

The Therapeutic Potential of CDK4/6 Inhibitors, Novel Cancer Drugs, in Kidney Diseases

Guangdong Provincial Key Laboratory of Autophagy and Major Chronic Non-Communicable Diseases, Key Laboratory of Prevention and Management of Chronic Kidney Diseases of Zhanjiang City, Institute of Nephrology, Affiliated Hospital of Guangdong Medical University, Zhanjiang 524001, China
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2023, 24(17), 13558; https://doi.org/10.3390/ijms241713558
Submission received: 12 July 2023
/
Revised: 27 August 2023
/
Accepted: 29 August 2023
/
Published: 31 August 2023
(This article belongs to the Special Issue Inflammation and Cancer 2023)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Inflammation is a crucial pathological feature in cancers and kidney diseases, playing a significant role in disease progression. Cyclin-dependent kinases CDK4 and CDK6 not only contribute to cell cycle progression but also participate in cell metabolism, immunogenicity and anti-tumor immune responses. Recently, CDK4/6 inhibitors have gained approval for investigational treatment of breast cancer and various other tumors. Kidney diseases and cancers commonly exhibit characteristic pathological features, such as the involvement of inflammatory cells and persistent chronic inflammation. Remarkably, CDK4/6 inhibitors have demonstrated impressive efficacy in treating non-cancerous conditions, including certain kidney diseases. Current studies have identified the renoprotective effect of CDK4/6 inhibitors, presenting a novel idea and potential direction for treating kidney diseases in the future. In this review, we briefly reviewed the cell cycle in mammals and the role of CDK4/6 in regulating it. We then provided an introduction to CDK4/6 inhibitors and their use in cancer treatment. Additionally, we emphasized the importance of these inhibitors in the treatment of kidney diseases. Collectively, growing evidence demonstrates that targeting CDK4 and CDK6 through CDK4/6 inhibitors might have therapeutic benefits in various cancers and kidney diseases and should be further explored in the future.
1. Introduction
The dysregulation of the cell cycle is closely associated with the development of various diseases, particularly cancer, where abnormal cell cycle activation is a hallmark of carcinogenesis. Moreover, dysregulated cell proliferation is also implicated in the pathogenesis of renal diseases, cardiovascular diseases and nervous system disorders. Consequently, there has been a considerable focus on targeting pathways that regulate the cell cycle across different disease disciplines for several years.
CDK4/6 inhibitors are a novel class of therapy that selectively targets the CDK4/6-cyclin D complex to regulate the cell cycle. In recent years, the FDA has approved several CDK4/6 inhibitors, such as palbociclib, ribociclib and abemaciclib, for the treatment of HR-positive breast cancer patients. These inhibitors have shown remarkable results and have significantly improved the options for breast cancer treatment [1,2,3,4]. The efficacy of CDK4/6 inhibitors is now being explored in other diseases, as it has been discovered that CDK4/6 may play a role in pathological conditions beyond cancer. Preliminary studies have already demonstrated a potential renal protective effect of CDK4/6 inhibitors. As research progresses, it is hoped that CDK4/6 inhibitors can be extended to treat a broader range of diseases, providing new therapeutic options for patients.
This review provides an overview of the cell cycle in eukaryotes and emphasizes the essential role of CDK4/6 in regulating this process. The application of CDK4/6 inhibitors in cancer treatment is discussed, with a particular focus on their potential in treating kidney diseases. Additionally, the review explores the various therapeutic strategies and challenges associated with using CDK4/6 inhibitors for treating kidney diseases. A deeper understanding of the mechanism and role of CDK4/6 inhibitors in kidney disease will aid in the development of more effective drugs for the treatment of these conditions.
2. Cell Cycle and Its Regulation
Cell division is a crucial process in biology that is tightly regulated by conserved signaling pathways, ensuring the accurate replication of DNA. In mammals, the cell cycle consists of four phases: G1, S, G2 and M, with an additional phase called G0, where cells exit the cycle and enter a quiescent state [5,6]. In the kidney, most cells are arrested in the G0 phase, but certain highly specialized cells, like neurons and cardiomyocytes, permanently withdraw from the cell cycle. The progression of the cell cycle is controlled by key components such as cyclins, cyclin-dependent kinases (CDKs) and cyclin-dependent kinase inhibitors (CKIs), which interact to form a precise regulatory network [7].
The discovery of CDK, a key regulator of cell cycle progression, dates back to the 1980s when it was first identified in sea urchin eggs [8]. The CDK family comprises serine/threonine kinases and can be categorized into subfamilies based on their evolutionary relationships [9,10]. Some CDKs, such as CDKs 1–6, 11 and 14–18, play a crucial role in regulating cell cycles, while others, including CDKs 7–13, 19 and 20, participate in transcription processes. Despite their structural similarities, each CDK has a specific function that is regulated by context-specific cyclins. The activation of CDKs is essential for the proper functioning of cell cycle proteins, and reciprocally, their interaction with these proteins significantly affects CDK activity. In addition to CDK-cyclins complexes, cell cycle protein-dependent protein kinase inhibitors (CKI) also contribute to the regulation of cell cycle progression. The CDK inhibitors consist of two main families: the INK4 family, which includes p16INK4a (Cdkn2a), p15INK4b (Cdkn2b), p18INK4c (Cdkn2c) and p19INK4d (Cdkn2d), specifically targeting CDK4/6-cyclin D complexes; and the Cip/Kip family members, including p21Cip1 (Cdkn1a), p27Kip1 (Cdkn1b) and p57Kip2 (Cdkn1c), which broadly inhibit CDK-cyclins complexes. These inhibitors play a crucial role in regulating cell cycle progression by disrupting the activity of CDKs and their associated cyclins.
2.1. Classical Regulation of G1/S Phase Transition
According to the classical regulatory pathway of cellular G1 to S phase transition, D-type cyclins are the regulators of CDK4/6, and their expression is increased in the early G1 phase in response to various mitotic stimuli [11]. The regulation of CDK4/6 activity is crucial for the inactivation of the retinoblastoma protein (Rb). When CDK4/6 forms a heterodimer with cyclin D, CDK4 and CDK6 are activated. Subsequently, the dimeric complex phosphorylates Rb1 and other Rb1-like “pocket” proteins, namely P130 and P107, which are respectively referred to as retinoblastoma-like proteins 1 and 2, at multiple sites [12,13,14]. In its hypophosphorylated state, Rb1 binds to the trans-activation domain of the E2F transcription factor family of proteins, thereby inhibiting the transcription of genes necessary for cell cycle progression [15,16].
Upon activation, CDK4/6 can phosphorylate Rb1, thereby relieving the repression of RB1 by E2F transcription factors and facilitating the dissociation of E2F from Rb1 [17,18]. The expression of genes essential for the transition to the S phase relies on E2F, which promotes DNA synthesis by activating various genes and facilitating the transcription of E-type proteins. Moreover, cyclin E plays a crucial role in the late G1 phase by activating CDK2 to form the CDK2-cyclin E complex. This complex then phosphorylates Rb1, thus releasing the inhibition of E2F and promoting cellular entry into the S phase [19,20,21,22,23] (Figure 1). Conversely, during the S-phase, CDK2 forms a complex with cell cycle protein A, while Rb1 undergoes transitional phosphorylation, thereby mediating the transcriptional control of DNA synthesis. Additionally, the CDK1-cyclin B complex regulates the progression through the M phase.
2.2. Non-Classical Regulation of G1/S Phase Transition
The classical understanding of G1/S transition is widely accepted, but the exact role of certain CDKs in this process may be more complex. Experiments using knockout mouse models for CDK4 and CDK6 challenge the traditional view of G1/S transition, which suggests a reliance on human cellular CDKs. Interestingly, certain cell types can enter the S phase without the presence of CDK 4 and CDK 6. For instance, mice can survive with a deficiency in either CDK4 or CDK6 alone. CDK4 deficiency typically leads to stunted growth and compromised reproductive and endocrine function [24,25,26], while CDK6 deficiency primarily affects the hematopoietic system, resulting in the reduced thymus and spleen cell function and a decreased number of peripheral blood erythrocytes [27]. Simultaneous knockout of CDK4 and CDK6, although causing embryonic mouse death, is considered to be a result of erythrocyte defects caused by CDK6 deficiency, leading to severe anemia [27].
However, in vitro experiments have shown that mouse embryonic fibroblasts (MEFs) with mutations in both CDK4 and CDK6 can still undergo cell cycle progression from G1 to S phase when stimulated by serum. This suggests that CDK4/6 is not necessarily required for the stimulation of cell proliferation in quiescent cells. Instead, it is believed that the phosphorylation of RB by an atypical CDK2-cyclin D complex may play a role in this process [27,28]. The discovery of these non-classical pathways challenges the traditional understanding of the G1 to S phase transition in the cell cycle, which has mainly focused on CDK4/6. Non-classical models propose that CDK2 can directly interact with cyclin E and cyclin D, forming a complex without requiring CDK4/6 activation. This complex then facilitates the cell cycle transition from the G1 to S phase [28,29,30] (Figure 2). However, the exact mechanisms underlying this process are still not fully understood.
In order to maintain the accuracy of DNA replication, mammals have developed a sophisticated quality control system comprising four distinct cell cycle checkpoints. These checkpoints play a vital role in ensuring the smooth progress of the cell cycle and regulating it with precision in eukaryotic cells. The transition from the G1 phase to the S phase is carefully controlled by the G1/S checkpoint. This initial checkpoint is responsible for detecting any DNA damage, halting the cell cycle for necessary DNA repair, and safeguarding the integrity of the genome [31]. The G1 phase represents a critical period during which cellular DNA begins replication after passing through the G1/S checkpoint and entering the S phase.
In addition, the S checkpoint follows, which is triggered by either DNA that has escaped the previous checkpoint or damaged DNA, effectively halting the progression of the cell cycle into the S phase [31]. The third checkpoint, referred to as the G2/M checkpoint, ensures the successful completion of mitosis [31]. The fourth checkpoint, known as the intermediate or spindle checkpoint, plays a crucial role in monitoring the cell cycle progression [31]. These checkpoints work together to establish a highly intricate and interconnected network, serving as a rigorous quality control mechanism that continuously scrutinizes and regulates the entire advancement of the cell cycle.
