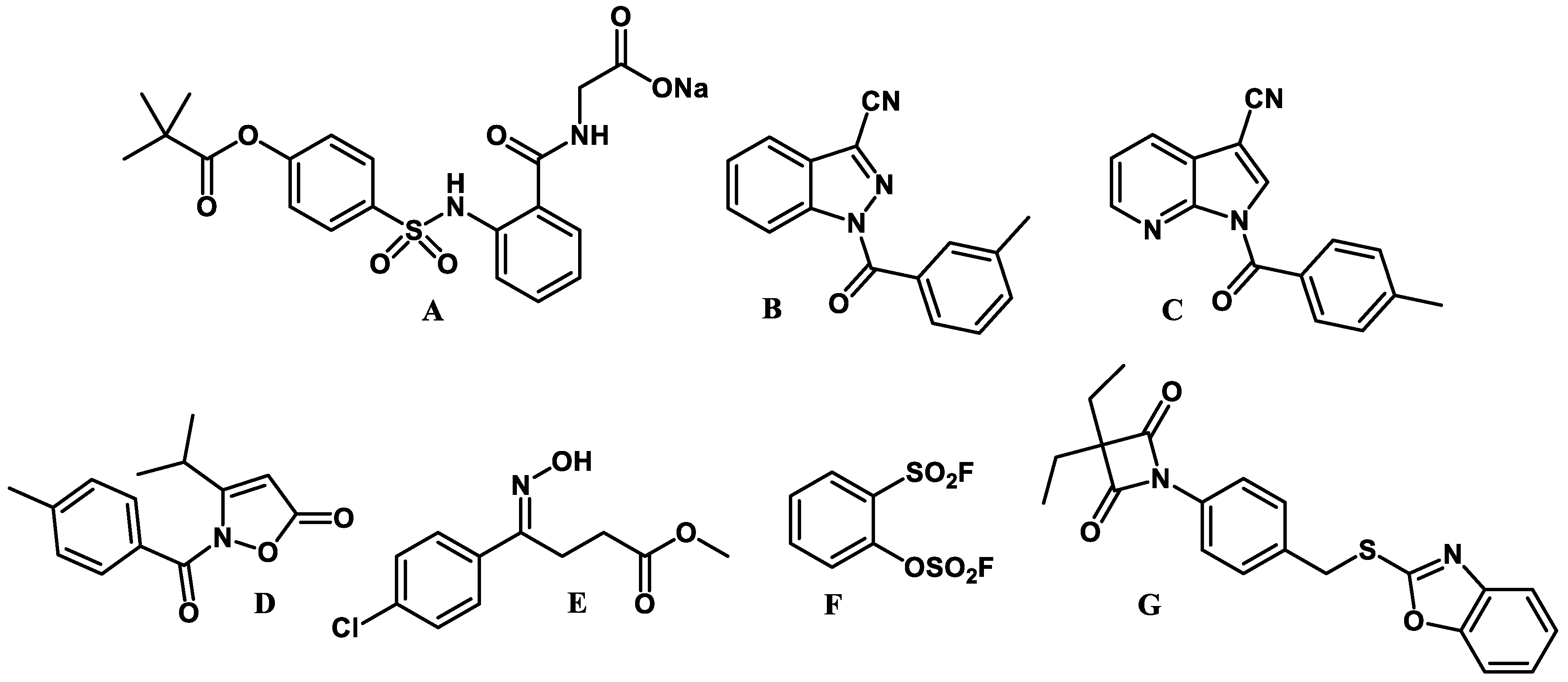
:1. Introduction
2. Results and Discussion
2.1. Chemical Synthesis
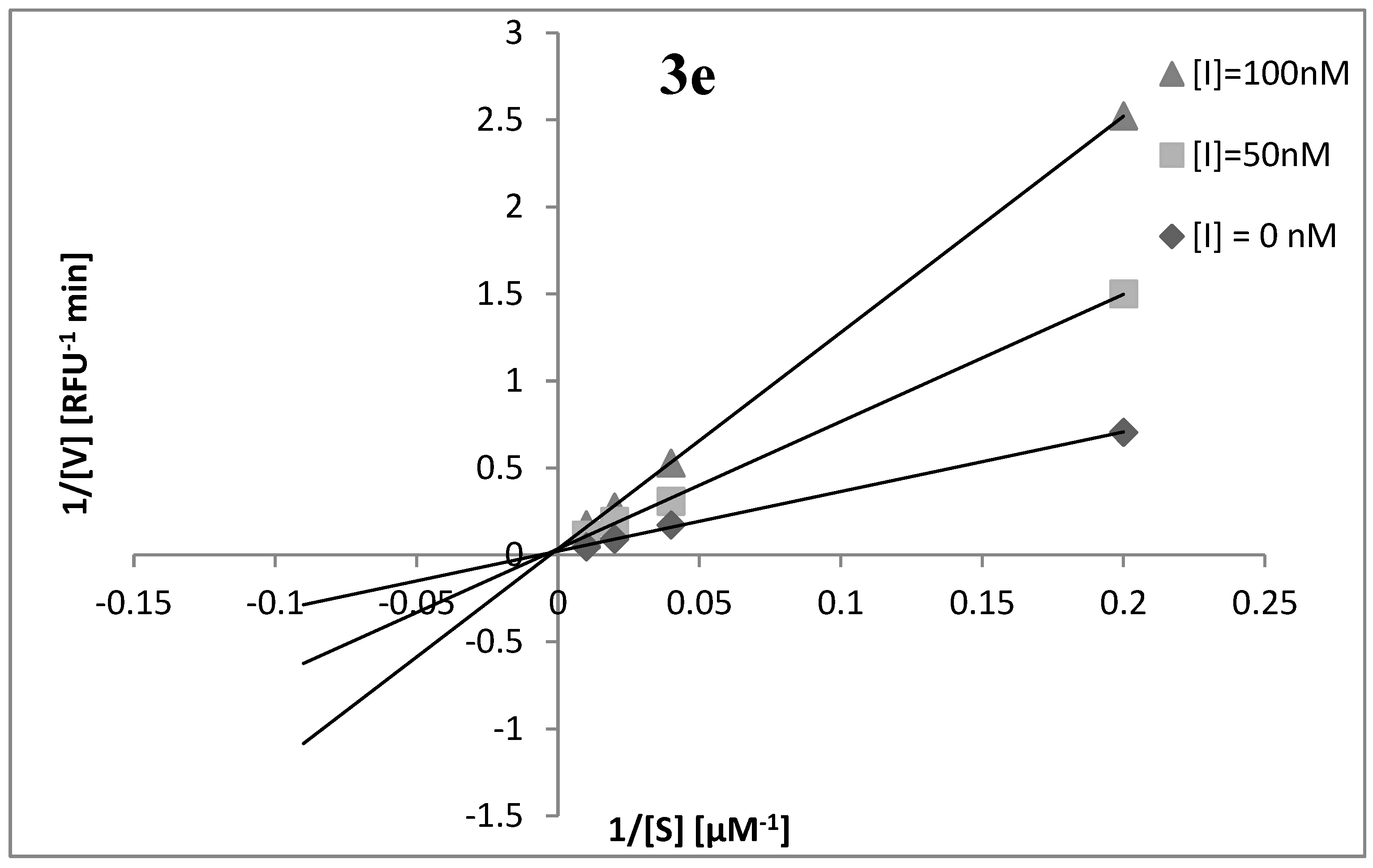
2.2. Elastase Inhibitory Activity and Kinetic Analysis
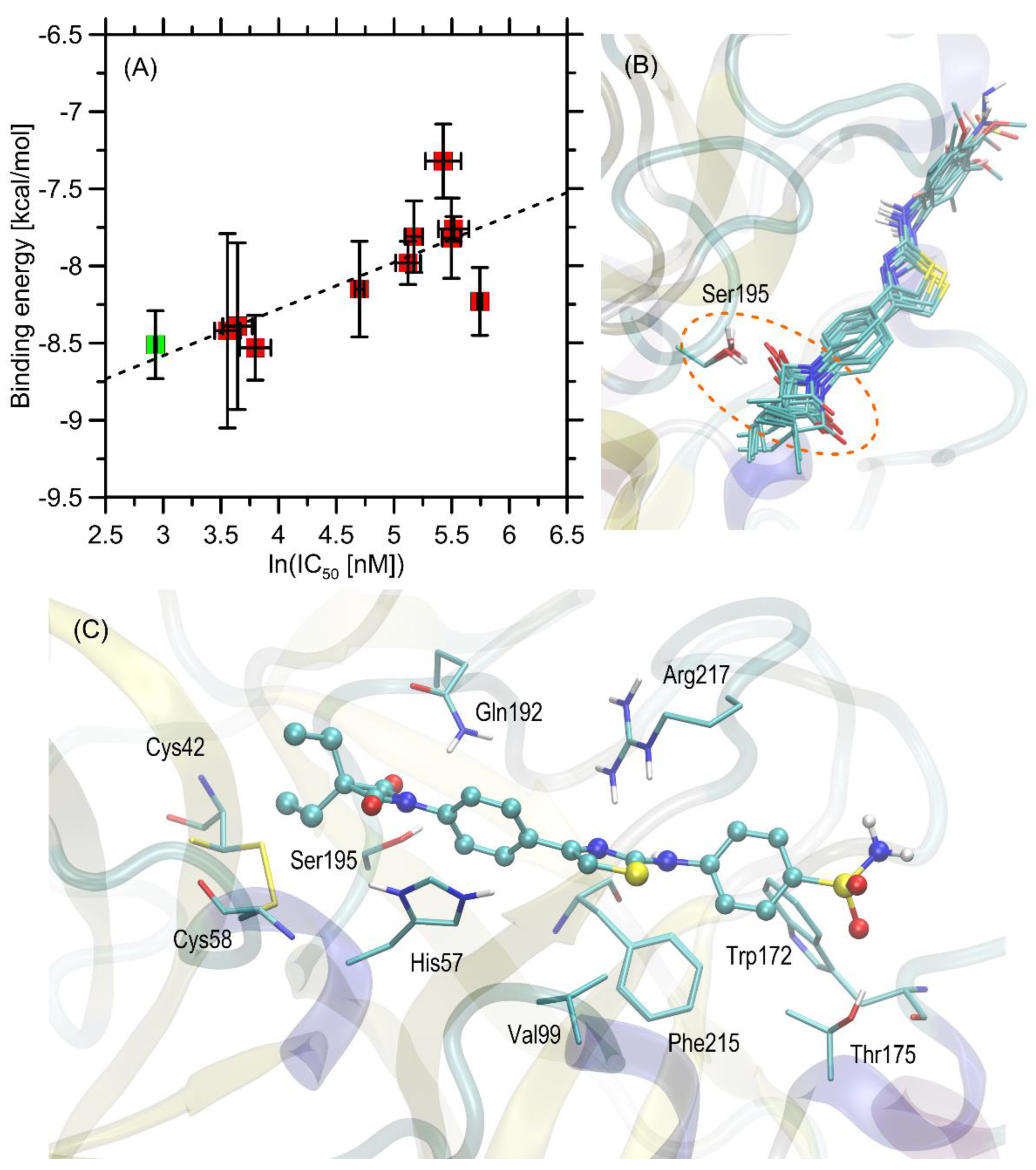
2.3. Molecular Docking Study
2.4. Antiproliferative Activity
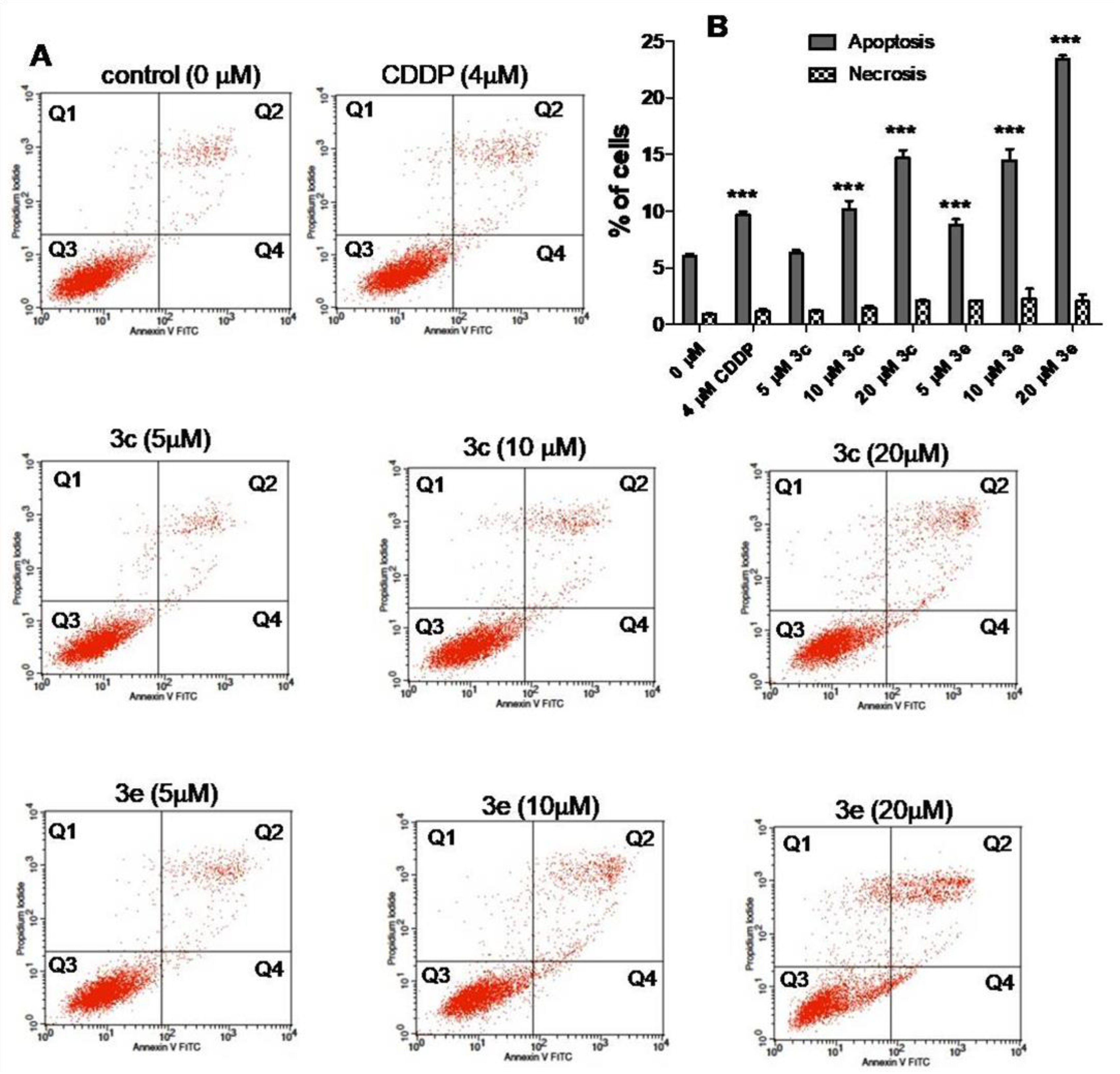
2.5. Apoptosis and Cell Cycle Assays
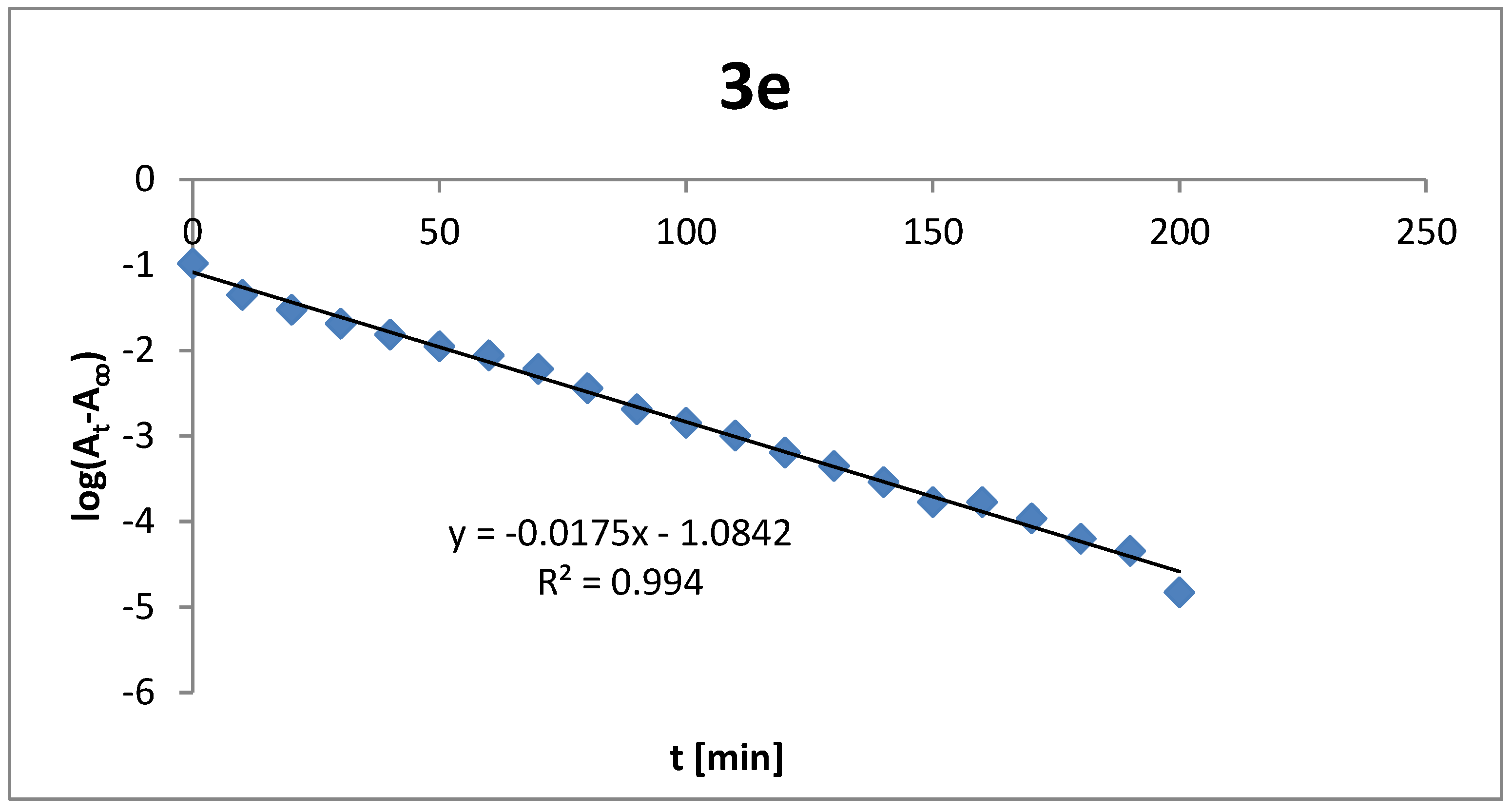
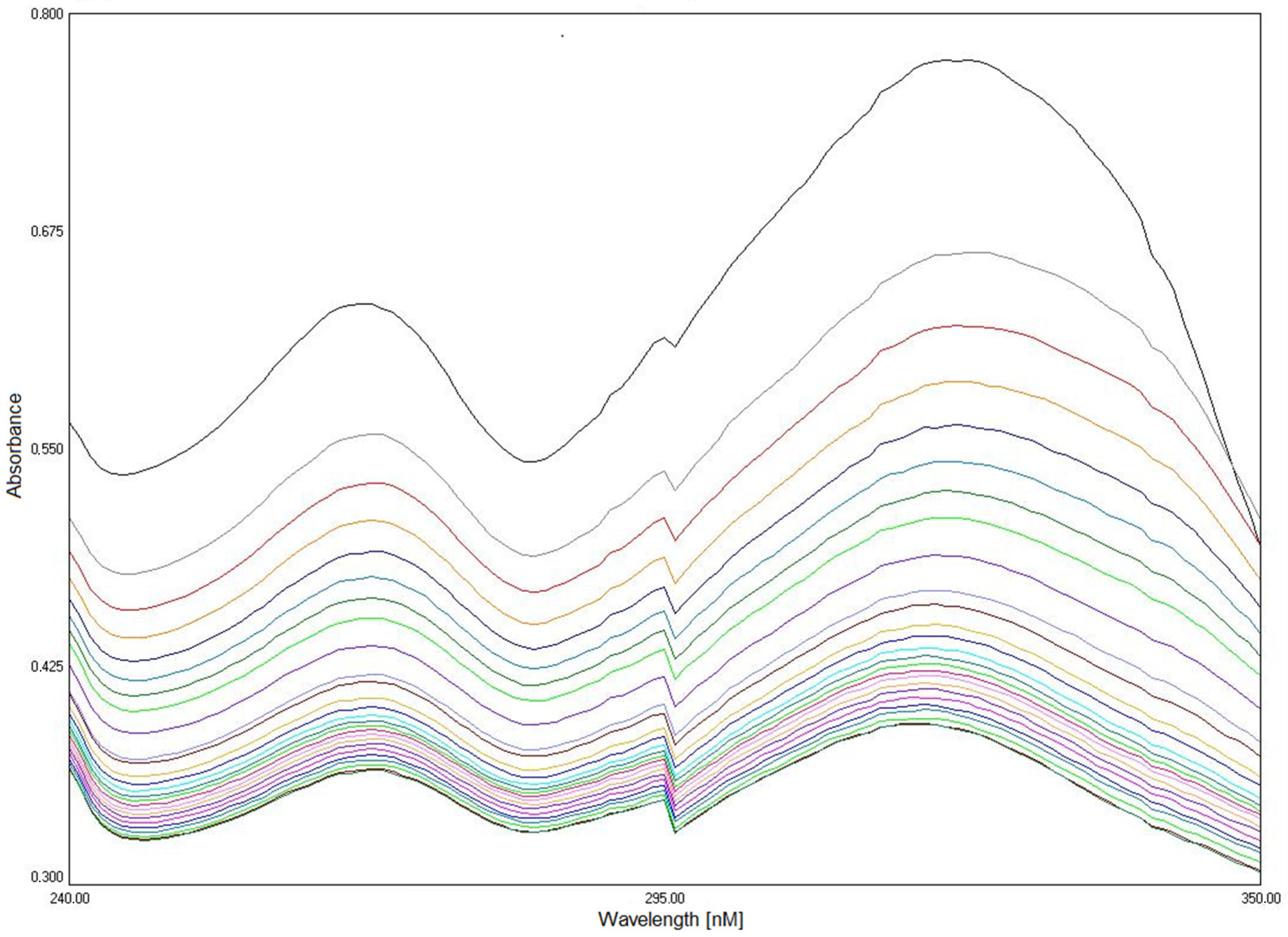
2.6. Compounds Stability
3. Materials and Methods
3.1. Chemistry
3.1.1. 1-(4-(2-Chloroacetyl)phenyl)-3,3-diethylazetidine-2,4-dione (2)
3.1.2. 1-(4-(2-(4-Chlorophenylamino)thiazol-4-yl)phenyl)-3,3-diethylazetidine-2,4-dione (3a) Typical Procedure
3.1.3. 3,3-Diethyl-1-(4-(2-(4-fluorophenylamino)thiazol-4-yl)phenyl)azetidine-2,4-dione (3b)
3.1.4. 3,3-Diethyl-1-(4-(2-(3,4,5-trimethoxyphenylamino)thiazol-4-yl)phenyl)azetidine-2,4-dione (3c)
3.1.5. 3,3-Diethyl-1-(4-(2-(perfluorophenylamino)thiazol-4-yl)phenyl)azetidine-2,4-dione (3d)
3.1.6. 4-(4-(4-(3,3-Diethyl-2,4-dioxoazetidin-1-yl)phenyl)thiazol-2-ylamino)benzenesulfo-namide (3e)
3.1.7. 1-(4-(2-(3,5-Dimethylphenylamino)thiazol-4-yl)phenyl)-3,3-diethylazetidine-2,4-dione (3f)
3.1.8. 1-(4-(2-(1H-Indazol-5-ylamino)thiazol-4-yl)phenyl)-3,3-diethylazetidine-2,4-dione (3g)
3.1.9. 3,3-Diethyl-1-(4-(2-(naphthalen-1-ylamino)thiazol-4-yl)phenyl)azetidine-2,4-dione (3h)
3.1.10. 3,3-Diethyl-1-(4-(2-(4-(trifluoromethyl)phenylamino)thiazol-4-yl)phenyl)-azetidine-2,4-dione (3i)
3.1.11. 3,3-Diethyl-1-(4-(2-(2,4,6-trichlorophenylamino)thiazol-4-yl)phenyl)azetidine-2,4-dione (3j)
3.2. Human Neutrophil Elastase Inhibition Assay
Kinetic Analysis of the Inhibition of Human Neutrophil Elastase
3.3. Molecular Docking Study
3.4. Antiproliferative Activity
3.5. Apoptosis and Cell Cycle Assays
3.6. Analysis of Compounds Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, W.L.; Downey, G.P. Leukocyte elastase: Physiological functions and role in acute lung injury. Am. J. Respir. Crit. Care Med. 2001, 64, 896–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, T.; Ekholm, K.; Gränse, M.; Lindahl, M.; Kozma, V.; Jungar, C.; Ottosson, T.; Falk-Håkansson, H.; Churg, A.; Wright, J.L.; et al. AZD9668: Pharmacological characterization of a novel oral inhibitor of neutrophil elastase. J. Pharmacol. Exp. Ther. 2011, 339, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dengler, V.; Downey, G.P.; Tuder, R.M.; Eltzschig, H.K.; Schmidt, E.P. Neutrophil intercellular communication in acute lung injury: Emerging roles of microparticles and gap junctions. