
Transcriptomic and Metabolomic Analyses Provide Insights into the Pathogenic Mechanism of the Rice False Smut Pathogen Ustilaginoidea virens
Abstract
:1. Introduction
2. Results
2.1. Evaluation of U. virens Pathogenicity
2.2. Transcriptome Test Results and Analysis
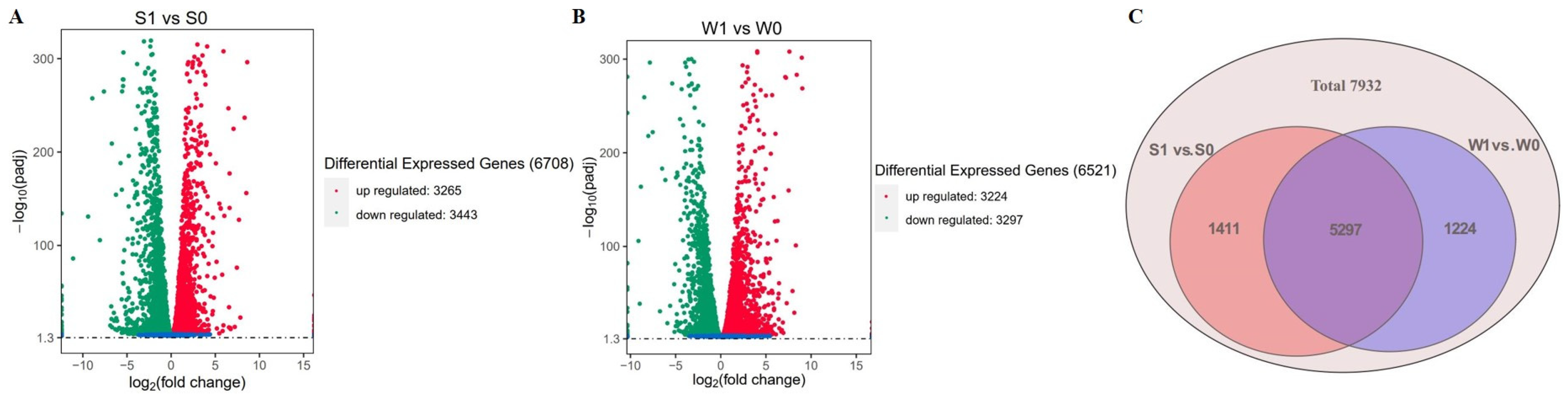
2.2.1. Summary of Transcriptome Data
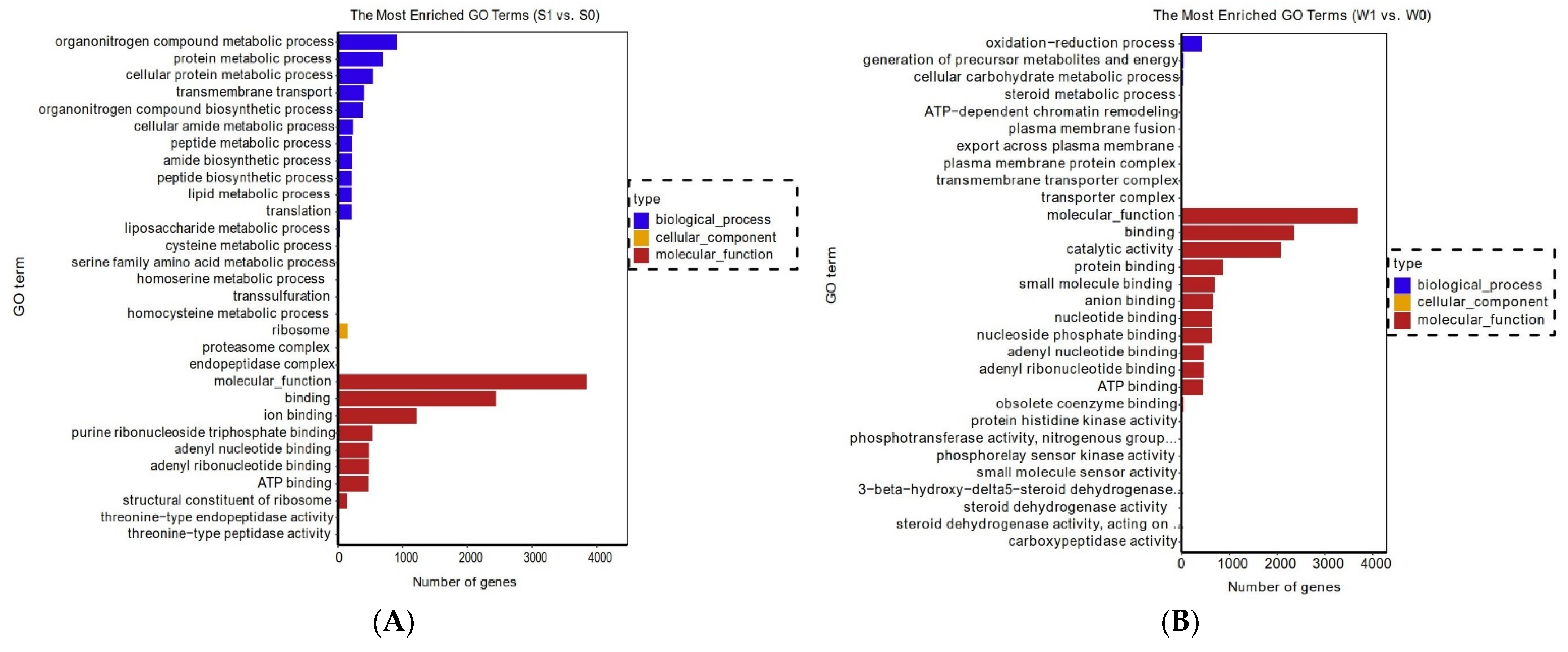
2.2.2. Enrichment Analysis of Functional Genes
2.2.3. Analysis of Pathogenicity-Related DEGs
2.2.4. RT-qPCR Validation of DEGs
2.3. Detection Results and Analysis of the Untargeted Metabolome
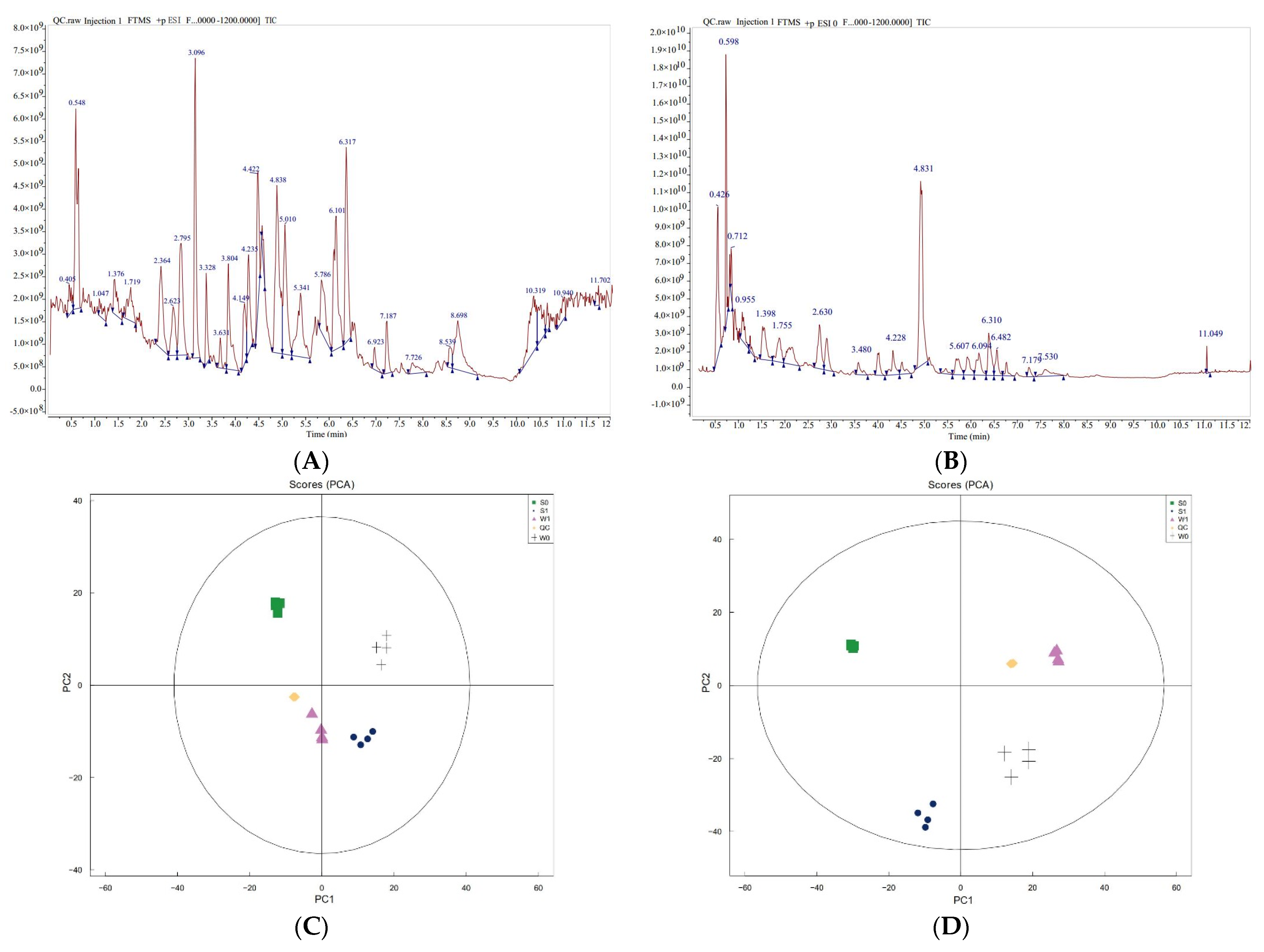
2.3.1. Quality Control and Statistical Analysis of Untargeted Metabolome Data
2.3.2. Qualitative and Quantitative Analyses of Metabolites
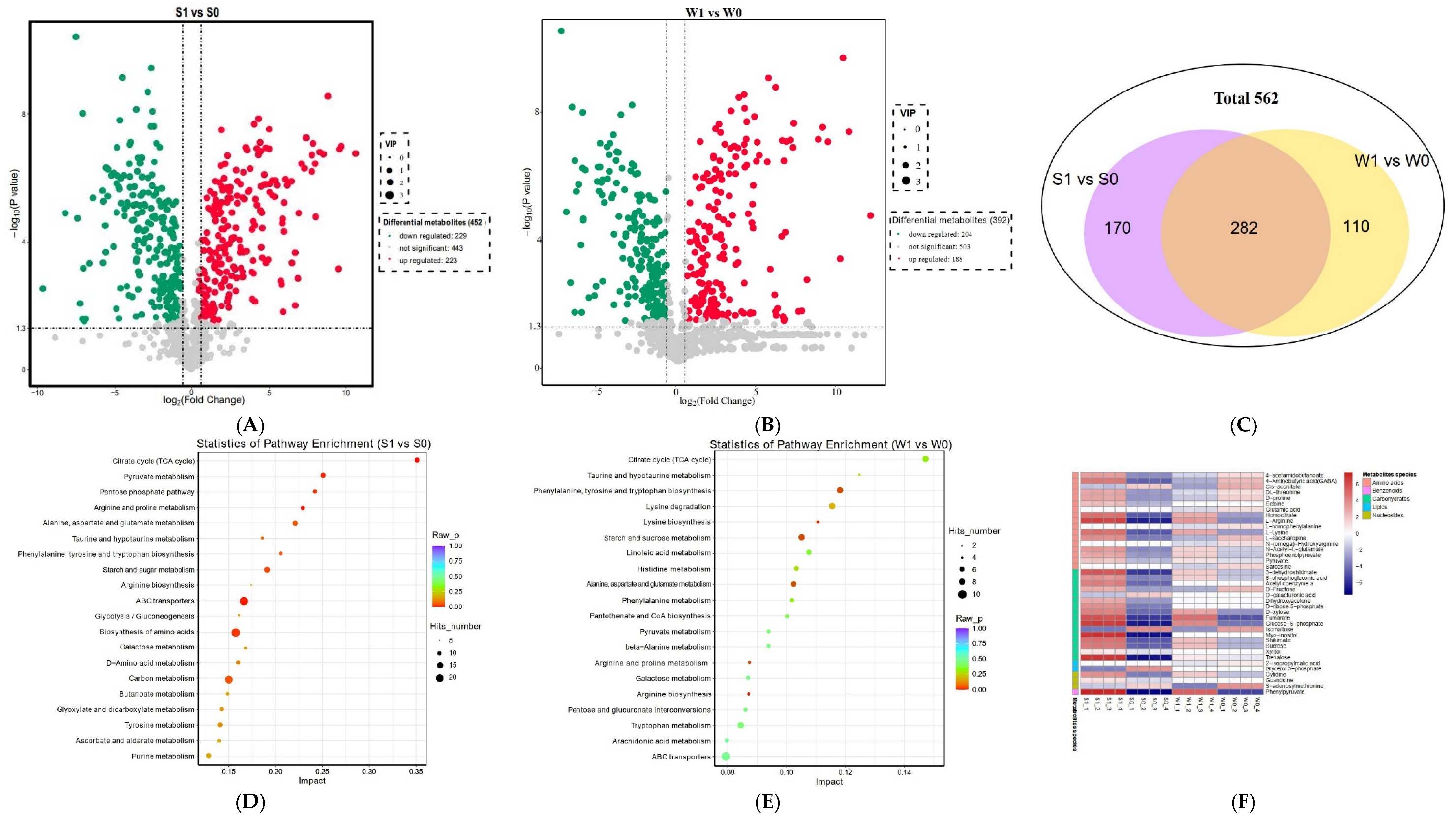
2.3.3. Differential Metabolite Analysis
2.4. Combined Analysis of Transcription and Metabolome
2.4.1. Correlation Analysis between DEGs and DAMs
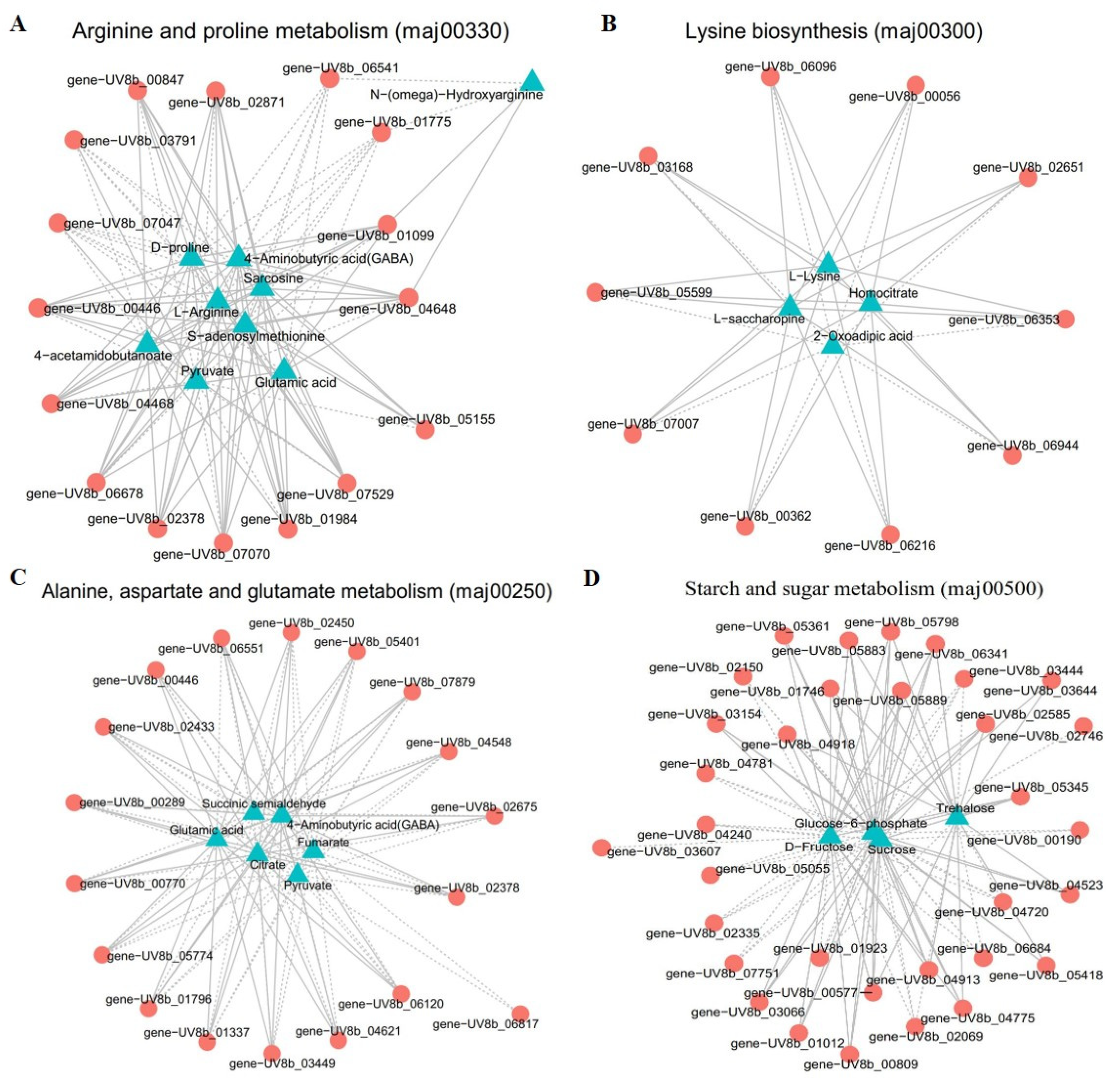
2.4.2. Correlation Analysis of DEGs and DAMs in Significantly Enriched Metabolic Pathways
3. Discussion
4. Materials and Methods
4.1. Materials and Fungal Inoculation
4.2. Transcriptome Sequencing and Analysis
4.3. Untargeted Metabolome Detection and Analysis
4.4. Combined Analysis of the Transcriptome and Metabolome
4.5. Validation of qRT-PCR
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tanaka, T.; Ashizawa, T.; Sonoda, R.; Tanaka, C. Villosiclava virens gen. nov., com. nov., teleomorph of Ustilaginoidea virens, the causal agent of rice false smut. Mycotaxon 2008, 106, 491–501. [Google Scholar]
- Dhua, U.; Dhua, S.R.; Sahu, R.K. Precise disease severity assessment for false smut disease of rice. J. Phytopathol. 2015, 163, 931–940. [Google Scholar] [CrossRef]
- Koyama, K.; Ominato, K.; Natori, S.; Tashiro, T.; Tsuruo, T. Cytotoxicity and antitumor activities of fungal bis-(naphtho-g-pyrone) derivatives. J. Pharm. Dyn. 1988, 11, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Ludueña, R.F.; Roach, M.C.; Prasad, V.; Banerjee, M.; Koiso, Y.; Li, Y.; Iwasaki, S. Interaction of ustiloxin a with bovine brain tubulin. Biochem. Pharmacol. 1994, 47, 1593–1599. [Google Scholar] [CrossRef]