3. CDK Inhibitors Have Emerged as Key Players in the Field of Cancer Therapeutics
CDK inhibitors are pharmacological agents that specifically target abnormal CDK activity in malignant cells. However, the first generation of clinically developed inhibitors faced limitations due to the need for selectivity, as CDKs have structural similarities [32,33]. An example of such an inhibitor is Flavopiridol, which targets multiple CDKs, including CDK1, CDK2, CDK7 and CDK9, along with CDK4/6. Unfortunately, clinical trials with Flavopiridol have shown disappointing results in patients with hematologic malignancies and solid tumors. Furthermore, studies on mouse embryos have indicated that the absence of CDK1 can impair development, suggesting that non-specific CDK inhibitors can have toxic effects on cellular growth and development [34]. CDK7, another target of these inhibitors, is also involved in cell cycle transition, but its exact function is not fully understood. Therefore, broad-spectrum CDK inhibitors with low selectivity often have toxic side effects, making it challenging to find safe and specific inhibitors.
Similarly, the highly sought-after drug roscovitine has proven ineffective in tests involving patients with triple-negative breast cancer, non-small cell lung cancer and other advanced solid tumors due to its limited effectiveness and toxicity [9]. Second-generation pan-CDK inhibitors, such as dinaciclib and SNS-032, have improved selectivity towards a lesser number of CDKs and decreased toxicity profiles. However, they still exhibit lower levels of inhibitory activity against CDK4/6 [33]. Given the underwhelming success of these first- and second-generation inhibitors, it is crucial to discover inhibitors that provide greater selectivity in targeting specific CDKs. Some progress has been made in targeting non-cell cycle CDKs, including CDK9 and CDK12 [35,36]. Nevertheless, the inhibitors that have made the most advancements and are currently in clinical use primarily focus on CDK4/6.
These inhibitors have predominantly been primarily developed through chemical screening and optimization, involving the incorporation of pyrido[2,3-d] pyrimidin-7-one compounds with a 2-amino pyridine side chain at the C2 position [37]. Consequently, CDK4/6 inhibitors such as palbociclib, ribociclib and abemaciclib, with potent efficacy and reduced toxicity, have quickly transitioned from research laboratories to clinical trials. In fact, all three of these drugs have gained FDA approval for the treatment of metastatic breast cancer. Compared to previous CDK inhibitors, these compounds exhibit high selectivity in inhibiting CDK4/6. They have more than a 100-fold affinity for CDK4 and CDK6 relative to other CDKs [38]. All three CDK4/6 inhibitors are administered orally, with palbociclib being the first to demonstrate clinical efficacy and extensively investigated in breast cancer treatment. Ribociclib bears a close structural resemblance to palbociclib, whereas abemaciclib deviates significantly in structure from the first two. Additionally, abemaciclib exhibits lower selectivity compared to palbociclib and ribociclib. However, it offers potential applications in central nervous system disorders such as brain cancer and secondary brain metastases due to its enhanced ability to crosse the blood–brain barrier more effectively [39].
4. CKD4/6 Inhibitors in the Treatment of ER-Positive Breast Cancer
ER-positive luminal breast cancer serves as the ideal model for investigating the effectiveness of CDK4/6 inhibitors. These inhibitors target the reliance of these cancer types on cyclin D1 for the initiation of the G1-to-S-phase transition. Previous clinical trials have established that endocrine anti-estrogen therapy is the cornerstone of systemic treatment for ER-positive breast cancer. However, resistance to endocrine therapy often arises when it fails to adequately control tumor progression. Various mechanisms have been proposed as contributors to endocrine resistance, including loss or mutations of the ER, alterations in ER pathways, dysregulation of cell cycle signaling molecules and activation of escape pathways [40,41]. The addition of CDK4/6 inhibitors to anti-estrogen therapy has been shown to improve progression-free survival (PFS), underscoring their potential as targeted and effective treatments for ER-positive breast cancer [42,43,44,45].
Currently, the FDA has approved the inhibition of CDK4/6 as a novel therapeutic approach for HR-positive breast cancer. Three CDK4/6 inhibitors, when used in combination with endocrine therapies, have shown significant efficacy and have partially overcome the limitations posed by endocrine resistance. Studies suggest that Palbociclib, a CDK4/6 inhibitor, can induce growth arrest in breast cancer cells that have developed acquired resistance to endocrine therapy [46]. The selection of the appropriate patient population for CDK4/6 inhibition in cancer therapy depends on the presence of functional Rb within the CDK4/6 axis. In most cases, tumors that retain functional Rb proteins, which are the primary targets of CDK4/6 inhibitors, during the development of endocrine resistance are suitable for CDK4/6 inhibition [47]. Understanding the exact pharmacological mechanism of action of CDK4/6 inhibitors is crucial to further research in this field.
When considering the differences in Rb-pathway behavior between ER-positive and ER-negative diseases, it is important to note that in ER-positive breast cancer, various factors contribute to the synergistic effect observed. Evidence suggests that ER-positive breast cancers typically maintain an Rb expression signature, which may play a role in this synergy [48]. Furthermore, activation of upstream signaling pathways such as RAS, mitogen-activated protein kinase (MAPK) and mammalian target of rapamycin (mTOR), and nuclear receptors (such as estrogen receptor (ER) in the mammary epithelium, regulates the cell cycle progression by promoting the formation of the CDK4/6-cyclin D complex. This, in turn, leads to uncontrolled cell proliferation [49,50,51,52,53,54]. CDK4/6 serves as a critical regulator of the cell cycle by forming a complex with cyclin D. This complex directly phosphorylates the RB, leading to the release of the transcription factor E2F and facilitating the transcription of genes involved in cell cycle progression [55]. Consequently, this process allows the cell cycle to advance from the G1 phase to the S phase, enabling DNA replication [56]. Furthermore, the precise pharmacological mechanisms of action of CDK4/6 inhibitors have been extensively investigated. Multiple explanations have been proposed regarding how these inhibitors function. The traditional model suggests that CDK4/6 inhibitors primarily block the activity of these kinases, particularly in ER-positive breast cancer (Figure 3a). Additionally, it has been postulated that CDK4/6 inhibitors indirectly inhibit the activity of CDK2, thereby impeding the progression from G1 to S phase [30,57,58] (Figure 3b,c). These findings emphasize the crucial role of CDK4/6 inhibitors in inhibiting the progression of ER-positive breast cancer.
However, despite the benefits of CDK4/6 inhibitors in controlling HR-positive breast cancer, not all patients experience a positive response to these drugs. This is because CDK4/6 inhibitors typically work downstream of endocrine therapy, and resistance can develop to both forms of treatment. This resistance may be caused by various factors, such as mutation or deletion of Rb [28,29]; overexpression of p16 in the presence of functional Rb [59]; amplification of CDK4, CDK6 [60,61,62], or CDK2 in the CDK2-cyclin E axis [28,63]; overexpression of E2F; and other dysregulations in the cell cycle and bypass mechanisms. Consequently, many patients who initially respond to CDK4/6 inhibitors eventually develop acquired drug resistance [64]. However, there is potential for improving treatment outcomes by combining CDK4/6 inhibitors with mTOR and other kinase and checkpoint inhibitors [65,66,67,68,69,70,71]. Hence, investigating and exploring the optimal combination regimen of CDK4/6 inhibitors with other endocrine therapies is crucial for enhancing drug sensitivity.
5. CDK 4/6 Inhibitors in the Treatment of Renal Diseases
In addition to their role in cancer treatment, CDK4/6 inhibitors have demonstrated beneficial effects in non-cancerous diseases as well. The use of CDK inhibitors in treating kidney disease was first reported in 1997, where the inhibition of the cell cycle showed improvement in embolic proliferative glomerulonephritis [72]. Subsequently, the application of CDK inhibitors expanded to other renal diseases, highlighting the antiproliferative effects on podocytes in crescentic glomerulonephritis. However, most of the research in this area has focused on CDK2 inhibitors [73]. As investigations progressed, it became evident that CDK4/6 inhibitors also exhibit promising efficacy in the treatment of renal disease. This review aims to delve into the mechanisms underlying the therapeutic effects of CDK4/6 inhibitors in various renal diseases.