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, J.; Dubin, A.; Potempa, J.; Watorek, W.; Kurdowska, A. Neutrophil proteinases. Ann. N. Y. Acad. Sci. 1991, 624, 81–86. [Google Scholar] [CrossRef]
- Zhou, X.; Dai, Q.; Huang, X. Neutrophils in acute lung injury. Front. Biosci. 2012, 17, 2278–2283. [Google Scholar] [CrossRef]
- Carrell, R.W.; Jeppsson, J.O.; Laurell, C.B.; Brennan, S.O.; Owen, M.C.; Vaughan, L.; Boswell, D.R. Structure and variation of human-1-antitrypsin. Nature 1982, 298, 329–334. [Google Scholar] [CrossRef]
- Thomson, R.C.; Ohlsson, K. Isolation, properties, and complete amino acid sequence of human secretory leukocyte protease inhibitor: A potent inhibitor of leukocyte elastase. Proc. Natl. Acad. Sci. USA 1986, 83, 6692–6696. [Google Scholar] [CrossRef] [Green Version]
- Reilly, C.F.; Travis, J. The degradation of human lung elastin by neutrophil proteinases. Biochim. Biophys. Acta 1980, 621, 147–157. [Google Scholar] [CrossRef]
- Moroy, G.; Alix, A.J.; Sapi, J.; Hornebeck, W.; Bourguet, E. Neutrophil elastase as a target in lung cancer. Anticancer Agents Med. Chem. 2012, 12, 565–579. [Google Scholar] [CrossRef]
- Sato, T.; Takahashi, S.; Mizumoto, T.; Harao, M.; Akuziki, M.; Takasugi, M.; Fukutomi, T.; Yamashita, J.I. Neutrophil elastase and cancer. Surg. Oncol. 2006, 15, 217–222. [Google Scholar] [CrossRef]
- Akizuki, M.; Fukutomi, T.; Takasugi, M.; Takahashi, S.; Sato, T.; Harao, M.; Mizumoto, T.; Yamashita, J. Prognostic significance of immunoreactive neutrophil elastase in human breast cancer: Long-term follow-up results in 313 patients. Neoplasia 2007, 9, 260–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerman, I.; Garcia-Hernandez, M.L.; Rangel-Moreno, J.; Chiriboga, L.; Pan, C.; Nastiuk, K.L.; Krolewski, J.J.; Sen, A.; Hammes, S.R. Infiltrating myeloid cells exert protumorigenic actions via neutrophil elastase. Mol. Cancer Res. 2017, 15, 1138–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerman, I.; Hammes, S.R. Neutrophil elastase in the tumor microenvironment. Steroids 2017, 133, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Vaguliene, N.; Zemaitis, M.; Lavinskiene, S.; Miliauskas, S.; Sakalauskas, R. Local and systemic neutrophil inflammation in patients with lung cancer and chronic obstructive pulmonary disease. BMC Immunol. 