
Acinar-to-Ductal Metaplasia (ADM): On the Road to Pancreatic Intraepithelial Neoplasia (PanIN) and Pancreatic Cancer
 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

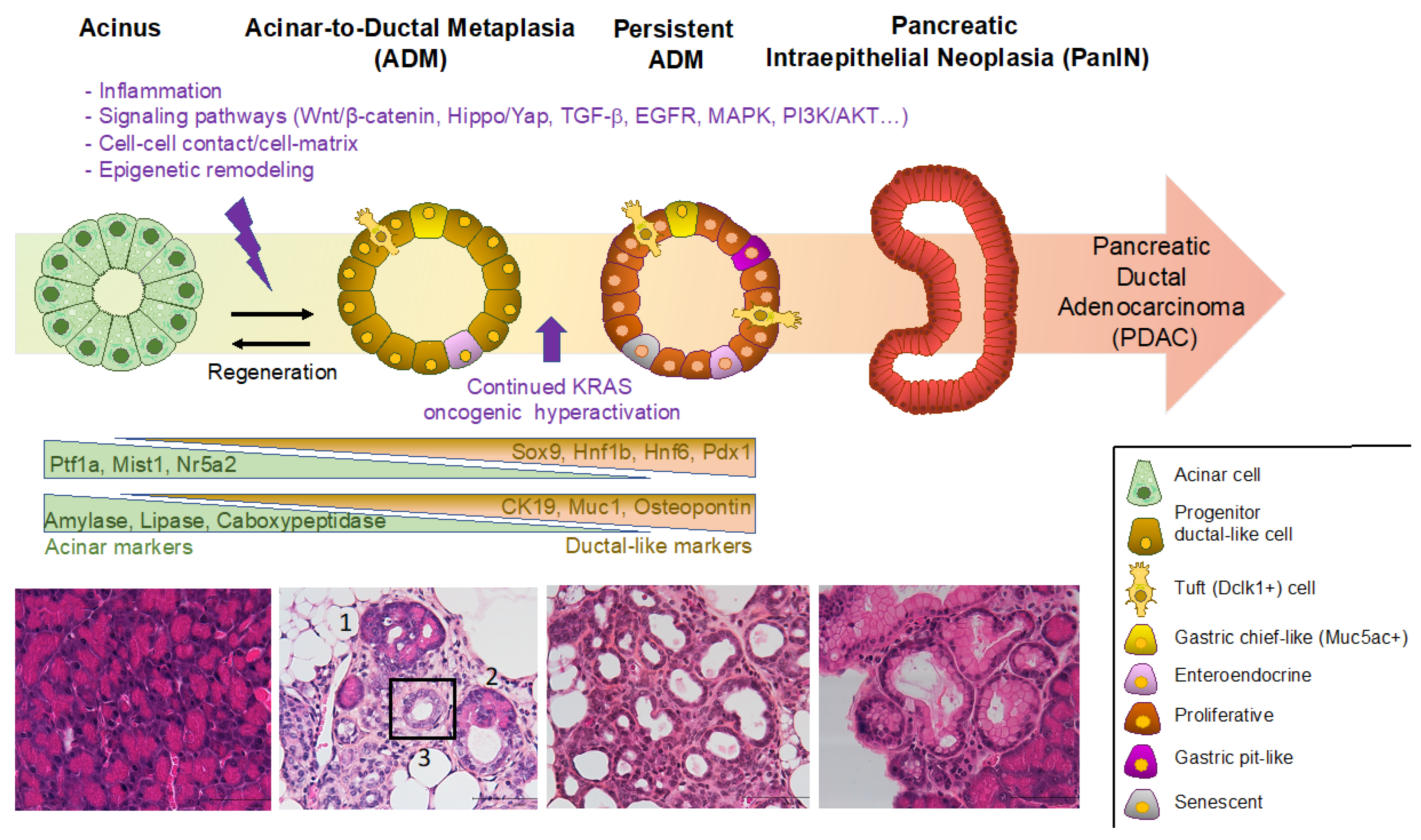
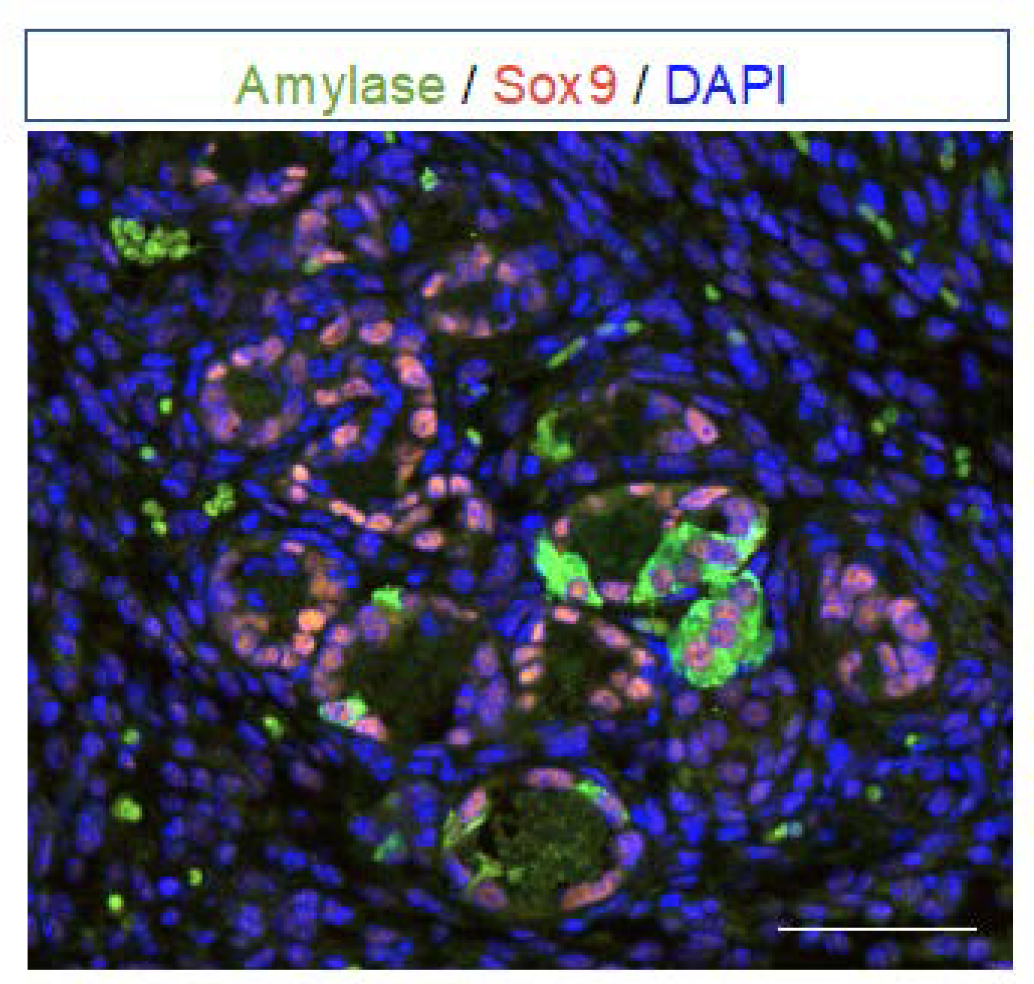
2. Definition of Acinar-to-Ductal Metaplasia (ADM) and Transcription Factors Involved
3. Factors Triggering ADM: Environmental and Cellular Insults
3.1. Pancreatitis and Inflammation
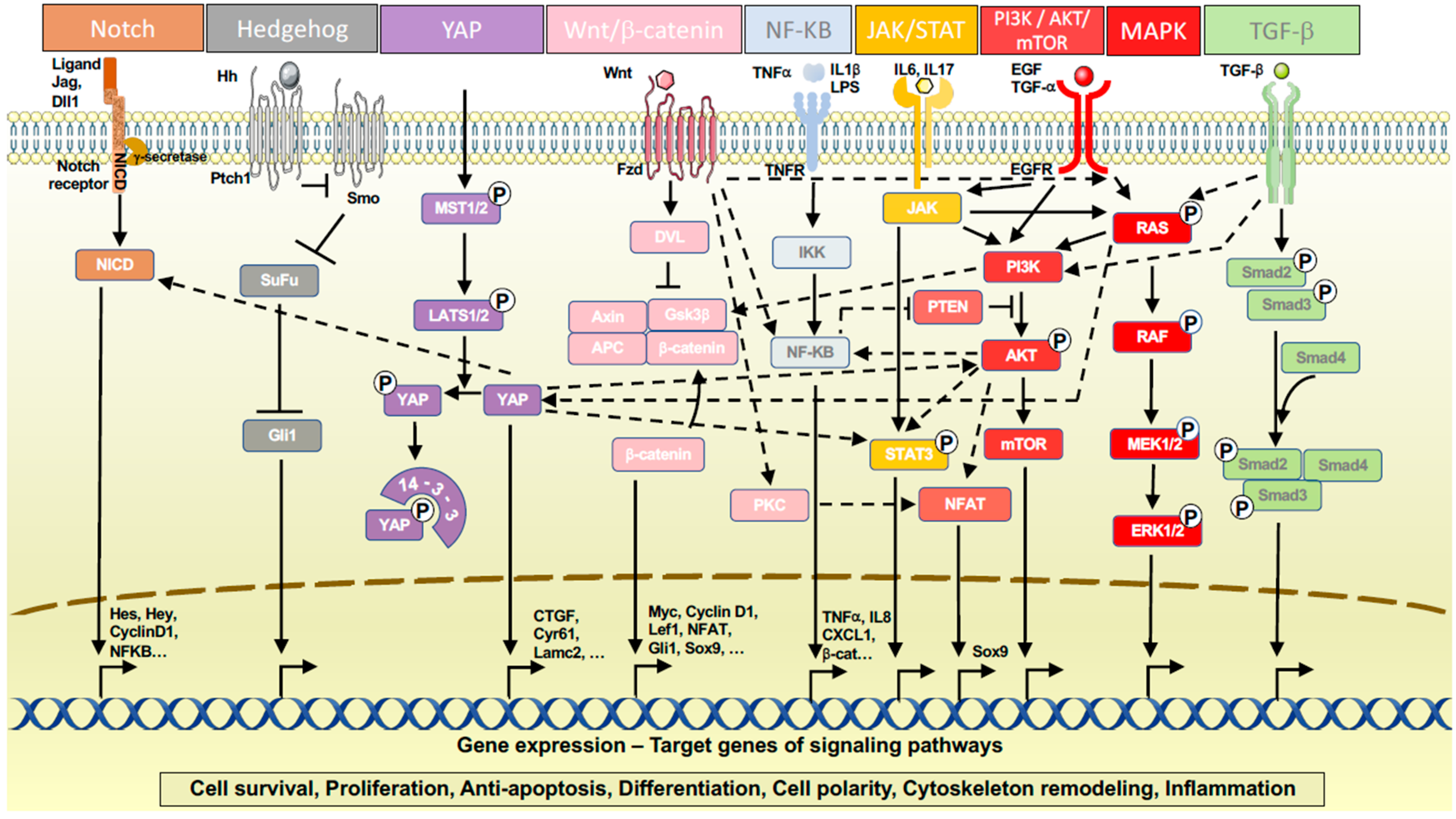
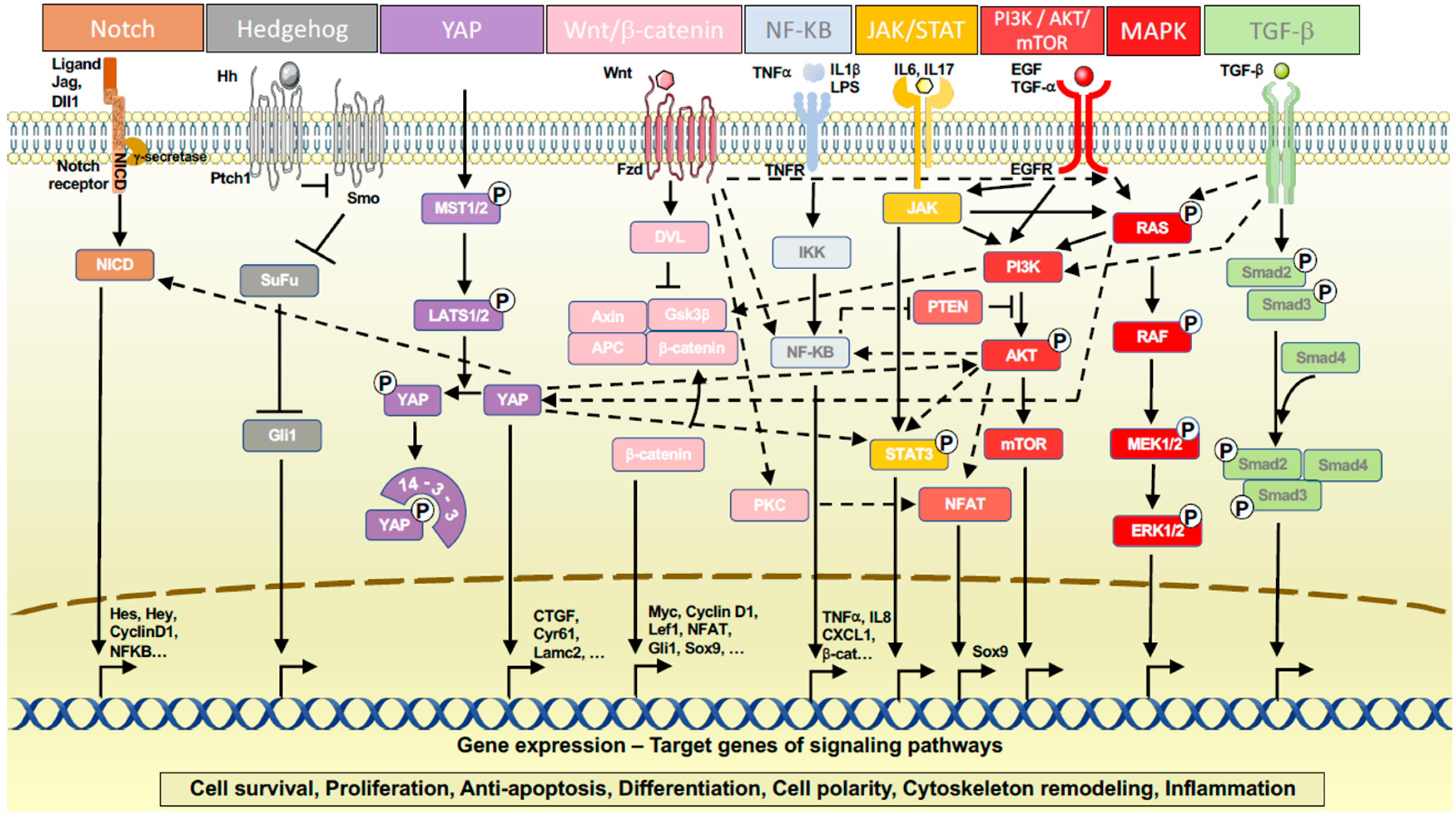
3.2. Signaling Pathways
3.3. Epigenetic Reprogramming
4. Progression of ADM to Pancreatic Intraepithelial Neoplasia (PanIN)
5. Heterogeneity of Metaplastic Cells Revealed by scRNA-seq
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, Y.; Furukawa, T.; Yachida, S.; Nishimura, M.; Seki, A.; Nonaka, K.; Aida, J.; Takubo, K.; Ishiwata, T.; Kimura, W.; et al. The Prevalence and Clinicopathological Characteristics of High-Grade Pancreatic Intraepithelial Neoplasia: Autopsy Study Evaluating the Entire Pancreatic Parenchyma. Pancreas 2017, 46, 658–664. [Google Scholar] [CrossRef]
- Quilichini, E.; Fabre, M.; Dirami, T.; Stedman, A.; De Vas, M.; Ozguc, O.; Pasek, R.C.; Cereghini, S.; Morillon, L.; Guerra, C.; et al. Pancreatic Ductal Deletion of Hnf1b Disrupts Exocrine Homeostasis, Leads to Pancreatitis, and Facilitates Tumorigenesis. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 487–511. [Google Scholar] [CrossRef] [Green Version]
- Puri, S.; Folias, A.E.; Hebrok, M. Plasticity and Dedifferentiation within the Pancreas: Development, Homeostasis, and Disease. Cell Stem. Cell 2015, 16, 18–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanger, B.Z.; Hebrok, M. Control of Cell Identity in Pancreas Development and Regeneration. Gastroenterology 2013, 144, 1170–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, J.C.; Sansom, O.J. Reserve Stem Cells: Differentiated Cells Reprogram to Fuel Repair, Metaplasia, and Neoplasia in the Adult Gastrointestinal Tract. Sci. Signal. 2015, 8, re8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, F.C.; Bankaitis, E.D.; Boyer, D.; Xu, X.; Van de Casteele, M.; Magnuson, M.A.; Heimberg, H.; Wright, C.V.E. Spatiotemporal Patterns of Multipotentiality in Ptf1a-Expressing Cells during Pancreas Organogenesis and Injury-Induced Facultative Restoration. Development 2013, 140, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Murtaugh, L.C.; Keefe, M.D. Regeneration and Repair of the Exocrine Pancreas. Annu. Rev. Physiol. 2015, 77, 229–249. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.N.; Cameron, E.; Garay, M.V.R.; Starkey, T.W.; Gianani, R.; Jensen, J. Recapitulation of Elements of Embryonic Development in Adult Mouse Pancreatic Regeneration. Gastroenterology 2005, 128, 728–741. [Google Scholar] [CrossRef]
- Mills, J.C.; Stanger, B.Z.; Sander, M. Nomenclature for Cellular Plasticity: Are the Terms as Plastic as the Cells Themselves? EMBO J. 2019, 38, e103148. [Google Scholar] [CrossRef]
- Willet, S.G.; Lewis, M.A.; Miao, Z.-F.; Liu, D.; Radyk, M.D.; Cunningham, R.L.; Burclaff, J.; Sibbel, G.; Lo, H.-Y.G.; Blanc, V.; et al. Regenerative Proliferation of Differentiated Cells by MTORC1-Dependent Paligenosis. EMBO J. 2018, 37, e98311. [Google Scholar] [CrossRef]
- Houbracken, I.; de Waele, E.; Lardon, J.; Ling, Z.; Heimberg, H.; Rooman, I.; Bouwens, L. Lineage Tracing Evidence for Transdifferentiation of Acinar to Duct Cells and Plasticity of Human Pancreas. Gastroenterology 2011, 141, 731–741.e4. [Google Scholar] [CrossRef]
- Baldan, J.; Houbracken, I.; Rooman, I.; Bouwens, L. Adult Human Pancreatic Acinar Cells Dedifferentiate into an Embryonic Progenitor-like State in 3D Suspension Culture. Sci. Rep. 2019, 9, 4040. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Akanuma, N.; Liu, C.; Naji, A.; Halff, G.A.; Washburn, W.K.; Sun, L.; Wang, P. TGF-Β1 Promotes Acinar to Ductal Metaplasia of Human Pancreatic Acinar Cells. Sci. Rep. 2016, 6, 30904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backx, E.; Wauters, E.; Baldan, J.; Van Bulck, M.; Michiels, E.; Heremans, Y.; De Paep, D.L.; Kurokawa, M.; Goyama, S.; Bouwens, L.; et al. MECOM Permits Pancreatic Acinar Cell Dedifferentiation Avoiding Cell Death under Stress Conditions. Cell Death Differ. 2021, 28, 2601–2615. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hakimjavadi, H.; Bray, J.K.; Perkins, C.; Gosling, A.; da Silva, L.; Bulut, G.; Ali, J.; Setiawan, V.W.; Campbell-Thompson, M.; et al. Transcriptional Profile of Human Pancreatic Acinar Ductal Metaplasia. Gastro. Hep. Adv. 2023, 2, 532–543. [Google Scholar] [CrossRef]
- Paoli, C.; Carrer, A. Organotypic Culture of Acinar Cells for the Study of Pancreatic Cancer Initiation. Cancers 2020, 12, 2606. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Cooper, B.; Gannon, M.; Ray, M.; MacDonald, R.J.; Wright, C.V.E. The Role of the Transcriptional Regulator Ptf1a in Converting Intestinal to Pancreatic Progenitors. Nat. Genet. 2002, 32, 128–134. [Google Scholar] [CrossRef]
- Masui, T.; Long, Q.; Beres, T.M.; Magnuson, M.A.; MacDonald, R.J. Early Pancreatic Development Requires the Vertebrate Suppressor of Hairless (RBPJ) in the PTF1 BHLH Complex. Genes Dev. 2007, 21, 2629–2643. [Google Scholar] [CrossRef] [Green Version]
- Schaffer, A.E.; Freude, K.K.; Nelson, S.B.; Sander, M. Nkx6 Transcription Factors and Ptf1a Function as Antagonistic Lineage Determinants in Multipotent Pancreatic Progenitors. Dev. Cell 2010, 18, 1022–1029. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.D.; Swift, G.H.; Peyton, M.J.; Hammer, R.E.; MacDonald, R.J. The Role of PTF1-P48 in Pancreatic Acinar Gene Expression. J. Biol. Chem. 2001, 276, 44018–44026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodolosse, A.; Chalaux, E.; Adell, T.; Hagège, H.; Skoudy, A.; Real, F.X. PTF1alpha/P48 Transcription Factor Couples Proliferation and Differentiation in the Exocrine Pancreas [corrected]. Gastroenterology 2004, 127, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Azevedo-Pouly, A.C.; Deering, T.G.; Hoang, C.Q.; DiRenzo, D.; Hess, D.A.; Konieczny, S.F.; Swift, G.H.; MacDonald, R.J. MIST1 and PTF1 Collaborate in Feed-Forward Regulatory Loops That Maintain the Pancreatic Acinar Phenotype in Adult Mice. Mol. Cell Biol. 2016, 36, 2945–2955. [Google Scholar] [CrossRef] [Green Version]