5.1. CDK4/6 Inhibitors and Acute Kidney Injury
Acute kidney injury (AKI) is a clinical condition characterized by a rapid decline in renal function, usually caused by acute ischemia, infection, or drug toxicity-induced death of renal tubular cells [74,75,76,77,78,79]. Under normal physiological conditions, renal tubular epithelial cells (RTECs) remain mostly quiescent. However, they possess a robust regenerative capacity, and their self-proliferation plays a crucial role in kidney injury repair, which involves the cell cycle [22]. Among the RTECs, the proximal S3 segment is particularly vulnerable to injury following acute ischemic or toxic damage. Previous studies have established the significant contribution of these cells in the development of kidney injury diseases [80,81,82,83,84,85,86]. This proliferative response is believed to play a critical role in kidney repair and regeneration [86,87]. Moreover, studies have demonstrated a protective effect of blocking or delaying cell cycle entry during AKI [88]. In fact, AKI often induces DNA damage, which activates ataxia telangiectasia mutated (ATM) or ataxia telangiectasia and Rad3-related (ATR) proteins. These proteins phosphorylate downstream targets, including p53 and checkpoint kinase 2 (CHK2), which belong to the phosphatidylinositol 3-kinase family. Consequently, p21, a CIP/KIP family cell cycle inhibitor, is generated, causing tubular epithelial cells to arrest in the G1 or G2/M phase [89,90,91]. p21 has been observed to be rapidly induced in proximal tubular cells in various experimental models of AKI, and its involvement in the assembly of the CDK4-cyclin D complex and its cytostatic effects have been identified [92,93,94]. Therefore, it is speculated that the involvement of p21/p27 in G1/S transitions serves as an adaptive mechanism. Moreover, it has been demonstrated that the depletion of p21 exacerbates cisplatin-induced acute kidney injury [94]. Several studies have indicated that enhancing p21 expression and reducing CDK2 levels may induce G1 arrest, thereby alleviating AKI by preventing apoptosis in proximal tubular cells [85,88,95,96,97]. However, the clinical utilization of CDK2 inhibitors is limited due to their toxic side effects, which can also impede other CDKs involved in DNA transcription and metabolic processes, resulting in G2/M arrest and S phase arrest, respectively. This can promote apoptosis and fibrosis [83,85,88,98]. The introduction of CDK4/6 inhibitors has shown promising renal protective effects with fewer adverse effects and improved inhibition of G1/S transition [33,99,100]. The presence of functional Rb1 in RTECs is believed to be associated with the ability of hypophosphorylated Rb to block cell cycle progression from G1 to S phase [99,101]. A mouse study investigated the impact of ribociclib on cisplatin-induced nephrotoxicity and suggested that it potentially mitigates renal damage at the early stages through an Rb1-dependent mechanism [99]. Administration of ribociclib resulted in reduced expression of phosphorylated Rb and displayed a protective effect against cisplatin-induced AKI, leading to improved renal function, decreased tubular injury and reduced levels of active/cleaved cysteines [99]. In vivo and in vitro experiments utilizing CDK4/6 inhibitors temporarily halted the traversal of the S phase, with cells re-entering the cell cycle after treatment. These inhibitors have demonstrated beneficial effects, including a reduction in the expression of inflammatory markers such as TNF and MCP-27, a significant decrease in macrophage infiltration, and a reduction in serum creatinine and urea nitrogen levels [102]. Additionally, another study provides strong evidence supporting the inhibition of CDK4/6 in preventing cell-cycle progression. Palbociclib and ribociclib have shown unique and potentially advantageous inhibitory activity on organic cation transporter-2 (OCT-2), in addition to their targeted inhibition of CDK4/6 activity [103] (Figure 4). Furthermore, an animal experiment confirmed that pretreatment with ribociclib in septic mice alleviated sepsis-induced AKI. The study also suggested the involvement of the mTOR/AKT pathway in the renal protective effect of ribociclib [104]. However, a study that examined the clinical features and histopathology associated with CDK4/6 inhibitors reported adverse events in patients with biopsy-confirmed AKI [105]. Clinical trials investigating CDK4/6 inhibitors have also reported elevated serum creatinine levels in some patients, though it remains difficult to determine whether the drug is the actual cause [105,106]. Elevated serum creatinine levels are believed to be the result of reversible inhibition of renal efflux transport proteins rather than acute kidney injury [106].
Further evidence supports the notion that reversible increases in serum creatinine, observed after treatment with abemaciclib, may be attributed to inhibition of renal transporters such as organic cation transporter-2 (OCT-2), multidrug, toxin extrusion-1 (MATE-1) and MATE2-K. Importantly, these increases do not affect the glomerular filtration rate [103,107] (Figure 4). The exact mechanism by which CDK4/6 inhibitors exert their renal protective effects remains unknown. Despite the potential for reversible creatinine elevation, the favorable pharmacological characteristics observed in numerous trials make CDK4/6 inhibitors promising candidates for AKI prevention.
5.2. CDK4/6 Inhibitors and Chronic Kidney Disease
Chronic kidney disease (CKD) is a long-term condition that occurs when there is a structural or functional abnormality in the kidneys lasting for more than three months. It has a detrimental impact on overall health, affecting multiple organ systems, especially the cardiovascular system and mineral–bone metabolism. This leads to various complications that are associated with increased morbidity and mortality, placing a significant socioeconomic burden on society [108,109]. Numerous epidemiological studies have demonstrated a close link between AKI and CKD. Those who survive AKI often go on to develop CKD, indicating a bidirectional relationship between these two conditions [110,111,112,113].
CKD is characterized by the presence of tubulointerstitial fibrosis (TIF), tubular atrophy, and the accumulation of extracellular matrix proteins. While increased cell cycle processes play a role in promoting repair after acute injury, dysregulation of renal tubular epithelial cells during this process can contribute to fibrosis [114,115,116]. Proximal tubular cells (PTCs) are the predominant cell type in the kidney and are crucial sites of injury. Therefore, much research has focused on understanding the transition from AKI to CKD, with particular emphasis on PTCs and resident fibroblasts, which are responsible for producing extracellular matrix proteins [91,114,117,118]. However, the exact mechanistic role of the cell cycle in CKD is still being investigated. Various studies have aimed to unravel the process of maladaptive repair that ultimately leads to the development of progressive fibrotic nephropathy [119,120].
The results of the two-week intervention with palbociclib in the UniNx/AngII and adenosine nephropathy CKD models suggest that palbociclib can effectively delay the progression of CKD. This is achieved by reducing proximal tubular cell death and inhibiting cell cycle progression in the G1/S phase. As a result, renal function is improved, and tubular injury and fibrosis are reduced. These beneficial effects may be mediated through the STAT3/IL-1β pathway, as illustrated in Figure 5 [121] (Figure 5). Previous studies have provided some evidence indicating that CDK4/6 might induce STAT3 activation through mechanisms involving the methyltransferase EZH3 or direct binding to the STAT2 promoter [122,123]. Additionally, it has been demonstrated that STAT3 plays a significant role in renal tubular interstitial fibrosis and becomes activated in response to injured renal tubules [124,125]. However, the exact mechanism by which CDK4/6 triggers STAT3 activation remains unclear. Additionally, further experiments have indicated that the deletion of the cyclin D1 actually worsens chronic kidney injury. This suggests that strategies aimed at preventing chronic kidney disease should prioritize the inhibition of CDK4/6 rather than cyclin D1 [121]. The observation that decreased expression of the CDK4/6 inhibitor p15 is closely associated with reduced eGFR suggests a causal relationship [121]. However, more information regarding the involvement of cell cycle processes in the G1/S transition of chronically injured tubules still needs to be provided.
According to the classical pathway, CDK4/6 binds to the cyclin D1 and phosphorylates Rb, leading to the release of E2F and promoting G1/S progression. It has been observed that palbociclib, a CDK4/6 inhibitor, cannot provide protection against the survival of proximal renal tubules in the absence of Rb. Retarding G1/S cell cycle progression has been shown to have a protective effect. Inhibition of CDK4/6 reduces the number of cells progressing to the S phase, which in turn decreases the population that undergoes G2/M arrest. This shift is associated with a more pro-fibrotic phenotype characterized by increased expression of TGF-β and CTGF [126,127]. In vitro studies have demonstrated that high concentrations (10 μmol/L) of palbociclib significantly impede the proliferative activity of human renal tubular epithelial cells (HK-2 cells). This inhibition is accompanied by an increase in the proportion of cells in the G0/G1 phase and upregulation of p16, p21 and p53 protein levels [127]. Senescent cells can secrete a variety of bioactive molecules known as senescence-associated secretory phenotypes (SASP), which can trigger chronic inflammatory responses and fibrosis. This process is believed to be a key mechanism behind aging-related organ fibrosis [128,129,130]. Mechanistic studies have shown that the increased severity of AKI in mice lacking the Smad7 gene is associated with heightened activation of TGF β/Smad3-p21 signaling [131]. This activation leads to sustained G1 cell cycle arrest and subsequent development of fibrosis. Contrary to previous findings suggesting that CDK4/6 inhibitors protect renal function, palbociclib may actually promote renal fibrosis by inducing cellular senescence in renal cells. While cell cycle arrest can be beneficial for repairing DNA damage or preventing abnormal cell divisions, prolonged arrest can contribute to the acquisition of a pro-fibrotic phenotype if PTCs are unable to re-enter the cell cycle [126]. Contrary to previous findings suggesting that CDK4/6 inhibitors protect renal function, palbociclib may actually promote renal fibrosis by inducing cellular senescence in renal cells. Therefore, the duration of CDK4/6 inhibition is crucial, as transient exposure to cell cycle inhibitors may have a protective effect. However, prolonged exposure could potentially lead to AKI or contribute to the progression of AKI to CKD [105]. Consequently, when using palbociclib in cancer patients with concomitant CKD, careful consideration should be given to the risk of progressive renal fibrosis and declining renal function.
5.3. CDK4/6 Inhibitors and Polycystic Kidney Disease
Dysregulation of CDK4 and CDK6 is highly prevalent in various human diseases, especially cancer. Inhibitors that target these kinases are currently being used to control the growth of tumors [100]. Previous studies have primarily focused on the Rb protein family, including Rb, P107 and P130, as the main targets of CDK4/6 [132]. However, in the case of polycystic kidney disease (PKD), researchers are exploring different substrates that could serve as new targets for CDK4/6 inhibitors. PKD is a common genetic disorder caused by mutations in the PKD1 and PKD2 genes, which encode polycystin-1 (PC-1) and polycystin-2 (PC-2). Autosomal dominant polycystic kidney disease (ADPKD) is the most common form of PKD [133,134,135]. Cysts form due to the excessive proliferation of the epithelial lining in the collecting ducts or renal tubules, which is a distinct pathological feature of ADPKD [134,136]. While the expansion rate of cysts in ADPKD is generally slower compared to tumor proliferation, the signaling and process alterations observed in ADPKD share similarities with those seen in cancer, such as dysregulation of the cell cycle [137]. SMYD2, a novel substrate of CDK4/6, is found to be overexpressed in renal tissues and cell lines derived from mice with ADPK [138]. Both CDK4/6 and SMYD2 dysregulation have also been reported in breast cancer [139]. Previous research suggests that the interaction between CDK4/6 and Rb occurs through phosphorylation, whereas SMYD2 interacts with Rb and methylates it. Moreover, it has been proposed that the binding of SMYD2 to CDK4/6 necessitates the presence of the (S)ET structural domain [140]. These findings hint at an inherent association between CDK4/6 and SMYD2, either directly or indirectly through Rb. The interaction between CDK4/6 and SMYD2 leads to the subsequent phosphorylation and activation of SMYD2, resulting in SMYD2-mediated modifications of histones, including methylation of histone H3 lysine 4 (H3K4) and histone H3 lysine 36 (H3K36), which in turn influences gene transcription [140] (Figure 6). However, there is limited research on the application of CDK4/6 inhibitors for the treatment of PKD. Targeting the overexpression of CDK4/6 and SMYD2 with inhibitors has shown promising results in partially restoring primary cilia in both tumor and cystic cells [140]. These findings further support the therapeutic potential of CDK4/6 inhibitors, either alone or in combination with other inhibitors, for the treatment of PKD.