2013, 14, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, K.K.; Wingate, H.; Yokota, T.; Liu, Y.; Mills, G.B.; Zhang, F.; Fang, B.; Su, C.-H.; Zhang, M.; Yi, M.; et al. Elafin, an inhibitor of elastase, is a prognostic indicator in breast cancer. Breast Cancer Res. 2013, 15, R3. [Google Scholar] [CrossRef] [Green Version]
- Goulet, B.; Markovic, Y.; Leduy, L.; Nepveu, A. Proteolytic processing of cut homeobox 1 by neutrophil elastase in the MV4; 11 myeloid leukemia cell line. Mol. Cancer Res. 2008, 6, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Houghton, A.M.; Rzymkiewicz, D.M.; Ji, H.; Gregory, A.D.; Egea, E.E.; Metz, H.E.; Stolz, D.B.; Land, S.R.; Marconcini, L.A.; Kliment, C.R.; et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med. 2010, 16, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Inada, M.; Yamashita, J.; Ogawa, M. Neutrophil elastase inhibitor (ONO-5046-Na) inhibits the growth of human lung cancer cell lines transplanted into severe combined immunodeficiency (scid) mice. Res. Commun. Mol. Pathol. Pharmacol. 1997, 97, 229–232. [Google Scholar]
- Crocetti, L.; Schepetkin, I.A.; Cilibrizzi, A.; Graziano, A.; Vergelli, C.; Giomi, D.; Khlebnikov, A.I.; Quinn, M.T.; Giovannoni, M.P. Optimization of N-benzoylindazole derivatives as inhibitors of human neutrophil elastase. J. Med. Chem. 2013, 56, 6259–6272. [Google Scholar] [CrossRef] [Green Version]
- Crocetti, L.; Schepetkin, I.A.; Ciciani, G.; Giovannoni, M.P.; Guerrini, G.; Iacovone, A.; Khlebnikov, A.I.; Kirpotina, L.N.; Quinn, M.T.; Vergelli, C. Synthesis and pharmacological evaluation of indole derivatives as deaza analogues of potent human neutrophil elastase inhibitors (HNE). Drug Dev. Res. 2016, 77, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Crocetti, L.; Giovannoni, M.P.; Schepetkin, I.A.; Quinn, M.T.; Khlebnikov, A.I.; Cantini, N.; Guerrini, G.; Iacovone, A.; Teodori, E.; Vergelli, C. 1H-pyrrolo[2,3-b]pyridine: A new scaffold for human neutrophil elastase (HNE) inhibitors. Bioorg. Med. Chem. 2018, 26, 5583–5595. [Google Scholar] [CrossRef]