- Koiso, Y.; Li, Y.; Iwasaki, S. Ustiloxins, antimitotic cyclic peptides from false smut balls on rice panicles caused by Ustilaginoidea virens. J. Antibiot. 1994, 47, 765–773. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.X.; Fan, J.; Fang, A.F.; Li, Y.J.; Tariqjaveed, M.; Li, D.Y.; Wang, W.M. Ustilaginoidea virens: Insights into an emerging rice pathogen. Annu. Rev. Phytopathol. 2020, 58, 363–385. [Google Scholar] [CrossRef]
- Chen, X.; Qiu, J.H.; Xiong, M.; Shu, Y.Z.; Huang, S.W.; Kou, Y.J. Research progress of rice false smut. Chinese Rice 2019, 25, 30–36. [Google Scholar]
- Zhou, Y.; Yu, J.; Pan, X.; Yu, M.; Du, Y.; Qi, Z.; Zhang, R.; Song, T.; Yin, X.; Liu, Y. Characterization of propiconazole field-resistant isolates of Ustilaginoidea virens. Pestic. Biochem. Phys. 2019, 153, 144–151. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, K.; Fang, A.; Han, Y.Q.; Yang, J.; Xue, M.F.; Sun, W. Specific adaptation of Ustilaginoidea virens in occupying host florets revealed by comparative and functional genomics. Nat. Commun. 2014, 5, 3849–3860. [Google Scholar] [CrossRef] [Green Version]
- Lv, B.; Zheng, L.; Liu, H.; Tang, J.; Hsiang, T.; Huang, J. Use of random T-DNA mutagenesis in identification of gene Uv PRO1, a regulator of conidiation, stress response, and virulence in Ustilaginoidea virens. Front Microbiol. 2016, 7, 2086–2099. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.W.; Wang, Y.; Han, Y.; Xu, J.R.; Wang, C.F. UvHOG1 is important for hyphal growth and stress responses in the rice false smut fungus Ustilaginoidea virens. Sci Rep. 2016, 6, 24824. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Xiong, M.; Jagernath, J.S.; Wang, C.C.; Qiu, J.H.; Shi, H.B.; Kou, Y.J. UvAtg8-mediated autophagy regulates fungal growth, stress responses, conidiation, and pathogenesis in Ustilaginoidea virens. Rice 2020, 13, 56. [Google Scholar] [CrossRef]