- Hoang, C.Q.; Hale, M.A.; Azevedo-Pouly, A.C.; Elsässer, H.P.; Deering, T.G.; Willet, S.G.; Pan, F.C.; Magnuson, M.A.; Wright, C.V.E.; Swift, G.H.; et al. Transcriptional Maintenance of Pancreatic Acinar Identity, Differentiation, and Homeostasis by PTF1A. Mol. Cell Biol. 2016, 36, 3033–3047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krah, N.M.; De La O, J.-P.; Swift, G.H.; Hoang, C.Q.; Willet, S.G.; Chen Pan, F.; Cash, G.M.; Bronner, M.P.; Wright, C.V.; MacDonald, R.J.; et al. The Acinar Differentiation Determinant PTF1A Inhibits Initiation of Pancreatic Ductal Adenocarcinoma. Elife 2015, 4, e07125. [Google Scholar] [CrossRef] [PubMed]
- Pin, C.L.; Rukstalis, J.M.; Johnson, C.; Konieczny, S.F. The BHLH Transcription Factor Mist1 Is Required to Maintain Exocrine Pancreas Cell Organization and Acinar Cell Identity. J. Cell Biol. 2001, 155, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Direnzo, D.; Hess, D.A.; Damsz, B.; Hallett, J.E.; Marshall, B.; Goswami, C.; Liu, Y.; Deering, T.; Macdonald, R.J.; Konieczny, S.F. Induced Mist1 Expression Promotes Remodeling of Mouse Pancreatic Acinar Cells. Gastroenterology 2012, 143, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Tran, T.; Rukstalis, J.M.; Sun, P.; Damsz, B.; Konieczny, S.F. Inhibition of Mist1 Homodimer Formation Induces Pancreatic Acinar-to-Ductal Metaplasia. Mol. Cell Biol. 2004, 24, 2673–2681. [Google Scholar] [CrossRef] [Green Version]
- Kowalik, A.S.; Johnson, C.L.; Chadi, S.A.; Weston, J.Y.; Fazio, E.N.; Pin, C.L. Mice Lacking the Transcription Factor Mist1 Exhibit an Altered Stress Response and Increased Sensitivity to Caerulein-Induced Pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1123–G1132. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Zhu, L.; Sun, Y.; Bettencourt, R.; Damsz, B.; Hruban, R.H.; Konieczny, S.F. Loss of the Acinar-Restricted Transcription Factor Mist1 Accelerates Kras-Induced Pancreatic Intraepithelial Neoplasia. Gastroenterology 2009, 136, 1368–1378. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; DiRenzo, D.; Qu, C.; Barney, D.; Miley, D.; Konieczny, S.F. Maintenance of Acinar Cell Organization Is Critical to Preventing Kras-Induced Acinar-Ductal Metaplasia. Oncogene 2013, 32, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Figura, G.; Morris, J.P.; Wright, C.V.E.; Hebrok, M. Nr5a2 Maintains Acinar Cell Differentiation and Constrains Oncogenic Kras-Mediated Pancreatic Neoplastic Initiation. Gut 2014, 63, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, J.L.; Dubois, C.L.; Schaffer, A.E.; Hao, E.; Shih, H.P.; Seymour, P.A.; Ma, J.; Sander, M. Sox9+ Ductal Cells Are Multipotent Progenitors throughout Development but Do Not Produce New Endocrine Cells in the Normal or Injured Adult Pancreas. Development 2011, 138, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.; Hebrok, M. Regulation of Cellular Identity in Cancer. Dev. Cell 2015, 35, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Prévot, P.-P.; Simion, A.; Grimont, A.; Colletti, M.; Khalaileh, A.; Van den Steen, G.; Sempoux, C.; Xu, X.; Roelants, V.; Hald, J.; et al. Role of the Ductal Transcription Factors HNF6 and Sox9 in Pancreatic Acinar-to-Ductal Metaplasia. Gut 2012, 61, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Miyatsuka, T.; Kaneto, H.; Shiraiwa, T.; Matsuoka, T.; Yamamoto, K.; Kato, K.; Nakamura, Y.; Akira, S.; Takeda, K.; Kajimoto, Y.; et al. Persistent Expression of PDX-1 in the Pancreas Causes Acinar-to-Ductal Metaplasia through Stat3 Activation. Genes Dev. 2006, 20, 1435–1440. [Google Scholar] [CrossRef] [Green Version]
- Gmyr, V.; Belaich, S.; Muharram, G.; Lukowiak, B.; Vandewalle, B.; Pattou, F.; Kerr-Conte, J. Rapid Purification of Human Ductal Cells from Human Pancreatic Fractions with Surface Antibody CA19-9. Biochem. Biophys. Res. Commun. 2004, 320, 27–33. [Google Scholar] [CrossRef]
- Inada, A.; Nienaber, C.; Fonseca, S.; Bonner-Weir, S. Timing and Expression Pattern of Carbonic Anhydrase II in Pancreas. Dev. Dyn. 2006, 235, 1571–1577. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, D.; Liu, T.; Liu, Y.H.; Guo, J.; Song, J.; Wu, Q.; Pan, Y.; Zhang, Y.; Guo, C.; et al. Expansion and Maintenance of CD133-Expressing Pancreatic Ductal Epithelial Cells by Inhibition of TGF-β Signaling. Stem. Cells Dev. 2019, 28, 1236–1252. [Google Scholar] [CrossRef]
- Kilic, G.; Wang, J.; Sosa-Pineda, B. Osteopontin Is a Novel Marker of Pancreatic Ductal Tissues and of Undifferentiated Pancreatic Precursors in Mice. Dev. Dyn. 2006, 235, 1659–1667. [Google Scholar] [CrossRef] [Green Version]
- Pan, F.C.; Wright, C. Pancreas Organogenesis: From Bud to Plexus to Gland. Dev. Dyn. 2011, 240, 530–565. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.P.; Wang, A.; Sander, M. Pancreas Organogenesis: From Lineage Determination to Morphogenesis. Annu. Rev. Cell Dev. Biol. 2013, 29, 81–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haumaitre, C.; Barbacci, E.; Jenny, M.; Ott, M.O.; Gradwohl, G.; Cereghini, S. Lack of TCF2/VHNF1 in Mice Leads to Pancreas Agenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 1490–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poll, A.V.; Pierreux, C.E.; Lokmane, L.; Haumaitre, C.; Achouri, Y.; Jacquemin, P.; Rousseau, G.G.; Cereghini, S.; Lemaigre, F.P. A VHNF1/TCF2-HNF6 Cascade Regulates the Transcription Factor Network That Controls Generation of Pancreatic Precursor Cells. Diabetes 2006, 55, 61–69. [Google Scholar] [CrossRef]