- Vergelli, C.; Schepetkin, I.A.; Crocetti, L.; Iacovone, A.; Giovannoni, M.P.; Guerrini, G.; Khlebnikov, A.I.; Ciattini, S.; Ciciani, G.; Quinn, M.T. Isoxazol-5(2H)-one: A new scaffold for potent human neutrophil elastase (HNE) inhibitors. J. Enzyme Inhib. Med. Chem. 2017, 32, 821–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasdemir, B.; Sacan, O.; Yasa, H.; Kucuk, H.B.; Yusufoglu, A.S.; Yanardag, R. Synthesis and elastase inhibition activities of novel aryl, substituted aryl, and heteroaryl oxime ester derivatives. Arch. Pharm. Chem. Life Sci. 2018, 351, 1700269. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Woehl, J.L.; Kitamura, S.; Santos-Martins, D.; Smedley, C.J.; Li, G.; Forli, S.; Moses, J.E.; Wolan, D.W.; Sharpless, K.B. SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc. Natl. Acad. Sci. USA 2019, 116, 18808–18814. [Google Scholar] [CrossRef] [Green Version]
- Donarska, B.; Świtalska, M.; Płaziński, W.; Wietrzyk, J.; Łączkowski, K.Z. Effect of the dichloro-substitution on antiproliferative activity of phthalimide-thiazole derivatives. Rational design, synthesis, elastase, caspase 3/7, and EGFR tyrosine kinase activity and molecular modeling study. Bioorg. Chem. 2021, 110, 104819. [Google Scholar] [CrossRef] [PubMed]
- Ebnöther, A.; Jucker, E.; Rissi, E.; Rutschmann, J.; Schreier, E.; Steiner, R.; Süess, R.; Vogel, A. Über Azetidin-2,4-dione (Malonimide). Helv. Chim. Acta 1959, 42, 918–955. [Google Scholar] [CrossRef]
- Mulchande, J.; Guedes, R.C.; Tsang, W.; Page, M.I.; Moreira, R.; Iley, J. Azetidine-2,4-diones (4-Oxo-β-lactams) as scafolds for designing elastase inhibitors. J. Med. Chem. 2008, 51, 1783–1790. [Google Scholar] [CrossRef]
- Mulchande, J.; Oliveira, R.; Carrasco, M.; Gouveia, L.; Guedes, R.C.; Iley, J.; Moreira, R. 4-Oxo-β-lactams (azetidine-2,4-diones) are potent and selective inhibitors of human leukocyte elastase. J. Med. Chem. 2010, 53, 241–253. [Google Scholar] [CrossRef]
- Mulchande, J.; Simões, S.I.; Gaspar, M.M.; Eleutério, C.V.; Oliveira, R.; Cruz, M.E.M.; Moreira, R.; Iley, J. Synthesis, stability, biochemical and pharmacokinetic properties of a new potent and selective 4-oxo-β-lactam inhibitor of human leukocyte elastase. J. Enzyme Inhib. Med. Chem. 2011, 26, 169–175. [Google Scholar] [CrossRef]