- Yu, J.J.; Yu, M.N.; Song, T.Q.; Cao, H.J.; Pan, X.Y.; Yong, M.L.; Qi, Z.Q.; Du, Y.; Zhang, R.S.; Yin, X.L.; et al. A Homeobox Transcription Factor UvHOX2 regulates chlamydospore formation, conidiogenesis, and pathogenicity in Ustilaginoidea virens. Front Microbiol. 2019, 10, 1071. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Yang, J.; Wang, Y.Q.; Li, G.B.; Li, Y.; Huang, F.; Wang, W.M. Current understanding on Villosiclava virens, a unique flower infecting fungus causing rice false smut disease. Mol. Plant Pathol. 2016, 17, 1321–1330. [Google Scholar] [CrossRef]
- Ashizawa, T.; Takahashi, M.; Arai, M.; Arie, T. Rice false pathogen, Ustilaginoidea virens, invades through small gap at the apex of a rice spikelet before heading. J. Gen. Plant Pathol. 2012, 78, 255–259. [Google Scholar] [CrossRef]
- Sana, T.R.; Fischer, S.; Wohlgemuth, G.; Katretar, A.; Jung, K.H.; Ronald, P.C.; Fiehn, O. Metabolomic and transcriptomic analysis of the rice response to the bacterial blight pathogen Xanthomonas oryzae pv. oryzae. Metabolomics 2010, 6, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, A.; Westrick, N.M.; Jain, S.; Piotrowski, J.S.; Ranjan, M.; Kessens, R.; Kabbage, M. Resistance against Sclerotinia sclerotiorum in soybean involves a reprogramming of the phenylpropanoid pathway and up-regulation of antifungal activity targeting ergosterol biosynthesis. Plant Biotechnol. J. 2019, 17, 1567–1581. [Google Scholar] [CrossRef] [Green Version]
- Almeida, J.; Perez-Fons, L.; Fraser, P.D. A transcriptomic, metabolomic and cellular approach to the physiological adaptation of tomato fruit to high temperature. Plant Cell Environ. 2021, 44, 2211–2229. [Google Scholar] [CrossRef]
- Xu, Q.; Fu, Y.Q.; Li, S.; Jiang, L.; Guan, R.F.; Huang, H. Integrated transcriptomic and metabolomic analysis of Rhizopus oryzae with different morphologies. Process Biochem. 2017, 10, 1359–1368. [Google Scholar] [CrossRef]
- Cai, X.Z.; Chen, M.Y.; Lu, Y.P.; Guo, Z.J.; Zeng, Z.H.; Liao, J.H.; Zeng, H. Metabolomics and transcriptomics unravel the mechanism of browning resistance in Agaricus bisporus. PLoS ONE. 2022, 17, e0255765. [Google Scholar] [CrossRef]