- De Vas, M.G.; Kopp, J.L.; Heliot, C.; Sander, M.; Cereghini, S.; Haumaitre, C. Hnf1b Controls Pancreas Morphogenesis and the Generation of Ngn3+ Endocrine Progenitors. Development 2015, 142, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Pinho, A.V.; Rooman, I.; Reichert, M.; De Medts, N.; Bouwens, L.; Rustgi, A.K.; Real, F.X. Adult Pancreatic Acinar Cells Dedifferentiate to an Embryonic Progenitor Phenotype with Concomitant Activation of a Senescence Programme That Is Present in Chronic Pancreatitis. Gut 2011, 60, 958–966. [Google Scholar] [CrossRef]
- Chuvin, N.; Vincent, D.F.; Pommier, R.M.; Alcaraz, L.B.; Gout, J.; Caligaris, C.; Yacoub, K.; Cardot, V.; Roger, E.; Kaniewski, B.; et al. Acinar-to-Ductal Metaplasia Induced by Transforming Growth Factor Beta Facilitates KRASG12D-Driven Pancreatic Tumorigenesis. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 263–282. [Google Scholar] [CrossRef] [Green Version]
- Seymour, P.A.; Freude, K.K.; Tran, M.N.; Mayes, E.E.; Jensen, J.; Kist, R.; Scherer, G.; Sander, M. SOX9 Is Required for Maintenance of the Pancreatic Progenitor Cell Pool. Proc. Natl. Acad. Sci. USA 2007, 104, 1865–1870. [Google Scholar] [CrossRef] [Green Version]
- Pierreux, C.E.; Poll, A.V.; Kemp, C.R.; Clotman, F.; Maestro, M.A.; Cordi, S.; Ferrer, J.; Leyns, L.; Rousseau, G.G.; Lemaigre, F.P. The Transcription Factor Hepatocyte Nuclear Factor-6 Controls the Development of Pancreatic Ducts in the Mouse. Gastroenterology 2006, 130, 532–541. [Google Scholar] [CrossRef]
- Mayerle, J.; Sendler, M.; Hegyi, E.; Beyer, G.; Lerch, M.M.; Sahin-Tóth, M. Genetics, Cell Biology, and Pathophysiology of Pancreatitis. Gastroenterology 2019, 156, 1951–1968.e1. [Google Scholar] [CrossRef] [Green Version]
- Marrache, F.; Tu, S.P.; Bhagat, G.; Pendyala, S.; Osterreicher, C.H.; Gordon, S.; Ramanathan, V.; Penz-Osterreicher, M.; Betz, K.S.; Song, Z.; et al. Overexpression of Interleukin-1beta in the Murine Pancreas Results in Chronic Pancreatitis. Gastroenterology 2008, 135, 1277–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Liu, Y.; Daniluk, J.; Gaiser, S.; Chu, J.; Wang, H.; Li, Z.-S.; Logsdon, C.D.; Ji, B. Activation of Nuclear Factor-ΚB in Acinar Cells Increases the Severity of Pancreatitis in Mice. Gastroenterology 2013, 144, 202–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrero-Andrés, A.; Panisello-Roselló, A.; Roselló-Catafau, J.; Folch-Puy, E. NLRP3 Inflammasome-Mediated Inflammation in Acute Pancreatitis. Int. J. Mol. Sci. 2020, 21, 5386. [Google Scholar] [CrossRef] [PubMed]
- Lerch, M.M.; Gorelick, F.S. Models of Acute and Chronic Pancreatitis. Gastroenterology 2013, 144, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Rebours, V.; Garteiser, P.; Ribeiro-Parenti, L.; Cavin, J.-B.; Doblas, S.; Pagé, G.; Bado, A.; Couvineau, A.; Ruszniewski, P.; Paradis, V.; et al. Obesity-Induced Pancreatopathy in Rats Is Reversible after Bariatric Surgery. Sci. Rep. 2018, 8, 16295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teper, Y.; Eibl, G. Pancreatic Macrophages: Critical Players in Obesity-Promoted Pancreatic Cancer. Cancers 2020, 12, 1946. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Döppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-Secreted Cytokines Drive Pancreatic Acinar-to-Ductal Metaplasia through NF-ΚB and MMPs. J. Cell Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef]
- Chung, K.M.; Singh, J.; Lawres, L.; Dorans, K.J.; Garcia, C.; Burkhardt, D.B.; Robbins, R.; Bhutkar, A.; Cardone, R.; Zhao, X.; et al. Endocrine-Exocrine Signaling Drives Obesity-Associated Pancreatic Ductal Adenocarcinoma. Cell 2020, 181, 832–847.e18. [Google Scholar] [CrossRef] [PubMed]
- Gukovsky, I.; Li, N.; Todoric, J.; Gukovskaya, A.; Karin, M. Inflammation, Autophagy, and Obesity: Common Features in the Pathogenesis of Pancreatitis and Pancreatic Cancer. Gastroenterology 2013, 144, 1199–1209.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Wu, X.; Holzer, R.G.; Lee, J.-H.; Todoric, J.; Park, E.-J.; Ogata, H.; Gukovskaya, A.S.; Gukovsky, I.; Pizzo, D.P.; et al. Loss of Acinar Cell IKKα Triggers Spontaneous Pancreatitis in Mice. J. Clin. Investig. 2013, 123, 2231–2243. [Google Scholar] [CrossRef] [Green Version]
- Gukovsky, I.; Gukovskaya, A.S. Impaired Autophagy Triggers Chronic Pancreatitis: Lessons from Pancreas-Specific Atg5 Knockout Mice. Gastroenterology 2015, 148, 501–505. [Google Scholar] [CrossRef] [Green Version]
- Diakopoulos, K.N.; Lesina, M.; Wörmann, S.; Song, L.; Aichler, M.; Schild, L.; Artati, A.; Römisch-Margl, W.; Wartmann, T.; Fischer, R.; et al. Impaired Autophagy Induces Chronic Atrophic Pancreatitis in Mice via Sex- and Nutrition-Dependent Processes. Gastroenterology 2015, 148, 626–638.e17. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, L.; Fagman, J.B.; Kim, J.Y.; Todoric, J.; Gukovsky, I.; Mackey, M.; Ellisman, M.H.; Karin, M. Basal Autophagy Maintains Pancreatic Acinar Cell Homeostasis and Protein Synthesis and Prevents ER Stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6166–E6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, R.L.; Staley, B.K.; Liou, A.; Hebrok, M. Numb Regulates Acinar Cell Dedifferentiation and Survival during Pancreatic Damage and Acinar-to-Ductal Metaplasia. Gastroenterology 2013, 145, 1088–1097.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendley, A.M.; Provost, E.; Bailey, J.M.; Wang, Y.J.; Cleveland, M.H.; Blake, D.; Bittman, R.W.; Roeser, J.C.; Maitra, A.; Reynolds, A.B.; et al. P120 Catenin Is Required for Normal Tubulogenesis but Not Epithelial Integrity in Developing Mouse Pancreas. Dev. Biol. 2015, 399, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Kaneta, Y.; Sato, T.; Hikiba, Y.; Sugimori, M.; Sue, S.; Kaneko, H.; Irie, K.; Sasaki, T.; Kondo, M.; Chuma, M.; et al. Loss of Pancreatic E-Cadherin Causes Pancreatitis-Like Changes and Contributes to Carcinogenesis. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 105–119. [Google Scholar] [CrossRef] [Green Version]