- Areias, L.R.P.; Ruivo, E.F.P.; Gonçalves, L.M.; Duarte, M.T.; André, V.; Moreira, R.; Lucas, S.D.; Guedes, R.C. A unified approach toward the rational design of selective low nanomolar human neutrophil elastase inhibitors. RSC Adv. 2015, 5, 51717–51721. [Google Scholar] [CrossRef]
- Ruivo, E.F.P.; Gonçalves, L.M.; Carvalho, L.A.R.; Guedes, R.V.; Hofbauer, S.; Brito, J.A.; Archer, M.; Moreira, R.; Lucas, S.D. Clickable 4-oxo-β-lactam-based selective probing for human neutrophil elastase related proteomes. ChemMedChem 2016, 11, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.; Marto, J.; Gonçalves, L.M.; Simões, S.; Félix, R.; Ascenso, A.; Lopes, F.; Ribeiro, H.M. Novel and modified neutrophil elastase inhibitor loaded in topical formulations for psoriasis management. Pharmaceutics 2020, 12, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Siqueiraa, L.R.P.; de Moraes Gomes, P.A.T.; de Lima Ferreira, L.P.; de Melo Rêgo, M.J.B.; Leite, A.C.L. Multi-target compounds acting in cancer progression: Focus on thiosemicarbazone, thiazole and thiazolidinone analogues. Eur. J. Med. Chem. 2019, 170, 237–260. [Google Scholar] [CrossRef] [PubMed]
- Chhabria, M.T.; Patel, S.; Modi, P.; Brahmkshatriya, P.S. Thiazole: A Review on Chemistry, Synthesis and Therapeutic Importance of its Derivatives. Curr. Top. Med. Chem. 2016, 16, 2841–2862. [Google Scholar] [CrossRef]
- Piechowska, K.; Świtalska, M.; Cytarska, J.; Jaroch, K.; Łuczykowski, K.; Chałupka, J.; Wietrzyk, J.; Misiura, K.; Bojko, B.; Kruszewski, S.; et al. Discovery of tropinone-thiazole derivatives as potent caspase 3/7 activators, and noncompetitive tyrosinase inhibitors with high antiproliferative activity: Rational design, one-pot tricomponent synthesis, and lipophilicity determination. Eur. J. Med. Chem. 2019, 175, 162–171. [Google Scholar] [CrossRef]
- Piechowska, K.; Mizerska-Kowalska, M.; Zdzisińska, B.; Cytarska, J.; Baranowska-Łączkowska, A.; Jaroch, K.; Łuczykowski, K.; Płaziński, W.; Bojko, B.; Kruszewski, S.; et al. Tropinone-derived alkaloids as potent anticancer agents: Synthesis, tyrosinase inhibition, mechanism of action, DFT calculation, and molecular docking studies. Int. J. Mol. Sci. 2020, 21, 9050. [Google Scholar] [CrossRef]