5.4. CDK4/6 Inhibitors and Nephritis
Since CDK2-cyclin E is known to play a crucial role in the cell cycle progression from the G1 to S phase, most previous studies investigating cell cycle inhibition in nephritis have focused on CDK2 inhibitors [141]. Tethered cell proliferation in Thy1 glomerulonephritis has been found to be associated with increased expression and activity of the CDK2-cyclin A complex [142]. The initiation of mesangial cell proliferation is linked to reduced levels of p27Kip1 during the peak of mesangial cell proliferation [142]. In a study conducted on rats with Thy1 glomerulonephritis, treatment with roscovitine showed beneficial effects on renal function, including increased creatinine clearance, elevated urinary excretion, decreased proteinuria and reduced hematuria [143].
In mice with systemic lupus erythematosus (SLE), the combination of seliciclib and low-dose methylprednisolone, administered after the onset of the disease, has shown increased efficacy in prolonging lifespan and reducing proteinuria and renal damage compared to treatment with either agent alone [144]. As a result, CDK2 inhibitors may hold promise as a practical therapeutic approach for treating diseases characterized by excessive cell proliferation in experimental glomerulonephritis [72]. However, their clinical use has been limited due to toxicity, lack of clinical activity and pharmacokinetic issues. CDK2 inhibitors have not been utilized in clinical studies. Consequently, there is growing interest in the development of inhibitors that target CDK4/6 with fewer side effects.
Treatment with the CDK4/6 inhibitor palbociclib has shown promise in reducing inflammation in the facial skin and lymph nodes of lupus-susceptible MRL-LPR female mice, leading to a decrease in inflammation [145]. However, a study examining the effects of palbociclib on bleomycin-induced pulmonary fibrosis found elevated levels of inflammatory cells in mice after treatment with the inhibitor [146]. Moreover, there have been reports of adverse reactions to palbociclib treatment, such as a patient with a history of psoriatic arthritis developing Stevens–Johnson syndrome-like skin lesions [147]. Additionally, progressive clinical case reports have indicated an association between the use of CDK4/6 inhibitors and Subacute Cutaneous Lupus Erythematosus (SCLE) [148,149,150]. Thus, while CDK4/6 inhibitors show potential for treating lupus and other conditions, their use may be accompanied by certain risks and side effects.
The notable characteristic of these skin lesions is the increased inflammation observed following palbociclib treatment, which contradicts previous findings. Initially, it was hypothesized that CDK4/6 inhibitors, like Palbociclib, would inhibit neutrophil activation [151]. However, the impact of palbociclib on the activation of various inflammatory cell types remains uncertain, and there is limited information regarding the role of CDK4/6 inhibitors in the development of nephritis, with the underlying mechanisms still unclear. Therefore, a more thorough examination is needed to carefully evaluate the benefits and drawbacks of CDK4/6 inhibitors for the treatment of nephritis.
6. Conclusions
CDK4/6 inhibitors have emerged as a promising class of cell cycle-regulating drugs that have demonstrated significant efficacy in the treatment of breast cancer and are currently undergoing preclinical and clinical trials. The therapeutic benefits of CDK4/6 inhibitors extend beyond their use in cancer treatment. In this article, we provide a comprehensive overview of the pivotal role of CDK4/6 in cell cycle processes, review the therapeutic potential of CDK4/6 inhibitors in cancer management, and explore the potential application and the possible side effects of these inhibitors in the treatment of kidney diseases. However, the utilization of CDK4/6 inhibitors for the treatment of renal diseases is currently underexplored, with existing research primarily limited to animal or in vitro cellular models. This highlights the necessity for additional clinical studies to further investigate their potential in this area. The precise mechanisms and specific roles of CDK4/6 inhibitors in treating various renal diseases, including AKI, CKD, PKD and other related nephritis, are still largely unknown. Future research should prioritize investigating the role and mechanism of CDK4/6 inhibitors in treating kidney diseases by utilizing animal models and conducting in vitro cell experiments. Furthermore, additional clinical studies are necessary to validate their effectiveness in various kidney diseases and assess any potential side effects. Combining CDK4/6 inhibitors with other endocrine therapies has demonstrated improved drug sensitivity and enhanced therapeutic outcomes. Therefore, further exploration is needed to identify optimal combination regimens. CDK4/6 inhibitors are anticipated to have broad applications in the treatment of not only various cancers but also kidney diseases. Therefore, further exploration is needed to identify optimal combination regimens. CDK4/6 inhibitors are anticipated to have broad applications in the treatment of not only various cancers but also kidney diseases.
Author Contributions
J.-X.T. and C.-W.Y. formulated and conceived this study. X.-B.L., Z.-C.D., R.Z., J.-X.T. and C.-W.Y. wrote the manuscript. All authors helped to interpret the results and approved the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Natural Science Foundation of Guangdong Province (2023A1515012187), Guangdong Provincial Key Laboratory of Autophagy and Major Chronic Non-communicable Diseases (2022B121203003), Discipline Construction Project of Guangdong Medical University (4SG21229G), Zhanjiang City Science and Technology Development Special Funds Competitive Allocation Project (2021A05083).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ADPKD | Autosomal dominant polycystic kidney disease |
| AKI | Acute kidney injury |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and rad3-related |
| CDK | Cell cycle-dependent kinase |
| CHK2 | Checkpoint kinase 2 |
| CKD | Chronic kidney disease |
| E2F | E2 promoter binding factor |
| ER | Estrogen receptor |
| FDA | Food and Drug Administration |
| H3K4 | Histone h3 lysine 4 |
| IL-1β | Interleukin 1β |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MATE-1 | Multidrug and toxin extrusion-1 |
| MATE2-k | Multidrug and toxin extrusion 2-k |
| MEFs | Mouse embryonic fibroblasts |
| mTOR | Mammalian target of rapamycin |
| NSCLC | Non-small cell lung cancer |
| OCT-2 | Organic cation transporter-2 |
| PC-1 | Polycystin-1 |
| PC-2 | Polycystin-2 |
| PFS | Progression-free survival |
| PKD | Polycystic kidney disease |
| PKD1 | Polycystin-1 |
| PKD2 | Polycystin-2 |
| PTCs | Proximal tubular epithelial cells |
| RAS | An oncogene |
| Rb | Retinoblastoma |
| RTECs | Renal tubular epithelial cells |
| SASP | Senescence-associated secretory phenotypes |
| SCLE | Subacute cutaneous lupus erythematosus |
| SLE | Systemic lupus erythematosus |
| SMYD2 | A histone/lysine methyltransferase |
| STAT2 | Signal transducer and activator of transcription 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TGF-β | Transforming growth factor β |
| TIF | Tubulointerstitial fibrosis |
| TNF | Tumor necrosis factor |
References
- Poratti, M.; Marzaro, G. Third-CDK inhibitors: A review on the synthesis and binding modes of Palbociclib, Ribociclib and Abemaciclib. Eur. J. Med. Chem. 2019, 172, 143–153. [Google Scholar] [CrossRef]
- Beaver, J.A.; Amiri-Kordestani, L.; Charlab, R.; Chen, W.; Palmby, T.; Tilley, A.; Zirkelbach, J.F.; Yu, J.; Liu, Q.; Zhao, L.; et al. FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4760–4766. [Google Scholar] [CrossRef]
- Syed, Y.Y. Ribociclib: First Global Approval. Drugs 2017, 77, 799–807. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef]
- Liu, L.; Michowski, W.; Kolodziejczyk, A.; Sicinski, P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 2019, 21, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Alonso, D.; Malumbres, M. Mammalian cell cycle cyclins. Semin. Cell Dev. Biol. 2020, 107, 28–35. [Google Scholar] [CrossRef]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.; Rosenthal, E.T.; Youngblom, J.; Distel, D.; Hunt, T. Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 1983, 33, 389–396. [Google Scholar] [CrossRef]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Peyressatre, M.; Prével, C.; Pellerano, M.; Morris, M.C. Targeting cyclin-dependent kinases in human cancers: From small molecules to Peptide inhibitors. Cancers 2015, 7, 179–237. [Google Scholar] [CrossRef]
- Sherr, C.J. D-type cyclins. Trends Biochem. Sci. 1995, 20, 187–190. [Google Scholar] [CrossRef]
- Matsushime, H.; Ewen, M.E.; Strom, D.K.; Kato, J.Y.; Hanks, S.K.; Roussel, M.F.; Sherr, C.J. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell 1992, 71, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Matsushime, H.; Hiebert, S.W.; Ewen, M.E.; Sherr, C.J. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993, 7, 331–342. [Google Scholar]