- Liboro, K.; Yu, S.R.; Lim, J.; So, Y.S.; Bahn, Y.S.; Eoh, H.; Park, H. Transcriptomic and metabolomic analysis revealed roles of yck2 in carbon metabolism and morphogenesis of Candida albicans. Front. Cell. Infect. Microbiol. 2021, 11, 636834. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Jin, J.; Wang, D.; Han, Y.; Zhu, R.S.; Zhu, Y.G.; Li, S. Cytological and transcriptional dynamics analysis of host plant revealed stage specific biological processes related to compatible rice Ustilaginoidea virens interaction. PLoS ONE 2014, 9, e91391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, R.T.; Chen, C.; Wang, J.; Liu, Y.; Zhao, L.Y.; Lu, D.H. Transcription profiling of rice panicle in response to crude toxin extract of Ustilaginoidea virens. Front. Microbiol. 2022, 13, 701489. [Google Scholar] [CrossRef] [PubMed]
- Rani, R.; Sharma, V.K. Diversity analysis of northern Indian isolates of Ustilaginoidea virens. Indian Phytopathol. 2018, 71, 33–42. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, H.; Xu, H.; Hei, R.; Zhang, S.; Jiang, C.; Xu, J.R. Independent losses and duplications of autophagy-related genes in fungal tree of life. Environ. Microbiol. 2019, 21, 226–243. [Google Scholar] [CrossRef] [Green Version]
- Rohde, J.; Heitman, J.; Cardenas, M.E. The TOR kinases link nutrient sensing to cell growth. J. Biol. Chem. 2001, 276, 9583–9586. [Google Scholar] [CrossRef] [Green Version]
- Rispail, N.; Soanes, D.M.; Ant, C.; Czajkowski, R.; Grünler, A.; Huguet, R.; Di Pietro, A. Comparative genomics of MAP kinase and calcium-calcineurin signaling components in plant and human pathogenic fungi. Fungal Genet. Biol. 2009, 46, 287–298. [Google Scholar] [CrossRef]
- Choi, J.; Jung, W.H.; Kronstad, J.W. The cAMP/protein kinase a signaling pathway in pathogenic basidiomycete fungi: Connections with iron homeostasis. J. Microbiol. 2015, 53, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zuo, R.F.; Zhu, Q.; Sun, Y.; Li, M.Y.; Dong, Y.H.; Ru, Y.Y.; Zhang, H.F.; Zheng, X.B.; Zhang, Z.G. MoLys2 is necessary for growth, conidiogenesis, lysine biosynthesis, and pathogenicity in Magnaporthe oryzae. Fungal Genet. Biol. 2014, 67, 51–57. [Google Scholar] [CrossRef]