- Łączkowski, K.Z.; Anusiak, J.; Świtalska, M.; Dzitko, K.; Cytarska, J.; Baranowska-Łączkowska, A.; Plech, T.; Paneth, A.; Wietrzyk, J.; Białczyk, J. Synthesis, molecular docking, ctDNA interaction, DFT calculation and evaluation of antiproliferative and anti-Toxoplasma gondii activities of 2,4-diaminotriazine-thiazole derivatives. Med. Chem. Res. 2018, 27, 1131–1148. [Google Scholar] [CrossRef] [Green Version]
- Łączkowski, K.Z.; Misiura, K.; Biernasiuk, A.; Malm, A.; Siwek, A.; Plech, T.; Ciok-Pater, E.; Skowron, K.; Gospodarek, E. Synthesis, in vitro biological screening and molecular docking studies of novel camphor-based thiazoles. Med. Chem. 2014, 10, 600–608. [Google Scholar] [CrossRef]
- Crocetti, L.; Giovannoni, M.P.; Cantini, N.; Guerrini, G.; Vergelli, C.; Schepetkin, I.A.; Khlebnikov, A.I.; Quinn, M.T. Novel sulfonamide analogs of sivelestat as potent human neutrophil elastase inhibitors. Front. Chem. 2020, 8, 795. [Google Scholar] [CrossRef]
- Nevozhay, D. Cheburator software for automatically calculating drug inhibitory concentrations from in vitro screening assays. PLoS ONE 2014, 9, e106186. [Google Scholar] [CrossRef] [Green Version]
- Stark, G.R.; Taylor, W.R. Analyzing the G2/M checkpoint. Methods Mol. Biol. 2004, 280, 51–82. [Google Scholar] [PubMed]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.; Yadav, R.; Jain, S.; Vaidya, A. Caspase-3: A primary target for natural and synthetic compounds for cancer therapy. Chem. Biol. Drug Des. 2021, 98, 144–165. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, J.H.; Song, Y.H.; Jeong, W.M.; Tan, X.; Uddin, Z.; Park, K.H. Human neutrophil elastase inhibitory alkaloids from Chelidonium majus L. J. Appl. Biol. Chem. 2015, 58, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R.J. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, L.V.; Shoemaker, R.H.; Paul, K.D.; Simon, R.M.; Tosini, S.; Skehan, P.; Sudiero, D.A.; Monks, A.; Boyd, M.R. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Nat. Cancer Inst. 1990, 82, 1113–1118. [Google Scholar] [CrossRef]
- Bramson, J.; McQuillan, A.; Aubin, R.; Alaoui-Jamali, M.; Batist, G.; Christodoulopoulos, G.; Panasci, L.C. Nitrogen mustard drug resistant B-cell chronic lymphocytic leukemia as an in vivo model for crosslinking agent resistance. Mut. Res. 1995, 336, 269–278. [Google Scholar] [CrossRef]