- Murtaugh, L.C.; Stanger, B.Z.; Kwan, K.M.; Melton, D.A. Notch Signaling Controls Multiple Steps of Pancreatic Differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 14920–14925. [Google Scholar] [CrossRef] [Green Version]
- Siveke, J.T.; Lubeseder-Martellato, C.; Lee, M.; Mazur, P.K.; Nakhai, H.; Radtke, F.; Schmid, R.M. Notch Signaling Is Required for Exocrine Regeneration after Acute Pancreatitis. Gastroenterology 2008, 134, 544–555. [Google Scholar] [CrossRef]
- Hidalgo-Sastre, A.; Brodylo, R.L.; Lubeseder-Martellato, C.; Sipos, B.; Steiger, K.; Lee, M.; von Figura, G.; Grünwald, B.; Zhong, S.; Trajkovic-Arsic, M.; et al. Hes1 Controls Exocrine Cell Plasticity and Restricts Development of Pancreatic Ductal Adenocarcinoma in a Mouse Model. Am. J. Pathol. 2016, 186, 2934–2944. [Google Scholar] [CrossRef] [Green Version]
- Hebrok, M.; Kim, S.K.; Melton, D.A. Notochord Repression of Endodermal Sonic Hedgehog Permits Pancreas Development. Genes Dev. 1998, 12, 1705–1713. [Google Scholar] [CrossRef] [Green Version]
- Cano, D.A.; Hebrok, M. Hedgehog Spikes Pancreas Regeneration. Gastroenterology 2008, 135, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Fendrich, V.; Esni, F.; Garay, M.V.R.; Feldmann, G.; Habbe, N.; Jensen, J.N.; Dor, Y.; Stoffers, D.; Jensen, J.; Leach, S.D.; et al. Hedgehog Signaling Is Required for Effective Regeneration of Exocrine Pancreas. Gastroenterology 2008, 135, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigante, E.D.; Caspary, T. Signaling in the Primary Cilium through the Lens of the Hedgehog Pathway. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e377. [Google Scholar] [CrossRef]
- Bangs, F.K.; Miller, P.; O’Neill, E. Ciliogenesis and Hedgehog Signalling Are Suppressed Downstream of KRAS during Acinar-Ductal Metaplasia in Mouse. Dis. Model Mech. 2020, 13, dmm044289. [Google Scholar] [CrossRef] [PubMed]
- Lodh, S.; O’Hare, E.A.; Zaghloul, N.A. Primary Cilia in Pancreatic Development and Disease. Birth Defects Res. C Embryo. Today 2014, 102, 139–158. [Google Scholar] [CrossRef] [Green Version]
- Cano, D.A.; Murcia, N.S.; Pazour, G.J.; Hebrok, M. Orpk Mouse Model of Polycystic Kidney Disease Reveals Essential Role of Primary Cilia in Pancreatic Tissue Organization. Development 2004, 131, 3457–3467. [Google Scholar] [CrossRef] [Green Version]
- Cano, D.A.; Sekine, S.; Hebrok, M. Primary Cilia Deletion in Pancreatic Epithelial Cells Results in Cyst Formation and Pancreatitis. Gastroenterology 2006, 131, 1856–1869. [Google Scholar] [CrossRef]
- Augereau, C.; Collet, L.; Vargiu, P.; Guerra, C.; Ortega, S.; Lemaigre, F.P.; Jacquemin, P. Chronic Pancreatitis and Lipomatosis Are Associated with Defective Function of Ciliary Genes in Pancreatic Ductal Cells. Hum. Mol. Genet. 2016, 25, 5017–5026. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Aegerter, P.; Nipper, M.; Ramjit, L.; Liu, J.; Wang, P. Hippo Signaling Pathway in Pancreas Development. Front. Cell Dev. Biol. 2021, 9, 663906. [Google Scholar] [CrossRef]
- Gao, T.; Zhou, D.; Yang, C.; Singh, T.; Penzo-Méndez, A.; Maddipati, R.; Tzatsos, A.; Bardeesy, N.; Avruch, J.; Stanger, B.Z. Hippo Signaling Regulates Differentiation and Maintenance in the Exocrine Pancreas. Gastroenterology 2013, 144, 1543–1553.e1. [Google Scholar] [CrossRef] [Green Version]
- Morvaridi, S.; Dhall, D.; Greene, M.I.; Pandol, S.J.; Wang, Q. Role of YAP and TAZ in Pancreatic Ductal Adenocarcinoma and in Stellate Cells Associated with Cancer and Chronic Pancreatitis. Sci. Rep. 2015, 5, 16759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellizzi, A.M.; Bloomston, M.; Zhou, X.-P.; Iwenofu, O.H.; Frankel, W.L. The MTOR Pathway Is Frequently Activated in Pancreatic Ductal Adenocarcinoma and Chronic Pancreatitis. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Kodama, T.; Sato, K.; Murai, K.; Yoshioka, T.; Shigekawa, M.; Yamada, R.; Hikita, H.; Sakamori, R.; Akita, H.; et al. Dysregulation of PI3K and Hippo Signaling Pathways Synergistically Induces Chronic Pancreatitis via CTGF Upregulation. J. Clin. Investig. 2021, 131, e143414. [Google Scholar] [CrossRef]
- Gruber, R.; Panayiotou, R.; Nye, E.; Spencer-Dene, B.; Stamp, G.; Behrens, A. YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-Regulation of JAK-STAT3 Signaling. Gastroenterology 2016, 151, 526–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Bian, Q.; Rong, D.; Wang, L.; Song, J.; Huang, H.-S.; Zeng, J.; Mei, J.; Wang, P.-Y. JAK/STAT Pathway: Extracellular Signals, Diseases, Immunity, and Therapeutic Regimens. Front. Bioeng. Biotechnol. 2023, 11, 1110765. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 Plays a Critical Role in KRAS-Induced Pancreatic Tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, A.; Wang, S.C.; Morris, J.P.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 Contribute to Pancreatic Ductal Adenocarcinoma Initiation and Progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [Green Version]
- Sawey, E.T.; Johnson, J.A.; Crawford, H.C. Matrix Metalloproteinase 7 Controls Pancreatic Acinar Cell Transdifferentiation by Activating the Notch Signaling Pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 19327–19332. [Google Scholar] [CrossRef] [Green Version]
- Crawford, H.C.; Scoggins, C.R.; Washington, M.K.; Matrisian, L.M.; Leach, S.D. Matrix Metalloproteinase-7 Is Expressed by Pancreatic Cancer Precursors and Regulates Acinar-to-Ductal Metaplasia in Exocrine Pancreas. J. Clin. Investig. 2002, 109, 1437–1444. [Google Scholar] [CrossRef]