- Wilson, R.A.; Fernandez, J.; Quispe, C.F.; Gradnigo, J.; Seng, A.; Moriyama, E.; Wright, J.D. Towards defining nutrient conditions encountered by the rice blast fungus during host infection. PLoS ONE 2012, 7, e47392. [Google Scholar] [CrossRef]
- Sattarova, R.S. Isolation, purification, and certain properties of β-1,3-glucanase from the fungus Verticillium dahliae. Chem. Nat. Compd. 2001, 37, 177–180. [Google Scholar] [CrossRef]
- Foster, A.J.; Jenkinson, J.M.; Taltot, N.J. Trehalose synthesis and metabolism are required at different stages of plant infection by Magnaporthe grisea. EMBO J. 2003, 22, 225–235. [Google Scholar] [CrossRef]
- Sharanabasav, H.; Pramesh, D.; Prasannakumar, M.K.; Chidanandappa, E.; Yadav, M.K.; Ngangkham, U.; Parivallal, B.; Raghavendra, B.T.; Manjunatha, S.K.; Sharma, S.K.; et al. Morpho-molecular and mating-type locus diversity of Ustilaginoidea virens: An incitant of false smut of rice from Southern parts of India. J. Appl. Microbiol. 2021, 131, 2372–2386. [Google Scholar] [CrossRef]
- Bag, M.K.; Ray, A.; Masurkar, P.; Devanna, B.N.; Parameswaran, C.; Baite, M.; Rath, P.C.; Nayak, A. Genetic diversity and population structure analysis of isolates of the rice false smut pathogen Ustilaginoidea virens in India. Plant Pathol. 2021, 70, 1085–1097. [Google Scholar] [CrossRef]
- Liu, X.H.; Lu, J.P.; Lin, F.C. Autophagy during conidiation, conidial germination and turgor generation in Magnaporthe grisea. Autophagy 2007, 3, 472–473. [Google Scholar] [CrossRef] [Green Version]
- Hamel, L.P.; Ellis, B.E. Mitogen-activated protein kinase signaling in plant-interacting fungi: Distinct messages from conserved messengers. Plant Cell 2012, 24, 1327–1351. [Google Scholar] [CrossRef] [Green Version]
- Akira, M.; Tsukada, M.; Wada, Y.; Ohsumi, Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 1997, 192, 245–250. [Google Scholar]