- Meyerson, M.; Harlow, E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol. Cell. Biol. 1994, 14, 2077–2086. [Google Scholar] [PubMed]
- Weintraub, S.J.; Prater, C.A.; Dean, D.C. Retinoblastoma protein switches the E2F site from positive to negative element. Nature 1992, 358, 259–261. [Google Scholar] [CrossRef]
- Hiebert, S.W.; Chellappan, S.P.; Horowitz, J.M.; Nevins, J.R. The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes Dev. 1992, 6, 177–185. [Google Scholar] [CrossRef]
- Goodrich, D.W.; Wang, N.P.; Qian, Y.W.; Lee, E.Y.; Lee, W.H. The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell 1991, 67, 293–302. [Google Scholar] [CrossRef]
- Harbour, J.W.; Luo, R.X.; Dei Santi, A.; Postigo, A.A.; Dean, D.C. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98, 859–869. [Google Scholar] [CrossRef]
- Baker, S.J.; Reddy, E.P. CDK4: A Key Player in the Cell Cycle, Development, and Cancer. Genes Cancer 2012, 3, 658–669. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Moonen, L.; D'Haese, P.C.; Vervaet, B.A. Epithelial Cell Cycle Behaviour in the Injured Kidney. Int. J. Mol. Sci. 2018, 19, 2038. [Google Scholar] [CrossRef]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 2014, 3, e02872. [Google Scholar] [CrossRef] [PubMed]
- Rane, S.G.; Dubus, P.; Mettus, R.V.; Galbreath, E.J.; Boden, G.; Reddy, E.P.; Barbacid, M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat. Genet. 1999, 22, 44–52. [Google Scholar] [CrossRef]
- Tsutsui, T.; Hesabi, B.; Moons, D.S.; Pandolfi, P.P.; Hansel, K.S.; Koff, A.; Kiyokawa, H. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27 (Kip1) activity. Mol. Cell. Biol. 1999, 19, 7011–7019. [Google Scholar] [CrossRef]
- Martín, J.; Hunt, S.L.; Dubus, P.; Sotillo, R.; Néhmé-Pélluard, F.; Magnuson, M.A.; Parlow, A.F.; Malumbres, M.; Ortega, S.; Barbacid, M. Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene 2003, 22, 5261–5269. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef]
- O'Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Watt, A.C.; Goel, S. Cellular mechanisms underlying response and resistance to CDK4/6 inhibitors in the treatment of hormone receptor-positive breast cancer. Breast Cancer Res. BCR 2022, 24, 17. [Google Scholar] [CrossRef]
- Johnson, D.G.; Walker, C.L. Cyclins and cell cycle checkpoints. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 295–312. [Google Scholar] [CrossRef]
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.S.; Zhong, W.Z.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010, 70, 3228–3238. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.F.; Cruz, C.; Greifenberg, A.K.; Dust, S.; Stover, D.G.; Chi, D.; Primack, B.; Cao, S.; Bernhardy, A.J.; Coulson, R.; et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 2016, 17, 2367–2381. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, Q.; Zhang, C.; Huang, Z.; Wang, T.; Wang, X.; Wang, X.; Xu, G.; Liu, Y.; Yang, S.; et al. Discovery of a highly potent, selective and novel CDK9 inhibitor as an anticancer drug candidate. Bioorg. Med. Chem. Lett. 2017, 27, 3231–3237. [Google Scholar] [CrossRef]
- VanderWel, S.N.; Harvey, P.J.; McNamara, D.J.; Repine, J.T.; Keller, P.R.; Quin, J., 3rd; Booth, R.J.; Elliott, W.L.; Dobrusin, E.M.; Fry, D.W.; et al. Pyrido[2,3-d]pyrimidin-7-ones as specific inhibitors of cyclin-dependent kinase 4. J. Med. Chem. 2005, 48, 2371–2387. [Google Scholar] [CrossRef]
- Tadesse, S.; Yu, M.; Mekonnen, L.B.; Lam, F.; Islam, S.; Tomusange, K.; Rahaman, M.H.; Noll, B.; Basnet, S.K.; Teo, T.; et al. Highly Potent, Selective, and Orally Bioavailable 4-Thiazol-N-(pyridin-2-yl)pyrimidin-2-amine Cyclin-Dependent Kinases 4 and 6 Inhibitors as Anticancer Drug Candidates: Design, Synthesis, and Evaluation. J. Med. Chem. 2017, 60, 1892–1915. [Google Scholar] [CrossRef]
- Raub, T.J.; Wishart, G.N.; Kulanthaivel, P.; Staton, B.A.; Ajamie, R.T.; Sawada, G.A.; Gelbert, L.M.; Shannon, H.E.; Sanchez-Martinez, C.; De Dios, A. Brain Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab. Dispos. Biol. Fate Chem. 2015, 43, 1360–1371. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef]
- Kuukasjärvi, T.; Kononen, J.; Helin, H.; Holli, K.; Isola, J. Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1996, 14, 2584–2589. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.L.; Winer, E.P.; et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1541–1547. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, C.; Dean, J.L.; Ertel, A.; Knudsen, K.E.; Aldaz, C.M.; Witkiewicz, A.K.; Clarke, R.; Knudsen, E.S. Therapeutically activating RB: Reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr.-Relat. Cancer 2011, 18, 333–345. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Ertel, A.; Dean, J.L.; Rui, H.; Liu, C.; Witkiewicz, A.K.; Knudsen, K.E.; Knudsen, E.S. RB-pathway disruption in breast cancer: Differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle (Georget. Tex.) 2010, 9, 4153–4163. [Google Scholar] [CrossRef]
- Altucci, L.; Addeo, R.; Cicatiello, L.; Germano, D.; Pacilio, C.; Battista, T.; Cancemi, M.; Petrizzi, V.B.; Bresciani, F.; Weisz, A. Estrogen induces early and timed activation of cyclin-dependent kinases 4, 5, and 6 and increases cyclin messenger ribonucleic acid expression in rat uterus. Endocrinology 1997, 138, 978–984. [Google Scholar] [CrossRef]
- Geum, D.; Sun, W.; Paik, S.K.; Lee, C.C.; Kim, K. Estrogen-induced cyclin D1 and D3 gene expressions during mouse uterine cell proliferation in vivo: Differential induction mechanism of cyclin D1 and D3. Mol. Reprod. Dev. 1997, 46, 450–458. [Google Scholar] [CrossRef]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef]
- Rodgers, J.T.; King, K.Y.; Brett, J.O.; Cromie, M.J.; Charville, G.W.; Maguire, K.K.; Brunson, C.; Mastey, N.; Liu, L.; Tsai, C.R.; et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G (Alert). Nature 2014, 510, 393–396. [Google Scholar] [CrossRef]
- Nair, B.C.; Vadlamudi, R.K. Regulation of hormonal therapy resistance by cell cycle machinery. Gene Ther. Mol. Biol. 2008, 12, 395. [Google Scholar] [PubMed]
- Spring, L.M.; Wander, S.A.; Andre, F.; Moy, B.; Turner, N.C.; Bardia, A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: Past, present, and future. Lancet 2020, 395, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Guiley, K.Z.; Stevenson, J.W.; Lou, K.; Barkovich, K.J.; Kumarasamy, V.; Wijeratne, T.U.; Bunch, K.L.; Tripathi, S.; Knudsen, E.S.; Witkiewicz, A.K.; et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019, 366, eaaw2106. [Google Scholar] [CrossRef] [PubMed]
- Pack, L.R.; Daigh, L.H.; Chung, M.; Meyer, T. Clinical CDK4/6 inhibitors induce selective and immediate dissociation of p21 from cyclin D-CDK4 to inhibit CDK2. Nat. Commun. 2021, 12, 3356. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.L.; McClendon, A.K.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Witkiewicz, A.K.; Knudsen, E.S. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012, 11, 2756–2761. [Google Scholar] [CrossRef]
- Condorelli, R.; Spring, L.; O'Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef]
- Olanich, M.E.; Sun, W.; Hewitt, S.M.; Abdullaev, Z.; Pack, S.D.; Barr, F.G. CDK4 Amplification Reduces Sensitivity to CDK4/6 Inhibition in Fusion-Positive Rhabdomyosarcoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4947–4959. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Etemadmoghadam, D.; Au-Yeung, G.; Wall, M.; Mitchell, C.; Kansara, M.; Loehrer, E.; Batzios, C.; George, J.; Ftouni, S.; Weir, B.A.; et al. Resistance to CDK2 inhibitors is associated with selection of polyploid cells in CCNE1-amplified ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 5960–5971. [Google Scholar] [CrossRef] [PubMed]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr.-Relat. Cancer 2019, 26, R15–R30. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Crafter, C.; Siersbaek, R.; Delpuech, O.; Curwen, J.O.; Carnevalli, L.S.; Staniszewska, A.D.; Polanska, U.M.; Cheraghchi-Bashi, A.; Lawson, M.; et al. Combined Inhibition of mTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-term Growth Inhibition in Estrogen Receptor-positive Breast Cancer. Mol. Cancer Ther. 2018, 17, 908–920. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kanaya, N.; Somlo, G.; Chen, S. Synergistic anti-cancer activity of CDK4/6 inhibitor palbociclib and dual mTOR kinase inhibitor MLN0128 in pRb-expressing ER-negative breast cancer. Breast Cancer Res. Treat. 2019, 174, 615–625. [Google Scholar] [CrossRef]
- Olmez, I.; Brenneman, B.; Xiao, A.; Serbulea, V.; Benamar, M.; Zhang, Y.; Manigat, L.; Abbas, T.; Lee, J.; Nakano, I.; et al. Combined CDK4/6 and mTOR Inhibition Is Synergistic against Glioblastoma via Multiple Mechanisms. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6958–6968. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef]