- Sidoryk, K.; Świtalska, M.; Wietrzyk, J.; Jaromin, A.; Piętka-Ottlik, M.; Cmoch, P.; Zagrodzka, J.; Szczepek, W.; Kaczmarek, L.; Peczyńska-Czoch, W. Synthesis and biological evaluation of new amino acid and dipeptide derivatives of neocryptolepine as anticancer agents. J. Med. Chem. 2012, 55, 5077–5087. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Khlebnikov, A.I.; Quinn, M.T. N-Benzoylpyrazoles are novel small-molecule inhibitors of human neutrophil elastase. J. Med. Chem. 2007, 50, 4928–4938. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thiazole Derivatives | IC50 ± SD [nM] | Dose [µM] | Km | Inhibition Type | Ki [nM] | Kis [nM] | |
|---|---|---|---|---|---|---|---|
| 3a | 227.29 ± 35.08 | 100.00 250.00 | 53.19 55.25 | 195.19 264.55 | mixed | 590.41 | 426.00 |
| 3b | 176.28 ± 13.49 | 100.00 250.00 | 63.29 15.95 | 280.95 112.31 | mixed | 72.15 | 65.50 |
| 3c | 38.25 ± 4.91 | 50.00 100.00 | 40.65 25.58 | 193.65 207.26 | mixed | 66.06 | 99.50 |
| 3d | 110.02 ± 4.41 | 100.00 250.00 | 30.49 10.59 | 126.00 90.08 | mixed | 138.22 | 49.33 |
| 3e | 35.02 ± 3.88 | 50.00 100.00 | 28.82 28.57 | 210.77 355.14 | mixed | 35.78 | 240.00 |
| 3f | 243.96 ± 19.40 | 100.00 250.00 | 44.64 37.31 | 186.93 204.42 | mixed | 144.40 | 426.00 |
| 3g | 312.19 ± 5.76 | 250.00 500.00 | 56.50 15.97 | 229.24 74.35 | mixed | 137.24 | 171.25 |
| 3h | 44.59 ± 6.04 | 50.00 100.00 | 29.15 15.04 | 190.67 184.09 | mixed | 33.94 | 46.50 |
| 3i | 167.57 ± 18.09 | 100.00 250.00 | 75.76 32.26 | 284.78 154.24 | mixed | 596.64 | 425.00 |
| 3j | 248.74 ± 32.76 | 250.00 500.00 | 24.63 6.88 | 132.14 71.54 | mixed | 209.35 | 38.00 |
| Sivelestat | 18.78 ± 0.13 | ─ | ─ | ─ | ─ | ─ | ─ |
| Compound | Binding Energy ± SD [kcal/mol] |
|---|---|
| 3a | −7.32 ± 0.24 |
| 3b | −7.81 ± 0.23 |
| 3c | −8.39 ± 0.54 |
| 3d | −8.15 ± 0.31 |
| 3e | −8.42 ± 0.63 |
| 3f | −7.82 ± 0.26 |
| 3g | −8.23 ± 0.22 |
| 3h | −8.53 ± 0.21 |
| 3i | −7.98 ± 0.14 |
| 3j | −7.76 ± 0.08 |
| Sivelestat | −8.51 ± 0.22 |
| Thiazole Derivatives | IC50 ± SD [µM] | ||||
|---|---|---|---|---|---|
| MV-4-11 | A-549 | MDA-MB-231 | UMUC-3 | Balb/3T3 | |
| 3a | 7.15 ± 0.94 | 7.01 ± 0.47 | 9.86 ± 0.94 | 10.68 ± 1.41 | 10.87 ± 1.18 |
| 3b | 5.92 ± 1.71 | 7.31 ± 0.46 | 9.34 ± 1.12 | 9.07 ± 0.49 | 11.49 ± 1.47 |
| 3c | 4.59 ± 2.03 | 6.40 ± 0.08 | 6.38 ± 0.71 | 6.90 ± 0.21 | 7.42 ± 1.04 |
| 3d | 5.40 ± 2.06 | 15.28 ± 4.98 | 72.09 ± 25.98 | 62.73 ± 9.98 | 46.52 ± 4.99 |
| 3e | 6.23 ± 0.42 | 6.32 ± 0.89 | 6.42 ± 1.70 | 7.49 ± 0.64 | 6.57 ± 0.21 |