- Perusina Lanfranca, M.; Zhang, Y.; Girgis, A.; Kasselman, S.; Lazarus, J.; Kryczek, I.; Delrosario, L.; Rhim, A.; Koneva, L.; Sartor, M.; et al. Interleukin 22 Signaling Regulates Acinar Cell Plasticity to Promote Pancreatic Tumor Development in Mice. Gastroenterology 2020, 158, 1417–1432.e11. [Google Scholar] [CrossRef]
- Gao, C.; Chen, G.; Zhang, D.H.; Zhang, J.; Kuan, S.-F.; Hu, W.; Esni, F.; Gao, X.; Guan, J.-L.; Chu, E.; et al. PYK2 Is Involved in Premalignant Acinar Cell Reprogramming and Pancreatic Ductal Adenocarcinoma Maintenance by Phosphorylating β-CateninY654. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 561–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtaugh, L.C. The What, Where, When and How of Wnt/β-Catenin Signaling in Pancreas Development. Organogenesis 2008, 4, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Morris, J.P.; Yan, W.; Schofield, H.K.; Gurney, A.; Simeone, D.M.; Millar, S.E.; Hoey, T.; Hebrok, M.; Pasca di Magliano, M. Canonical Wnt Signaling Is Required for Pancreatic Carcinogenesis. Cancer Res 2013, 73, 4909–4922. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.P.; Cano, D.A.; Sekine, S.; Wang, S.C.; Hebrok, M. Beta-Catenin Blocks Kras-Dependent Reprogramming of Acini into Pancreatic Cancer Precursor Lesions in Mice. J. Clin. Investig. 2010, 120, 508–520. [Google Scholar] [CrossRef] [Green Version]
- Keefe, M.D.; Wang, H.; De La O, J.-P.; Khan, A.; Firpo, M.A.; Murtaugh, L.C. β-Catenin Is Selectively Required for the Expansion and Regeneration of Mature Pancreatic Acinar Cells in Mice. Dis. Model Mech. 2012, 5, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- Nagashio, Y.; Ueno, H.; Imamura, M.; Asaumi, H.; Watanabe, S.; Yamaguchi, T.; Taguchi, M.; Tashiro, M.; Otsuki, M. Inhibition of Transforming Growth Factor Beta Decreases Pancreatic Fibrosis and Protects the Pancreas against Chronic Injury in Mice. Lab. Investig. 2004, 84, 1610–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wildi, S.; Kleeff, J.; Mayerle, J.; Zimmermann, A.; Böttinger, E.P.; Wakefield, L.; Büchler, M.W.; Friess, H.; Korc, M. Suppression of Transforming Growth Factor Beta Signalling Aborts Caerulein Induced Pancreatitis and Eliminates Restricted Stimulation at High Caerulein Concentrations. Gut 2007, 56, 685–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Waele, E.; Wauters, E.; Ling, Z.; Bouwens, L. Conversion of Human Pancreatic Acinar Cells toward a Ductal-Mesenchymal Phenotype and the Role of Transforming Growth Factor β and Activin Signaling. Pancreas 2014, 43, 1083–1092. [Google Scholar] [CrossRef]
- Korc, M.; Friess, H.; Yamanaka, Y.; Kobrin, M.S.; Buchler, M.; Beger, H.G. Chronic Pancreatitis Is Associated with Increased Concentrations of Epidermal Growth Factor Receptor, Transforming Growth Factor Alpha, and Phospholipase C Gamma. Gut 1994, 35, 1468–1473. [Google Scholar] [CrossRef] [Green Version]
- Navas, C.; Hernández-Porras, I.; Schuhmacher, A.J.; Sibilia, M.; Guerra, C.; Barbacid, M. EGF Receptor Signaling Is Essential for K-Ras Oncogene-Driven Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 22, 318–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; Delgiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF Receptor Is Required for KRAS-Induced Pancreatic Tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.-M.; Singh, G.; Koenig, A.; Liou, G.-Y.; Storz, P.; Zhang, J.-S.; Regul, L.; Nagarajan, S.; Kühnemuth, B.; Johnsen, S.A.; et al. NFATc1 Links EGFR Signaling to Induction of Sox9 Transcription and Acinar-Ductal Transdifferentiation in the Pancreas. Gastroenterology 2015, 148, 1024–1034.e9. [Google Scholar] [CrossRef] [Green Version]
- Means, A.L.; Ray, K.C.; Singh, A.B.; Washington, M.K.; Whitehead, R.H.; Harris, R.C.; Wright, C.V.E.; Coffey, R.J.; Leach, S.D. Overexpression of Heparin-Binding EGF-like Growth Factor in Mouse Pancreas Results in Fibrosis and Epithelial Metaplasia. Gastroenterology 2003, 124, 1020–1036. [Google Scholar] [CrossRef] [PubMed]
- Means, A.L.; Meszoely, I.M.; Suzuki, K.; Miyamoto, Y.; Rustgi, A.K.; Coffey, R.J.; Wright, C.V.E.; Stoffers, D.A.; Leach, S.D. Pancreatic Epithelial Plasticity Mediated by Acinar Cell Transdifferentiation and Generation of Nestin-Positive Intermediates. Development 2005, 132, 3767–3776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.; Weber, C.K.; Bressau, F.; Greten, F.R.; Stagge, V.; Ebert, M.; Leach, S.D.; Adler, G.; Schmid, R.M. Transgenic Overexpression of Amphiregulin Induces a Mitogenic Response Selectively in Pancreatic Duct Cells. Gastroenterology 2002, 122, 1898–1912. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, X.; Hamdan, F.H.; Likhobabina, A.; Patil, S.; Aperdannier, L.; Sen, M.; Traub, J.; Neesse, A.; Fischer, A.; et al. NFATc1 Is a Central Mediator of EGFR-Induced ARID1A Chromatin Dissociation During Acinar Cell Reprogramming. Cell Mol. Gastroenterol. Hepatol. 2023, 15, 1219–1246. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP Kinase Signalling Pathways in Cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.D.; Williams, J.A. Cholecystokinin Rapidly Activates Mitogen-Activated Protein Kinase in Rat Pancreatic Acini. Am. J. Physiol. 1994, 267, G401–G408. [Google Scholar] [CrossRef]
- Duan, R.D.; Zheng, C.F.; Guan, K.L.; Williams, J. Activation of MAP Kinase Kinase (MEK) and Ras by Cholecystokinin in Rat Pancreatic Acini. Am. J. Physiol. 1995, 268, G1060–G1065. [Google Scholar] [CrossRef]
- Mazzon, E.; Impellizzeri, D.; Di Paola, R.; Paterniti, I.; Esposito, E.; Cappellani, A.; Bramanti, P.; Cuzzocrea, S. Effects of Mitogen-Activated Protein Kinase Signaling Pathway Inhibition on the Development of Caerulein-Induced Acute Pancreatitis in Mice. Pancreas 2012, 41, 560–570. [Google Scholar] [CrossRef]