- Yu, J.; Yan, J.; Guo, Q.; Chi, Z.; Tang, B.; Zheng, B.; Yu, J.; Yin, T.; Cheng, Z.; Wu, X.; et al. Genetic Aberrations in the CDK4 Pathway Are Associated with Innate Resistance to PD-1 Blockade in Chinese Patients with Non-Cutaneous Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6511–6523. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Pippin, J.W.; Qu, Q.; Meijer, L.; Shankland, S.J. Direct in vivo inhibition of the nuclear cell cycle cascade in experimental mesangial proliferative glomerulonephritis with Roscovitine, a novel cyclin-dependent kinase antagonist. J. Clin. Investig. 1997, 100, 2512–2520. [Google Scholar] [CrossRef]
- Nabel, E.G. CDKs and CKIs: Molecular targets for tissue remodelling. Nat. Rev. Drug Discov. 2002, 1, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.U.; Coresh, J.; Devuyst, O.; Johnson, R.J.; Köttgen, A.; Levey, A.S.; Levin, A. Evolving importance of kidney disease: From subspecialty to global health burden. Lancet 2013, 382, 158–169. [Google Scholar] [CrossRef]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Saikumar, P.; Venkatachalam, M.A. Role of apoptosis in hypoxic/ischemic damage in the kidney. Semin. Nephrol. 2003, 23, 511–521. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef]
- Poston, J.T.; Koyner, J.L. Sepsis associated acute kidney injury. BMJ (Clin. Res. Ed.) 2019, 364, k4891. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Bobulescu, I.A. Renal lipid metabolism and lipotoxicity. Curr. Opin. Nephrol. Hypertens. 2010, 19, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Price, P.M.; Megyesi, J.; Saf Irstein, R.L. Cell cycle regulation: Repair and regeneration in acute renal failure. Kidney Int. 2004, 66, 509–514. [Google Scholar] [CrossRef]
- Megyesi, J.; Safirstein, R.L.; Price, P.M. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J. Clin. Investig. 1998, 101, 777–782. [Google Scholar] [CrossRef]
- Price, P.M.; Safirstein, R.L.; Megyesi, J. Protection of renal cells from cisplatin toxicity by cell cycle inhibitors. Am. J. Physiol. Ren. Physiol. 2004, 286, F378–F384. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Czerniak, S.; DiRocco, D.P.; Hasnain, W.; Cheema, R.; Bonventre, J.V. Repair of injured proximal tubule does not involve specialized progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9226–9231. [Google Scholar] [CrossRef]
- Kusaba, T.; Lalli, M.; Kramann, R.; Kobayashi, A.; Humphreys, B.D. Differentiated kidney epithelial cells repair injured proximal tubule. Proc. Natl. Acad. Sci. USA 2014, 111, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Price, P.M.; Safirstein, R.L.; Megyesi, J. The cell cycle and acute kidney injury. Kidney Int. 2009, 76, 604–613. [Google Scholar] [CrossRef]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef]
- Kellum, J.A.; Chawla, L.S. Cell-cycle arrest and acute kidney injury: The light and the dark sides. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2016, 31, 16–22. [Google Scholar] [CrossRef]
- De Chiara, L.; Conte, C.; Antonelli, G.; Lazzeri, E. Tubular Cell Cycle Response upon AKI: Revising Old and New Paradigms to Identify Novel Targets for CKD Prevention. Int. J. Mol. Sci. 2021, 22, 11093. [Google Scholar] [CrossRef]
- Cheng, M.; Olivier, P.; Diehl, J.A.; Fero, M.; Roussel, M.F.; Roberts, J.M.; Sherr, C.J. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999, 18, 1571–1583. [Google Scholar] [CrossRef]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef]
- Nishioka, S.; Nakano, D.; Kitada, K.; Sofue, T.; Ohsaki, H.; Moriwaki, K.; Hara, T.; Ohmori, K.; Kohno, M.; Nishiyama, A. The cyclin-dependent kinase inhibitor p21 is essential for the beneficial effects of renal ischemic preconditioning on renal ischemia/reperfusion injury in mice. Kidney Int. 2014, 85, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Megyesi, J.; Andrade, L.; Vieira, J.M., Jr.; Safirstein, R.L.; Price, P.M. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int. 2001, 60, 2164–2172. [Google Scholar] [CrossRef]
- Yu, F.; Megyesi, J.; Safirstein, R.L.; Price, P.M. Involvement of the CDK2-E2F1 pathway in cisplatin cytotoxicity in vitro and in vivo. Am. J. Physiol. Ren. Physiol. 2007, 293, F52–F59. [Google Scholar] [CrossRef] [PubMed]
- Hodeify, R.; Tarcsafalvi, A.; Megyesi, J.; Safirstein, R.L.; Price, P.M. Cdk2-dependent phosphorylation of p21 regulates the role of Cdk2 in cisplatin cytotoxicity. Am. J. Physiol. Ren. Physiol. 2011, 300, F1171–F1179. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 1770–1783. [Google Scholar] [CrossRef]
- Kim, J.Y.; Jayne, L.A.; Bai, Y.; Feng, M.; Clark, M.A.; Chung, S.; Christman, J.W.; Cianciolo, R.E.; Pabla, N.S. Ribociclib mitigates cisplatin-associated kidney injury through retinoblastoma-1 dependent mechanisms. Biochem. Pharmacol. 2020, 177, 113939. [Google Scholar] [CrossRef]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef]
- DiRocco, D.P.; Bisi, J.; Roberts, P.; Strum, J.; Wong, K.K.; Sharpless, N.; Humphreys, B.D. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F379–F388. [Google Scholar] [CrossRef]
- Pabla, N.; Gibson, A.A.; Buege, M.; Ong, S.S.; Li, L.; Hu, S.; Du, G.; Sprowl, J.A.; Vasilyeva, A.; Janke, L.J.; et al. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc. Natl. Acad. Sci. USA 2015, 112, 5231–5236. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gong, X.; Jia, J.; Li, G. Protective effect and mechanism of Ribociclib on sepsis induced-acute kidney injury. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2020, 32, 204–209. [Google Scholar] [PubMed]
- Gupta, S.; Caza, T.; Herrmann, S.M.; Sakhiya, V.C.; Jhaveri, K.D. Clinicopathologic Features of Acute Kidney Injury Associated With CDK4/6 Inhibitors. Kidney Int. Rep. 2022, 7, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.E.; Mok, K.; Kiely, B.E.; Nguyen, R.; Moylan, E. Association between ribociclib and changes in creatinine in patients with hormone receptor positive metastatic breast cancer. Intern. Med. J. 2019, 49, 1438–1442. [Google Scholar] [CrossRef]
- Chappell, J.C.; Turner, P.K.; Pak, Y.A.; Bacon, J.; Chiang, A.Y.; Royalty, J.; Hall, S.D.; Kulanthaivel, P.; Bonventre, J.V. Abemaciclib Inhibits Renal Tubular Secretion Without Changing Glomerular Filtration Rate. Clin. Pharmacol. Ther. 2019, 105, 1187–1195. [Google Scholar] [CrossRef]
- Levey, A.S.; Eckardt, K.U.; Tsukamoto, Y.; Levin, A.; Coresh, J.; Rossert, J.; De Zeeuw, D.; Hostetter, T.H.; Lameire, N.; Eknoyan, G. Definition and classification of chronic kidney disease: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2005, 67, 2089–2100. [Google Scholar] [CrossRef]
- Wang, A.Y.; Akizawa, T.; Bavanandan, S.; Hamano, T.; Liew, A.; Lu, K.C.; Lumlertgul, D.; Oh, K.H.; Zhao, M.H.; Ka-Shun Fung, S.; et al. 2017 Kidney Disease: Improving Global Outcomes (KDIGO) Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update Implementation: Asia Summit Conference Report. Kidney Int. Rep. 2019, 4, 1523–1537. [Google Scholar] [CrossRef]
- Hsu, R.K.; Hsu, C.Y. The Role of Acute Kidney Injury in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 283–292. [Google Scholar] [CrossRef]
- Zuk, A.; Bonventre, J.V. Recent advances in acute kidney injury and its consequences and impact on chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2019, 28, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. JASN 2015, 26, 1765–1776. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, C. From AKI to CKD: Maladaptive Repair and the Underlying Mechanisms. Int. J. Mol. Sci. 2022, 23, 10880. [Google Scholar] [CrossRef]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef]
- Fiorentino, M.; Grandaliano, G.; Gesualdo, L.; Castellano, G. Acute Kidney Injury to Chronic Kidney Disease Transition. Contrib. Nephrol. 2018, 193, 45–54. [Google Scholar] [PubMed]
- Guzzi, F.; Cirillo, L.; Roperto, R.M.; Romagnani, P.; Lazzeri, E. Molecular Mechanisms of the Acute Kidney Injury to Chronic Kidney Disease Transition: An Updated View. Int. J. Mol. Sci. 2019, 20, 4941. [Google Scholar] [CrossRef]
- Kumar, S. Cellular and molecular pathways of renal repair after acute kidney injury. Kidney Int. 2018, 93, 27–40. [Google Scholar] [CrossRef]
- Zuk, A.; Bonventre, J.V. Acute Kidney Injury. Annu. Rev. Med. 2016, 67, 293–307. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef]
- Yu, S.M.; Bonventre, J.V. Acute kidney injury and maladaptive tubular repair leading to renal fibrosis. Curr. Opin. Nephrol. Hypertens. 2020, 29, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Osaki, Y.; Manolopoulou, M.; Ivanova, A.V.; Vartanian, N.; Mignemi, M.P.; Kern, J.; Chen, J.; Yang, H.; Fogo, A.B.; Zhang, M.; et al. Blocking cell cycle progression through CDK4/6 protects against chronic kidney disease. JCI Insight 2022, 7, e158754. [Google Scholar] [CrossRef]
- Sundar, V.; Vimal, S.; Sai Mithlesh, M.S.; Dutta, A.; Tamizhselvi, R.; Manickam, V. Transcriptional cyclin-dependent kinases as the mediators of inflammation-a review. Gene 2021, 769, 145200. [Google Scholar] [CrossRef]