| 3f | 5.89 ± 1.67 | 7.28 ± 1.07 | 12.48 ± 1.79 | 19.70 ± 5.49 | 23.83 ± 4.53 |
| 3g | 5.38 ± 0.46 | 5.59 ± 0.16 | 6.19 ± 0.42 | 7.89 ± 0.23 | 6.03 ± 0.46 |
| 3h | 6.64 ± 1.59 | 9.20 ± 1.36 | 14.64 ± 3.17 | 13.31 ± 1.81 | 22.44 ± 8.62 |
| 3i | 5.47 ± 1.52 | 6.42 ± 0.72 | 11.65 ± 3.70 | 10.67 ± 3.05 | 10.13 ± 0.87 |
| 3j | 5.11 ± 1.42 | 6.21 ± 0.73 | 14.30 ± 3.45 | 25.29 ± 9.94 | 34.48 ± 22.72 |
| cisplatin | 1.33 ± 0.17 | 4.09 ± 1.00 | 19.43 ± 6.80 | 1.73 ± 0.17 | 8.04 ± 3.16 |
| Thiazole Derivatives | Cell Lines/Calculated Selectivity Index SI | |||
|---|---|---|---|---|
| MV-4-11 | A-549 | MDA-MB-231 | UMUC-3 | |
| 3a | 1.52 | 1.55 | 1.10 | 1.02 |
| 3b | 1.94 | 1.57 | 1.23 | 1.27 |
| 3c | 1.62 | 1.16 | 1.16 | 1.07 |
| 3d | 8.61 | 3.04 | 0.64 | 0.74 |
| 3e | 1.05 | 1.04 | 1.02 | 0.88 |
| 3f | 4.04 | 3.27 | 1.90 | 1.21 |
| 3g | 1.12 | 1.08 | 0.97 | 0.76 |
| 3h | 3.38 | 2.44 | 1.53 | 1.69 |
| 3i | 1.85 | 1.58 | 0.87 | 0.95 |
| 3j | 6.75 | 5.55 | 2.41 | 1.36 |
| Thiazole Derivatives | λ [nm] | k [min−1] | t1/2 [min] |
|---|---|---|---|
| 3c | 293 | 0.015 | 46.2 |
| 3e | 262, 322 | 0.0175 | 39.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donarska, B.; Świtalska, M.; Wietrzyk, J.; Płaziński, W.; Mizerska-Kowalska, M.; Zdzisińska, B.; Łączkowski, K.Z. Discovery of New 3,3-Diethylazetidine-2,4-dione Based Thiazoles as Nanomolar Human Neutrophil Elastase Inhibitors with Broad-Spectrum Antiproliferative Activity. Int. J. Mol. Sci. 2022, 23, 7566. https://doi.org/10.3390/ijms23147566
Donarska B, Świtalska M, Wietrzyk J, Płaziński W, Mizerska-Kowalska M, Zdzisińska B, Łączkowski KZ. Discovery of New 3,3-Diethylazetidine-2,4-dione Based Thiazoles as Nanomolar Human Neutrophil Elastase Inhibitors with Broad-Spectrum Antiproliferative Activity. International Journal of Molecular Sciences. 2022; 23(14):7566. https://doi.org/10.3390/ijms23147566
Chicago/Turabian StyleDonarska, Beata, Marta Świtalska, Joanna Wietrzyk, Wojciech Płaziński, Magdalena Mizerska-Kowalska, Barbara Zdzisińska, and Krzysztof Z. Łączkowski. 2022. "Discovery of New 3,3-Diethylazetidine-2,4-dione Based Thiazoles as Nanomolar Human Neutrophil Elastase Inhibitors with Broad-Spectrum Antiproliferative Activity" International Journal of Molecular Sciences 23, no. 14: 7566. https://doi.org/10.3390/ijms23147566