- Collins, M.A.; Yan, W.; Sebolt-Leopold, J.S.; Pasca di Magliano, M. MAPK Signaling Is Required for Dedifferentiation of Acinar Cells and Development of Pancreatic Intraepithelial Neoplasia in Mice. Gastroenterology 2014, 146, 822–834.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbrook, C.J.; Wen, H.-J.; Ruggeri, J.M.; Takeuchi, K.K.; Zhang, Y.; di Magliano, M.P.; Crawford, H.C. Mitogen-Activated Protein Kinase Kinase Activity Maintains Acinar-to-Ductal Metaplasia and Is Required for Organ Regeneration in Pancreatitis. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habbe, N.; Shi, G.; Meguid, R.A.; Fendrich, V.; Esni, F.; Chen, H.; Feldmann, G.; Stoffers, D.A.; Konieczny, S.F.; Leach, S.D.; et al. Spontaneous Induction of Murine Pancreatic Intraepithelial Neoplasia (MPanIN) by Acinar Cell Targeting of Oncogenic Kras in Adult Mice. Proc. Natl. Acad. Sci. USA 2008, 105, 18913–18918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Shi, G.; Schmidt, C.M.; Hruban, R.H.; Konieczny, S.F. Acinar Cells Contribute to the Molecular Heterogeneity of Pancreatic Intraepithelial Neoplasia. Am. J. Pathol. 2007, 171, 263–273. [Google Scholar] [CrossRef] [Green Version]
- De La O, J.-P.; Emerson, L.L.; Goodman, J.L.; Froebe, S.C.; Illum, B.E.; Curtis, A.B.; Murtaugh, L.C. Notch and Kras Reprogram Pancreatic Acinar Cells to Ductal Intraepithelial Neoplasia. Proc. Natl. Acad. Sci. USA 2008, 105, 18907–18912. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, H.; Zogopoulos, G.; Shao, Q.; Dong, K.; Lv, F.; Nwilati, K.; Gui, X.-Y.; Cuggia, A.; Liu, J.-L.; et al. Reg Proteins Promote Acinar-to-Ductal Metaplasia and Act as Novel Diagnostic and Prognostic Markers in Pancreatic Ductal Adenocarcinoma. Oncotarget 2016, 7, 77838–77853. [Google Scholar] [CrossRef]
- Zhang, H.; Corredor, A.L.G.; Messina-Pacheco, J.; Li, Q.; Zogopoulos, G.; Kaddour, N.; Wang, Y.; Shi, B.-Y.; Gregorieff, A.; Liu, J.-L.; et al. REG3A/REG3B Promotes Acinar to Ductal Metaplasia through Binding to EXTL3 and Activating the RAS-RAF-MEK-ERK Signaling Pathway. Commun. Biol. 2021, 4, 688. [Google Scholar] [CrossRef]
- Baer, R.; Cintas, C.; Therville, N.; Guillermet-Guibert, J. Implication of PI3K/Akt Pathway in Pancreatic Cancer: When PI3K Isoforms Matter? Adv. Biol. Regul. 2015, 59, 19–35. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The Emerging Mechanisms of Isoform-Specific PI3K Signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Baer, R.; Cintas, C.; Dufresne, M.; Cassant-Sourdy, S.; Schönhuber, N.; Planque, L.; Lulka, H.; Couderc, B.; Bousquet, C.; Garmy-Susini, B.; et al. Pancreatic Cell Plasticity and Cancer Initiation Induced by Oncogenic Kras Is Completely Dependent on Wild-Type PI 3-Kinase P110α. Genes Dev. 2014, 28, 2621–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eser, S.; Reiff, N.; Messer, M.; Seidler, B.; Gottschalk, K.; Dobler, M.; Hieber, M.; Arbeiter, A.; Klein, S.; Kong, B.; et al. Selective Requirement of PI3K/PDK1 Signaling for Kras Oncogene-Driven Pancreatic Cell Plasticity and Cancer. Cancer Cell 2013, 23, 406–420. [Google Scholar] [CrossRef] [Green Version]
- Payne, S.N.; Maher, M.E.; Tran, N.H.; Van De Hey, D.R.; Foley, T.M.; Yueh, A.E.; Leystra, A.A.; Pasch, C.A.; Jeffrey, J.J.; Clipson, L.; et al. PIK3CA Mutations Can Initiate Pancreatic Tumorigenesis and Are Targetable with PI3K Inhibitors. Oncogenesis 2015, 4, e169. [Google Scholar] [CrossRef] [Green Version]
- Stanger, B.Z.; Stiles, B.; Lauwers, G.Y.; Bardeesy, N.; Mendoza, M.; Wang, Y.; Greenwood, A.; Cheng, K.; McLaughlin, M.; Brown, D.; et al. Pten Constrains Centroacinar Cell Expansion and Malignant Transformation in the Pancreas. Cancer Cell 2005, 8, 185–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, R.; Calvopina, J.H.; Kim, C.; Wang, Y.; Dawson, D.W.; Donahue, T.R.; Dry, S.; Wu, H. PTEN Loss Accelerates KrasG12D-Induced Pancreatic Cancer Development. Cancer Res. 2010, 70, 7114–7124. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Liou, G.-Y.; Schmitt, D.M.; Storz, P.; Zhang, J.-S.; Billadeau, D.D. Glycogen Synthase Kinase-3β Ablation Limits Pancreatitis-Induced Acinar-to-Ductal Metaplasia. J. Pathol. 2017, 243, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; Yang, J.W.; Yang, V.W.; Bialkowska, A.B. Krüppel-like Factor 5, Increased in Pancreatic Ductal Adenocarcinoma, Promotes Proliferation, Acinar-to-Ductal Metaplasia, Pancreatic Intraepithelial Neoplasia, and Tumor Growth in Mice. Gastroenterology 2018, 154, 1494–1508.e13. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Wang, L.; Yan, Y.; Jia, Z.; Gagea, M.; Li, Z.; Zuo, X.; Kong, X.; Huang, S.; Xie, K. KLF4 Is Essential for Induction of Cellular Identity Change and Acinar-to-Ductal Reprogramming during Early Pancreatic Carcinogenesis. Cancer Cell 2016, 29, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Quilichini, E.; Haumaitre, C. Implication of Epigenetics in Pancreas Development and Disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 883–898. [Google Scholar] [CrossRef] [Green Version]
- Wauters, E.; Sanchez-Arévalo Lobo, V.J.; Pinho, A.V.; Mawson, A.; Herranz, D.; Wu, J.; Cowley, M.J.; Colvin, E.K.; Njicop, E.N.; Sutherland, R.L.; et al. Sirtuin-1 Regulates Acinar-to-Ductal Metaplasia and Supports Cancer Cell Viability in Pancreatic Cancer. Cancer Res. 2013, 73, 2357–2367. [Google Scholar] [CrossRef] [Green Version]
- Bombardo, M.; Saponara, E.; Malagola, E.; Chen, R.; Seleznik, G.M.; Haumaitre, C.; Quilichini, E.; Zabel, A.; Reding, T.; Graf, R.; et al. Class I Histone Deacetylase Inhibition Improves Pancreatitis Outcome by Limiting Leukocyte Recruitment and Acinar-to-Ductal Metaplasia. Br. J. Pharmacol. 2017, 174, 3865–3880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanika, G.; Khan, S.; Jena, G. Sodium Butyrate Ameliorates L-Arginine-Induced Pancreatitis and Associated Fibrosis in Wistar Rat: Role of Inflammation and Nitrosative Stress. J. Biochem. Mol. Toxicol. 2015, 29, 349–359. [Google Scholar] [CrossRef]