- Müller, A.; Dickmanns, A.; Resch, C.; Schäkel, K.; Hailfinger, S.; Dobbelstein, M.; Schulze-Osthoff, K.; Kramer, D. The CDK4/6-EZH2 pathway is a potential therapeutic target for psoriasis. J. Clin. Investig. 2020, 130, 5765–5781. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.; Paladugu, P.; Das, B.; He, J.C.; Mallipattu, S.K. Targeting STAT3 signaling in kidney disease. Am. J. Physiol. Ren. Physiol. 2019, 316, F1151–F1161. [Google Scholar] [CrossRef]
- Bienaimé, F.; Muorah, M.; Yammine, L.; Burtin, M.; Nguyen, C.; Baron, W.; Garbay, S.; Viau, A.; Broueilh, M.; Blanc, T.; et al. Stat3 Controls Tubulointerstitial Communication during CKD. J. Am. Soc. Nephrol. JASN 2016, 27, 3690–3705. [Google Scholar] [CrossRef]
- Wynn, T.A. Fibrosis under arrest. Nat. Med. 2010, 16, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.S.; Liang, S.; Li, D.Y.; Wen, J.H.; Tang, J.X.; Liu, H.F. Cell Cycle Dysregulation and Renal Fibrosis. Front. Cell Dev. Biol. 2021, 9, 714320. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef]
- Fu, S.; Tang, Y.; Huang, X.R.; Feng, M.; Xu, A.P.; Lan, H.Y. Smad7 protects against acute kidney injury by rescuing tubular epithelial cells from the G1 cell cycle arrest. Clin. Sci. 2017, 131, 1955–1969. [Google Scholar] [CrossRef] [PubMed]
- Ewen, M.E.; Sluss, H.K.; Sherr, C.J.; Matsushime, H.; Kato, J.; Livingston, D.M. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell 1993, 73, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Ghata, J.; Cowley, B.D., Jr. Polycystic Kidney Disease. Compr. Physiol. 2017, 7, 945–975. [Google Scholar]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.C.; Torres, V.E. Polycystic kidney disease. Annu. Rev. Med. 2009, 60, 321–337. [Google Scholar] [CrossRef]
- Ma, M. Cilia and polycystic kidney disease. Semin. Cell Dev. Biol. 2021, 110, 139–148. [Google Scholar] [CrossRef]
- Seeger-Nukpezah, T.; Geynisman, D.M.; Nikonova, A.S.; Benzing, T.; Golemis, E.A. The hallmarks of cancer: Relevance to the pathogenesis of polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 515–534. [Google Scholar] [CrossRef]
- Li, L.X.; Fan, L.X.; Zhou, J.X.; Grantham, J.J.; Calvet, J.P.; Sage, J.; Li, X. Lysine methyltransferase SMYD2 promotes cyst growth in autosomal dominant polycystic kidney disease. J. Clin. Investig. 2017, 127, 2751–2764. [Google Scholar] [CrossRef]
- Dean, J.L.; Thangavel, C.; McClendon, A.K.; Reed, C.A.; Knudsen, E.S. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 2010, 29, 4018–4032. [Google Scholar] [CrossRef]
- Li, L.X.; Zhou, J.X.; Wang, X.; Zhang, H.; Harris, P.C.; Calvet, J.P.; Li, X. Cross-talk between CDK4/6 and SMYD2 regulates gene transcription, tubulin methylation, and ciliogenesis. Sci. Adv. 2020, 6, eabb3154. [Google Scholar] [CrossRef]
- Gladden, A.B.; Diehl, J.A. Cell cycle progression without cyclin E/CDK2: Breaking down the walls of dogma. Cancer Cell 2003, 4, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Shankland, S.J.; Hugo, C.; Coats, S.R.; Nangaku, M.; Pichler, R.H.; Gordon, K.L.; Pippin, J.; Roberts, J.M.; Couser, W.G.; Johnson, R.J. Changes in cell-cycle protein expression during experimental mesangial proliferative glomerulonephritis. Kidney Int. 1996, 50, 1230–1239. [Google Scholar] [CrossRef]
- Chiara, M.; Menegatti, E.; Di Simone, D.; Davit, A.; Bellis, D.; Sferch, D.; De Rosa, G.; Giachino, O.; Sena, L.M.; Roccatello, D. Mycophenolate mofetil and roscovitine decrease cyclin expression and increase p27 (kip1) expression in anti Thy1 mesangial proliferative nephritis. Clin. Exp. Immunol. 2005, 139, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Casiraghi, F.; Conti, S.; Corna, D.; Rottoli, D.; Cavinato, R.A.; Remuzzi, G.; Benigni, A. Cyclin-dependent kinase inhibition limits glomerulonephritis and extends lifespan of mice with systemic lupus. Arthritis Rheum. 2007, 56, 1629–1637. [Google Scholar] [CrossRef]
- Zhang, Y.; Jin, B.; Miller, H.D.; Ge, D.; Zhang, X.; You, Z. CDK4/6 inhibitor palbociclib reduces inflammation in lupus-prone mice. Am. J. Clin. Exp. Urol. 2021, 9, 32–43. [Google Scholar] [PubMed]
- Birnhuber, A.; Egemnazarov, B.; Biasin, V.; Bonyadi Rad, E.; Wygrecka, M.; Olschewski, H.; Kwapiszewska, G.; Marsh, L.M. CDK4/6 inhibition enhances pulmonary inflammatory infiltration in bleomycin-induced lung fibrosis. Respir. Res. 2020, 21, 167. [Google Scholar] [CrossRef]
- Karagounis, T.; Vallurupalli, M.; Nathan, N.; Nazarian, R.; Vedak, P.; Spring, L.; Chen, S.T. Stevens-Johnson syndrome-like eruption from palbociclib in a patient with metastatic breast cancer. JAAD Case Rep. 2018, 4, 452–454. [Google Scholar] [CrossRef]
- Pinard, J.; Patel, M.; Granter, S.R.; Vleugels, R.A.; Merola, J.F. Subacute Cutaneous Lupus Erythematosus Induced by Palbociclib. J. Cutan. Med. Surg. 2018, 22, 341–343. [Google Scholar] [CrossRef]
- Freedman, J.B.; Herskovitz, I.; Maderal, A.D. Chronic cutaneous lupus erythematosus (discoid lupus) induced by palbociclib. Int. J. Dermatol. 2020, 59, e216–e218. [Google Scholar] [CrossRef]
- Russell-Goldman, E.; Nazarian, R.M. Subacute cutaneous lupus erythematosus with positive anti-Ro antibodies following palbociclib and letrozole treatment: A case report and literature review. J. Cutan. Pathol. 2020, 47, 654–658. [Google Scholar] [CrossRef]
- Amulic, B.; Knackstedt, S.L.; Abu Abed, U.; Deigendesch, N.; Harbort, C.J.; Caffrey, B.E.; Brinkmann, V.; Heppner, F.L.; Hinds, P.W.; Zychlinsky, A. Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev. Cell 2017, 43, 449–462. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Model diagram of CDK4/6 and G1/S phase transition. Quiescent cells in the G0 or early G1 phase exhibit hypophosphorylated retinoblastoma protein (RB1), which inhibits the transcriptional activity of the E2F. The INK4 protein p16 acts as a suppressor of CDK4/6 activation. The mitogenic and estrogen receptor signaling upregulates the transcription of cyclin D. The cyclin D forms a complex with CDK4/6 to phosphorylate RB, partially activating the E2F-family proteins and leading to the transcription of cyclin A and cyclin E, as well as CDK2. RB phosphorylation also induces chromatin remodeling that promotes transcription. CDK4/6–cyclin D complexes sequester CIP/KIP proteins, diminishing their inhibitory effect on CDK2 and lowering the threshold for CDK2 activation by cyclin E. With increasing cyclin E levels, cyclin E binds with CDK2 to hyperphosphorylate RB, establishing a positive feedback loop through E2F. This loop releases and fully activates E2F, driving the cell from G1 to S phase.
Figure 1.
Model diagram of CDK4/6 and G1/S phase transition. Quiescent cells in the G0 or early G1 phase exhibit hypophosphorylated retinoblastoma protein (RB1), which inhibits the transcriptional activity of the E2F. The INK4 protein p16 acts as a suppressor of CDK4/6 activation. The mitogenic and estrogen receptor signaling upregulates the transcription of cyclin D. The cyclin D forms a complex with CDK4/6 to phosphorylate RB, partially activating the E2F-family proteins and leading to the transcription of cyclin A and cyclin E, as well as CDK2. RB phosphorylation also induces chromatin remodeling that promotes transcription. CDK4/6–cyclin D complexes sequester CIP/KIP proteins, diminishing their inhibitory effect on CDK2 and lowering the threshold for CDK2 activation by cyclin E. With increasing cyclin E levels, cyclin E binds with CDK2 to hyperphosphorylate RB, establishing a positive feedback loop through E2F. This loop releases and fully activates E2F, driving the cell from G1 to S phase.

Figure 2.
Model diagram of non-classical CDK4/6 and G1/S transition. CDK2 is active during early G1 through direct formation of complexes with cyclins E and potentially cyclin D. Both CDK4/6 and CDK2 phosphorylate RB and drive the G1-S phase transition.
Figure 2.
Model diagram of non-classical CDK4/6 and G1/S transition. CDK2 is active during early G1 through direct formation of complexes with cyclins E and potentially cyclin D. Both CDK4/6 and CDK2 phosphorylate RB and drive the G1-S phase transition.

Figure 3.
Model diagram of G1/S cyclin-dependent kinase inhibition by CDK4/6 inhibitors. a. CDK4/6 inhibitors inhibit active CDK4/6-cyclin D-p21/p27 allosteric enzymes and block RB phosphorylation of CDK4/6; b. CDK4/6 inhibitors bind to monomeric CDK4/6 and prevent the formation of CDK4/6-cyclin D-p21/p27 trimer. Free p21 then binds to and inhibits the cyclin E/CDK2, preventing phosphorylation of RB; c. CDK4/6 inhibitors directly inhibit the catalytic activity of CDK4/6, which then displaces p21, freeing it to inhibit cyclin E/CDK2 further.
Figure 3.
Model diagram of G1/S cyclin-dependent kinase inhibition by CDK4/6 inhibitors. a. CDK4/6 inhibitors inhibit active CDK4/6-cyclin D-p21/p27 allosteric enzymes and block RB phosphorylation of CDK4/6; b. CDK4/6 inhibitors bind to monomeric CDK4/6 and prevent the formation of CDK4/6-cyclin D-p21/p27 trimer. Free p21 then binds to and inhibits the cyclin E/CDK2, preventing phosphorylation of RB; c. CDK4/6 inhibitors directly inhibit the catalytic activity of CDK4/6, which then displaces p21, freeing it to inhibit cyclin E/CDK2 further.