- Nguyen, L.N.; Bormann, J.; Le, G.T.T.; Stärkel, C.; Olsson, S.; Nosanchuk, J.D.; Schäfer, W. Autophagy-related lipase FgATG15 of Fusarium graminearum is important for lipid turnover and plant infection. Fungal Genet. Biol. 2011, 48, 217–224. [Google Scholar] [CrossRef]
- Li, F.; Vierstra, R.D. Autophagy: A multifaceted intracellular system for bulk and selective recycling. Trends Plant Sci. 2012, 17, 526–537. [Google Scholar] [CrossRef]
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated phosphorylation of atg13 inhibits starvation-induced autophagy. J. Biol. Chem. 2016, 291, 6026–6035. [Google Scholar] [CrossRef] [Green Version]
- Thines, E.; Weber, R.W.; Talbot, N.J. MAP kinase and protein kinase A—Dependent mobilization of triacylglycerol and glycogen during appressorium turgor Generation by Magnaporthe grisea. Plant Cell 2000, 12, 1703–1718. [Google Scholar] [PubMed] [Green Version]
- Zhao, X.; Xu, J.R. A mitogen-activated protein kinase cascade regulating infection-related morphogenesis in Magnaporthe grisea. Plant Cell. 2005, 17, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Xiong, Y.; Zhu, W.; Wang, N.; Yang, G.; Peng, F. Colletotrichum higginsianum mitogen-activated protein kinase ChMK1: Role in growth, cell wall integrity, colony melanization, and pathogenicity. Front. Microbiol. 2016, 8, 01212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron, O.; Wang, M.; Lin, L.; Batool, W.; Lin, B.; Shabbir, A.; Tang, W. MoGLN2 is important for vegetative growth, conidiogenesis, maintenance of cell wall integrity and pathogenesis of Magnaporthe oryzae. J. Fungi 2021, 7, 463. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; van Kan, J.A.L.; Bailey, A.M.; Bailey, A.M.; Forter, G.D. Inadvertent gene silencing of argininosuccinate synthase (bcass1) in Botrytis cinerea by the pLOB1vector system. Mol. Plant Pathol. 2010, 11, 613–624. [Google Scholar]