- da Silva, L.; Jiang, J.; Perkins, C.; Atanasova, K.R.; Bray, J.K.; Bulut, G.; Azevedo-Pouly, A.; Campbell-Thompson, M.; Yang, X.; Hakimjavadi, H.; et al. Pharmacological Inhibition and Reversal of Pancreatic Acinar Ductal Metaplasia. Cell Death Discov. 2022, 8, 378. [Google Scholar] [CrossRef] [PubMed]
- Eisses, J.F.; Criscimanna, A.; Dionise, Z.R.; Orabi, A.I.; Javed, T.A.; Sarwar, S.; Jin, S.; Zhou, L.; Singh, S.; Poddar, M.; et al. Valproic Acid Limits Pancreatic Recovery after Pancreatitis by Inhibiting Histone Deacetylases and Preventing Acinar Redifferentiation Programs. Am. J. Pathol. 2015, 185, 3304–3315. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Romero, C.; Rooman, I.; Skoudy, A.; Guerra, C.; Molero, X.; González, A.; Iglesias, M.; Lobato, T.; Bosch, A.; Barbacid, M.; et al. The Epigenetic Regulators Bmi1 and Ring1B Are Differentially Regulated in Pancreatitis and Pancreatic Ductal Adenocarcinoma. J. Pathol. 2009, 219, 205–213. [Google Scholar] [CrossRef]
- Benitz, S.; Regel, I.; Reinhard, T.; Popp, A.; Schäffer, I.; Raulefs, S.; Kong, B.; Esposito, I.; Michalski, C.W.; Kleeff, J. Polycomb Repressor Complex 1 Promotes Gene Silencing through H2AK119 Mono-Ubiquitination in Acinar-to-Ductal Metaplasia and Pancreatic Cancer Cells. Oncotarget 2016, 7, 11424–11433. [Google Scholar] [CrossRef] [Green Version]
- Benitz, S.; Straub, T.; Mahajan, U.M.; Mutter, J.; Czemmel, S.; Unruh, T.; Wingerath, B.; Deubler, S.; Fahr, L.; Cheng, T.; et al. Ring1b-Dependent Epigenetic Remodelling Is an Essential Prerequisite for Pancreatic Carcinogenesis. Gut 2019, 68, 2007–2018. [Google Scholar] [CrossRef]
- Johnson, C.L.; Mehmood, R.; Laing, S.W.; Stepniak, C.V.; Kharitonenkov, A.; Pin, C.L. Silencing of the Fibroblast Growth Factor 21 Gene Is an Underlying Cause of Acinar Cell Injury in Mice Lacking MIST1. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E916–E928. [Google Scholar] [CrossRef]
- Mallen-St Clair, J.; Soydaner-Azeloglu, R.; Lee, K.E.; Taylor, L.; Livanos, A.; Pylayeva-Gupta, Y.; Miller, G.; Margueron, R.; Reinberg, D.; Bar-Sagi, D. EZH2 Couples Pancreatic Regeneration to Neoplastic Progression. Genes Dev. 2012, 26, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, M.; Fukuda, A.; Roy, N.; Hiramatsu, Y.; Leonhardt, L.; Kakiuchi, N.; Hoyer, K.; Ogawa, S.; Goto, N.; Ikuta, K.; et al. The BRG1/SOX9 Axis Is Critical for Acinar Cell-Derived Pancreatic Tumorigenesis. J. Clin. Investig. 2018, 128, 3475–3489. [Google Scholar] [CrossRef]
- Livshits, G.; Alonso-Curbelo, D.; Morris, J.P.; Koche, R.; Saborowski, M.; Wilkinson, J.E.; Lowe, S.W. Arid1a Restrains Kras-Dependent Changes in Acinar Cell Identity. Elife 2018, 7, e35216. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Goga, A.; He, Y.; Silva, P.N.; Hirt, C.K.; Herrmanns, K.; Guccini, I.; Godbersen, S.; Schwank, G.; Stoffel, M. MiR-802 Suppresses Acinar-to-Ductal Reprogramming During Early Pancreatitis and Pancreatic Carcinogenesis. Gastroenterology 2022, 162, 269–284. [Google Scholar] [CrossRef]
- Brune, K.; Abe, T.; Canto, M.; O’Malley, L.; Klein, A.P.; Maitra, A.; Volkan Adsay, N.; Fishman, E.K.; Cameron, J.L.; Yeo, C.J.; et al. Multifocal Neoplastic Precursor Lesions Associated with Lobular Atrophy of the Pancreas in Patients Having a Strong Family History of Pancreatic Cancer. Am. J. Surg. Pathol. 2006, 30, 1067–1076. [Google Scholar] [PubMed]
- Klatte, D.C.F.; Wallace, M.B.; Löhr, M.; Bruno, M.J.; van Leerdam, M.E. Hereditary Pancreatic Cancer. Best Pract. Res. Clin. Gastroenterol. 2022, 58–59, 101783. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, M.; Werther, M.; Bockholt, A.; Scheurer, M.; Rüschoff, J.; Dietmaier, W.; Ghadimi, B.M.; Heinmöller, E. Genomic Instability at Both the Base Pair Level and the Chromosomal Level Is Detectable in Earliest PanIN Lesions in Tissues of Chronic Pancreatitis. Pancreas 2010, 39, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Kibe, S.; Ohuchida, K.; Ando, Y.; Takesue, S.; Nakayama, H.; Abe, T.; Endo, S.; Koikawa, K.; Okumura, T.; Iwamoto, C.; et al. Cancer-Associated Acinar-to-Ductal Metaplasia within the Invasive Front of Pancreatic Cancer Contributes to Local Invasion. Cancer Lett. 2019, 444, 70–81. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Hruban, R.H.; Adsay, N.V.; Albores-Saavedra, J.; Anver, M.R.; Biankin, A.V.; Boivin, G.P.; Furth, E.E.; Furukawa, T.; Klein, A.; Klimstra, D.S.; et al. Pathology of Genetically Engineered Mouse Models of Pancreatic Exocrine Cancer: Consensus Report and Recommendations. Cancer Res. 2006, 66, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.-F.; Dubois, C.L.; Morris, J.P.; Pan, F.C.; Akiyama, H.; Wright, C.V.E.; Jensen, K.; et al. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Pekala, K.R.; Ma, X.; Kropp, P.A.; Petersen, C.P.; Hudgens, C.W.; Chung, C.H.; Shi, C.; Merchant, N.B.; Maitra, A.; Means, A.L.; et al. Loss of HNF6 Expression Correlates with Human Pancreatic Cancer Progression. Lab. Investig. 2014, 94, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Grimont, A.; Pinho, A.V.; Cowley, M.J.; Augereau, C.; Mawson, A.; Giry-Laterrière, M.; Van den Steen, G.; Waddell, N.; Pajic, M.; Sempoux, C.; et al. SOX9 Regulates ERBB Signalling in Pancreatic Cancer Development. Gut 2015, 64, 1790–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.P.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the Twisted Developmental Biology of Pancreatic Ductal Adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef]
- De La O, J.-P.; Murtaugh, L.C. Notch and Kras in Pancreatic Cancer: At the Crossroads of Mutation, Differentiation and Signaling. Cell Cycle 2009, 8, 1860–1864. [Google Scholar] [CrossRef] [Green Version]