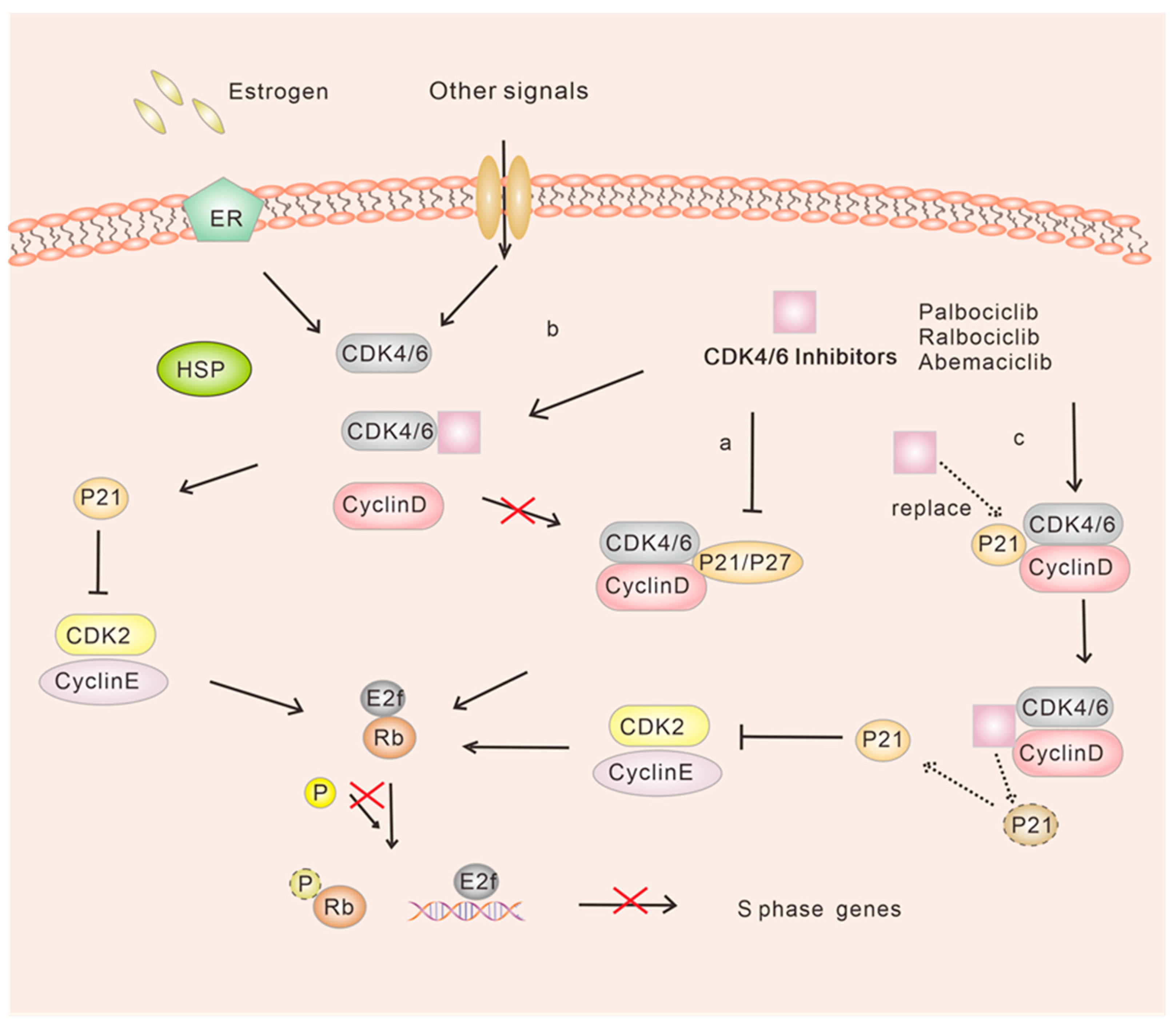
Figure 4.
Model diagram of the effect of CDK4/6 inhibitors on AKI. In acute kidney injury, CDK4/6 inhibitors may delay G1 entry to the S phase through different pathways by inhibiting CDK4/6 activity and other substrates, such as OCT-2 and MATE.
Figure 4.
Model diagram of the effect of CDK4/6 inhibitors on AKI. In acute kidney injury, CDK4/6 inhibitors may delay G1 entry to the S phase through different pathways by inhibiting CDK4/6 activity and other substrates, such as OCT-2 and MATE.
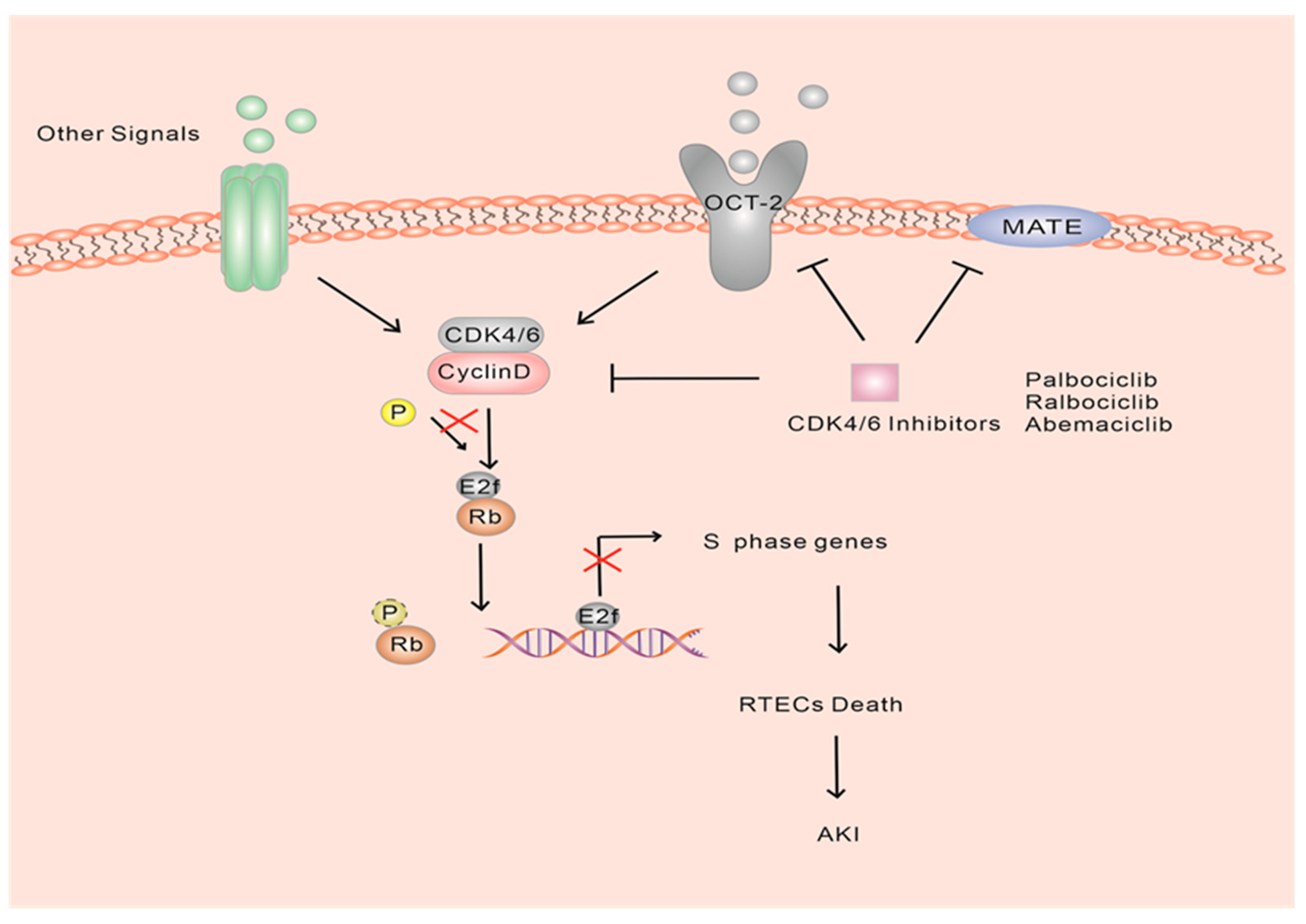
Figure 5.
Model diagram of the effect of CDK4/6 inhibitors on CKD. In chronic kidney injury, CDK4/6 may induce STAT3 activation either through the methyltransferase EZH3 or by directly binding to the STAT2 and possibly block cell cycle progression in G1/S phase through a pathway involving STAT3/IL-1β. But, the precise mechanism by which CDK4/6 initiates STAT3 activation remains unclear.
Figure 5.
Model diagram of the effect of CDK4/6 inhibitors on CKD. In chronic kidney injury, CDK4/6 may induce STAT3 activation either through the methyltransferase EZH3 or by directly binding to the STAT2 and possibly block cell cycle progression in G1/S phase through a pathway involving STAT3/IL-1β. But, the precise mechanism by which CDK4/6 initiates STAT3 activation remains unclear.

Figure 6.
Model diagram of the effect of CDK4/6 inhibitors on PDK. SMYD2 is the new substrate of CDK4/6, which exhibits overexpression in renal tissues and cell lines derived from mice with autosomal dominant PKD. The interaction between SMYD2 and CDK4/6 may be dependent on the presence of the (S)ET structural domain. CDK4/6 interacts with SMYD2, resulting in the phosphorylation and activation of SMYD2. This activation leads to SMYD2-mediated methylation of histones, including histone H3 lysine 4 (H3K4) and histone H3 lysine 36 (H3K36), thereby influencing S gene transcription. However, much research is still needed to investigate and confirm the mechanism of action of CDK4/6 inhibitors in the treatment of PDK.
Figure 6.
Model diagram of the effect of CDK4/6 inhibitors on PDK. SMYD2 is the new substrate of CDK4/6, which exhibits overexpression in renal tissues and cell lines derived from mice with autosomal dominant PKD. The interaction between SMYD2 and CDK4/6 may be dependent on the presence of the (S)ET structural domain. CDK4/6 interacts with SMYD2, resulting in the phosphorylation and activation of SMYD2. This activation leads to SMYD2-mediated methylation of histones, including histone H3 lysine 4 (H3K4) and histone H3 lysine 36 (H3K36), thereby influencing S gene transcription. However, much research is still needed to investigate and confirm the mechanism of action of CDK4/6 inhibitors in the treatment of PDK.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liang, X.-B.; Dai, Z.-C.; Zou, R.; Tang, J.-X.; Yao, C.-W. The Therapeutic Potential of CDK4/6 Inhibitors, Novel Cancer Drugs, in Kidney Diseases. Int. J. Mol. Sci. 2023, 24, 13558. https://doi.org/10.3390/ijms241713558
AMA Style
Liang X-B, Dai Z-C, Zou R, Tang J-X, Yao C-W. The Therapeutic Potential of CDK4/6 Inhibitors, Novel Cancer Drugs, in Kidney Diseases. International Journal of Molecular Sciences. 2023; 24(17):13558. https://doi.org/10.3390/ijms241713558
Chicago/Turabian StyleLiang, Xuan-Bing, Zhi-Cheng Dai, Rong Zou, Ji-Xin Tang, and Cui-Wei Yao. 2023. "The Therapeutic Potential of CDK4/6 Inhibitors, Novel Cancer Drugs, in Kidney Diseases" International Journal of Molecular Sciences 24, no. 17: 13558. https://doi.org/10.3390/ijms241713558
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.