- Zhang, Y.; Shi, H.; Liang, S.; Ning, G.; Xu, N.; Lu, J.; Lin, F. MoARG1, MoARG5, 6 and MoARG7 involved in arginine biosynthesis are essential for growth, conidiogenesis, sexual reproduction, and pathogenicity in Magnaporthe oryzae. Microbiol. Res. 2015, 180, 11–22. [Google Scholar] [CrossRef]
- Seong, K.Y.; Hou, Z.; Tracy, M.; Kistler, H.C.; Xu, J.R. Random insertional mutagenesis identifies genes associated with virulence in the wheat scab fungus Fusarium graminearum. Phytopathology 2005, 95, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Thevelein, J.M. Regulation of trehalose mobilization in fungi. Microbiol. Rev. 1984, 48, 42–59. [Google Scholar] [CrossRef]
- Iordachescu, M.; Imai, R. Trehalose biosynthesis in response to abiotic stresses. J. Integr. Plant Biol. 2008, 50, 1223–1229. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Gustafson, D.L.; Long, M.E.; Bradshaw, E.L.; Merz, A.L.; Kerzic, P.J. P450 induction alters paclitaxel pharmacokinetics and tissue distribution with multiple dosing. Cancer Chemother. Pharmacol. 2005, 56, 248–254. [Google Scholar] [CrossRef]
- Kaling, M.; Schmidt, A.; Moritz, F.; Rosenkranz, M.; Witting, M.; Kasper, K.; Polle, A. Mycorrhiza-triggered transcriptomic and metabolomic networks impinge on herbivore fitness. Plant Physiol. 2018, 176, 2639–2656. [Google Scholar] [CrossRef] [Green Version]
- Boccard, J.; Rutledge Douglas, N.A. Consensus orthogonal partial least squares discriminant analysis (OPLS-DA) strategy for multiblock Omics data fusion. Anal. Chim. Acta 2013, 769, 30–39. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using realtime quantitative PCR and the 2−△△CT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primers(5′–3′) | Log2FC (RNA-Seq) | Log2FC (RT-qPCR) |
|---|---|---|---|
| UV8b_04520 | F-ATGGCTCCCCGAGGAGGCTTTG R-TCAGTCCGCCTCTGCCAGCTGCATGA | 3.34251 | 2.93451 |
| UV8b_06544 | F-ATGGCGAGTCGTCAAGATGGAT R-TTATGCTGATCCATGCGATGGTA | 2.95398 | 2.56875 |
| UV8b_07283 | F-ATGCCCAAGCGCCTACTCCGT R-CGGAGTCTGCTGCAGCCAAACCCTA | 2.97542 | 2.28723 |
| UV8b_07350 | F-ATGGGGACTTCTCCTGCCACCGAGT R-TCATGAGAAGCGGCGCTGGACGCCT | 3.32256 | 3.01291 |