- Carrière, C.; Young, A.L.; Gunn, J.R.; Longnecker, D.S.; Korc, M. Acute Pancreatitis Markedly Accelerates Pancreatic Cancer Progression in Mice Expressing Oncogenic Kras. Biochem. Biophys. Res. Commun. 2009, 382, 561–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.-C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Pasca di Magliano, M. Oncogenic Kras Is Required for Both the Initiation and Maintenance of Pancreatic Cancer in Mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Maitra, A.; Ghosh, B.; Zechner, U.; Argani, P.; Iacobuzio-Donahue, C.A.; Sriuranpong, V.; Iso, T.; Meszoely, I.M.; Wolfe, M.S.; et al. Notch Mediates TGF Alpha-Induced Changes in Epithelial Differentiation during Pancreatic Tumorigenesis. Cancer Cell 2003, 3, 565–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, Y.; Kodama, Y.; Shiokawa, M.; Matsumori, T.; Marui, S.; Kuriyama, K.; Kuwada, T.; Sogabe, Y.; Kakiuchi, N.; Tomono, T.; et al. Hes1 Plays an Essential Role in Kras-Driven Pancreatic Tumorigenesis. Oncogene 2019, 38, 4283–4296. [Google Scholar] [CrossRef] [Green Version]
- Loncle, C.; Bonjoch, L.; Folch-Puy, E.; Lopez-Millan, M.B.; Lac, S.; Molejon, M.I.; Chuluyan, E.; Cordelier, P.; Dubus, P.; Lomberk, G.; et al. IL17 Functions through the Novel REG3β-JAK2-STAT3 Inflammatory Pathway to Promote the Transition from Chronic Pancreatitis to Pancreatic Cancer. Cancer Res. 2015, 75, 4852–4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, G.-Y.; Döppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS-Induced Expression of ICAM-1 in Pancreatic Acinar Cells Causes Attraction of Macrophages to Expedite the Formation of Precancerous Lesions. Cancer Discov. 2015, 5, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling Mechanisms of P53-Mediated Tumour Suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Biankin, A.V.; Kench, J.G.; Morey, A.L.; Lee, C.S.; Biankin, S.A.; Head, D.R.; Hugh, T.B.; Henshall, S.M.; Sutherland, R.L. Overexpression of P21(WAF1/CIP1) Is an Early Event in the Development of Pancreatic Intraepithelial Neoplasia. Cancer Res. 2001, 61, 8830–8837. [Google Scholar] [PubMed]
- Mello, S.S.; Flowers, B.M.; Mazur, P.K.; Lee, J.J.; Müller, F.; Denny, S.K.; Ferreira, S.; Hanson, K.; Kim, S.K.; Greenleaf, W.J.; et al. Multifaceted Role for P53 in Pancreatic Cancer Suppression. Proc. Natl. Acad. Sci. USA 2023, 120, e2211937120. [Google Scholar] [CrossRef] [PubMed]
- Cui Zhou, D.; Jayasinghe, R.G.; Chen, S.; Herndon, J.M.; Iglesia, M.D.; Navale, P.; Wendl, M.C.; Caravan, W.; Sato, K.; Storrs, E.; et al. Spatially Restricted Drivers and Transitional Cell Populations Cooperate with the Microenvironment in Untreated and Chemo-Resistant Pancreatic Cancer. Nat. Genet. 2022, 54, 1390–1405. [Google Scholar] [CrossRef]
- Schlesinger, Y.; Yosefov-Levi, O.; Kolodkin-Gal, D.; Granit, R.Z.; Peters, L.; Kalifa, R.; Xia, L.; Nasereddin, A.; Shiff, I.; Amran, O.; et al. Single-Cell Transcriptomes of Pancreatic Preinvasive Lesions and Cancer Reveal Acinar Metaplastic Cells’ Heterogeneity. Nat. Commun. 2020, 11, 4516. [Google Scholar] [CrossRef]
- Delgiorno, K.E.; Hall, J.C.; Takeuchi, K.K.; Pan, F.C.; Halbrook, C.J.; Washington, M.K.; Olive, K.P.; Spence, J.R.; Sipos, B.; Wright, C.V.E.; et al. Identification and Manipulation of Biliary Metaplasia in Pancreatic Tumors. Gastroenterology 2014, 146, 233–244.e5. [Google Scholar] [CrossRef] [Green Version]
- DelGiorno, K.E.; Naeem, R.F.; Fang, L.; Chung, C.-Y.; Ramos, C.; Luhtala, N.; O’Connor, C.; Hunter, T.; Manor, U.; Wahl, G.M. Tuft Cell Formation Reflects Epithelial Plasticity in Pancreatic Injury: Implications for Modeling Human Pancreatitis. Front. Physiol. 2020, 11, 88. [Google Scholar] [CrossRef]
- Bailey, J.M.; Alsina, J.; Rasheed, Z.A.; McAllister, F.M.; Fu, Y.-Y.; Plentz, R.; Zhang, H.; Pasricha, P.J.; Bardeesy, N.; Matsui, W.; et al. DCLK1 Marks a Morphologically Distinct Subpopulation of Cells with Stem Cell Properties in Preinvasive Pancreatic Cancer. Gastroenterology 2014, 146, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westphalen, C.B.; Takemoto, Y.; Tanaka, T.; Macchini, M.; Jiang, Z.; Renz, B.W.; Chen, X.; Ormanns, S.; Nagar, K.; Tailor, Y.; et al. Dclk1 Defines Quiescent Pancreatic Progenitors That Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem. Cell 2016, 18, 441–455. [Google Scholar] [CrossRef] [Green Version]
- Maruno, T.; Fukuda, A.; Goto, N.; Tsuda, M.; Ikuta, K.; Hiramatsu, Y.; Ogawa, S.; Nakanishi, Y.; Yamaga, Y.; Yoshioka, T.; et al. Visualization of Stem Cell Activity in Pancreatic Cancer Expansion by Direct Lineage Tracing with Live Imaging. Elife 2021, 10, e55117. [Google Scholar] [CrossRef]
- Lu, Q.; Feng, H.; Chen, H.; Weygant, N.; Du, J.; Yan, Z.; Cao, Z. Role of DCLK1 in Oncogenic Signaling (Review). Int. J. Oncol. 2022, 61, 137. [Google Scholar] [CrossRef]
- Ma, Z.; Lytle, N.K.; Chen, B.; Jyotsana, N.; Novak, S.W.; Cho, C.J.; Caplan, L.; Ben-Levy, O.; Neininger, A.C.; Burnette, D.T.; et al. Single-Cell Transcriptomics Reveals a Conserved Metaplasia Program in Pancreatic Injury. Gastroenterology 2022, 162, 604–620.e20. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marstrand-Daucé, L.; Lorenzo, D.; Chassac, A.; Nicole, P.; Couvelard, A.; Haumaitre, C. Acinar-to-Ductal Metaplasia (ADM): On the Road to Pancreatic Intraepithelial Neoplasia (PanIN) and Pancreatic Cancer. Int. J. Mol. Sci. 2023, 24, 9946. https://doi.org/10.3390/ijms24129946
Marstrand-Daucé L, Lorenzo D, Chassac A, Nicole P, Couvelard A, Haumaitre C. Acinar-to-Ductal Metaplasia (ADM): On the Road to Pancreatic Intraepithelial Neoplasia (PanIN) and Pancreatic Cancer. International Journal of Molecular Sciences. 2023; 24(12):9946. https://doi.org/10.3390/ijms24129946
Chicago/Turabian StyleMarstrand-Daucé, Louis, Diane Lorenzo, Anaïs Chassac, Pascal Nicole, Anne Couvelard, and Cécile Haumaitre. 2023. "Acinar-to-Ductal Metaplasia (ADM): On the Road to Pancreatic Intraepithelial Neoplasia (PanIN) and Pancreatic Cancer" International Journal of Molecular Sciences 24, no. 12: 9946. https://doi.org/10.3390/ijms24129946