| UV8b_05641 | F-ATGGACGGTCCGAGATCGTCCTTG R-GATGTTGCCTTCGTTGGACAGCA | 2.41345 | 2.13214 |
| UV8b_01480 | F-ATGGCAGGCGGCATGGGTCCCC R-TTAGCAGTATGACTCGTTAGGC | 3.90424 | 3.45192 |
| UV8b_06216 | F-ATGGCAAATGCAAACTCCATCTA R-TCAGGTAATCTGGGCAATAACC | 1.63942 | 1.02376 |
| UV8b_07418 | F-ATGTCCAGTGACACCAGTATTAC R-AATTCTATGGTACCGAATTTGCCGT | 2.56132 | 1.98124 |
| UV8b_04169 | F-ATGGCGGCAACGGTAGCCCAGTCG R-TCAAAACTCCTCGAAGTACCGG | −3.60581 | −3.97812 |
| UV8b_00876 | F-ATGGCGCAACAACAGCAGGTC R-CCTTCCCTGACATATATGAGCA | −3.27163 | −3.51234 |
| Species of Metabolites | Number of Metabolites |
|---|---|
| Organic acids and derivatives | 186 |
| Lipids and lipid-like molecules | 133 |
| Organoheterocyclic compounds | 100 |
| Benzenoids | 86 |
| Organic oxygen compounds | 81 |
| Phenylpropanoids and polyketides | 31 |
| Nucleosides, nucleotides, and analogues | 28 |
| Organic nitrogen compounds | 25 |
| Other | 13 |
| Alkaloids and derivatives | 8 |
| Organosulfur compounds | 2 |
| Lignans, neolignans and related compounds | 2 |
| Homogeneous non-metal compounds | 2 |
| Hydrocarbon derivatives | 1 |
| Total | 698 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, R.; Wang, J.; Chen, C.; Liu, Y.; Zhao, L.; Lu, D. Transcriptomic and Metabolomic Analyses Provide Insights into the Pathogenic Mechanism of the Rice False Smut Pathogen Ustilaginoidea virens. Int. J. Mol. Sci. 2023, 24, 10805. https://doi.org/10.3390/ijms241310805
Fu R, Wang J, Chen C, Liu Y, Zhao L, Lu D. Transcriptomic and Metabolomic Analyses Provide Insights into the Pathogenic Mechanism of the Rice False Smut Pathogen Ustilaginoidea virens. International Journal of Molecular Sciences. 2023; 24(13):10805. https://doi.org/10.3390/ijms241310805
Chicago/Turabian StyleFu, Rongtao, Jian Wang, Cheng Chen, Yao Liu, Liyu Zhao, and Daihua Lu. 2023. "Transcriptomic and Metabolomic Analyses Provide Insights into the Pathogenic Mechanism of the Rice False Smut Pathogen Ustilaginoidea virens" International Journal of Molecular Sciences 24, no. 13: 10805. https://doi.org/10.3390/ijms241310805