
Shake It Up Baby Now: The Changing Focus on TWIST1 and Epithelial to Mesenchymal Transition in Cancer and Other Diseases

Abstract
:1. Introduction
2. Beyond Cancers: Additional Pathologies Associated with TWIST1
3. TWIST1, EMT, and Cancer Stem Cell Phenotypes
4. TWIST1, EMT, and Drug Response in Cancer
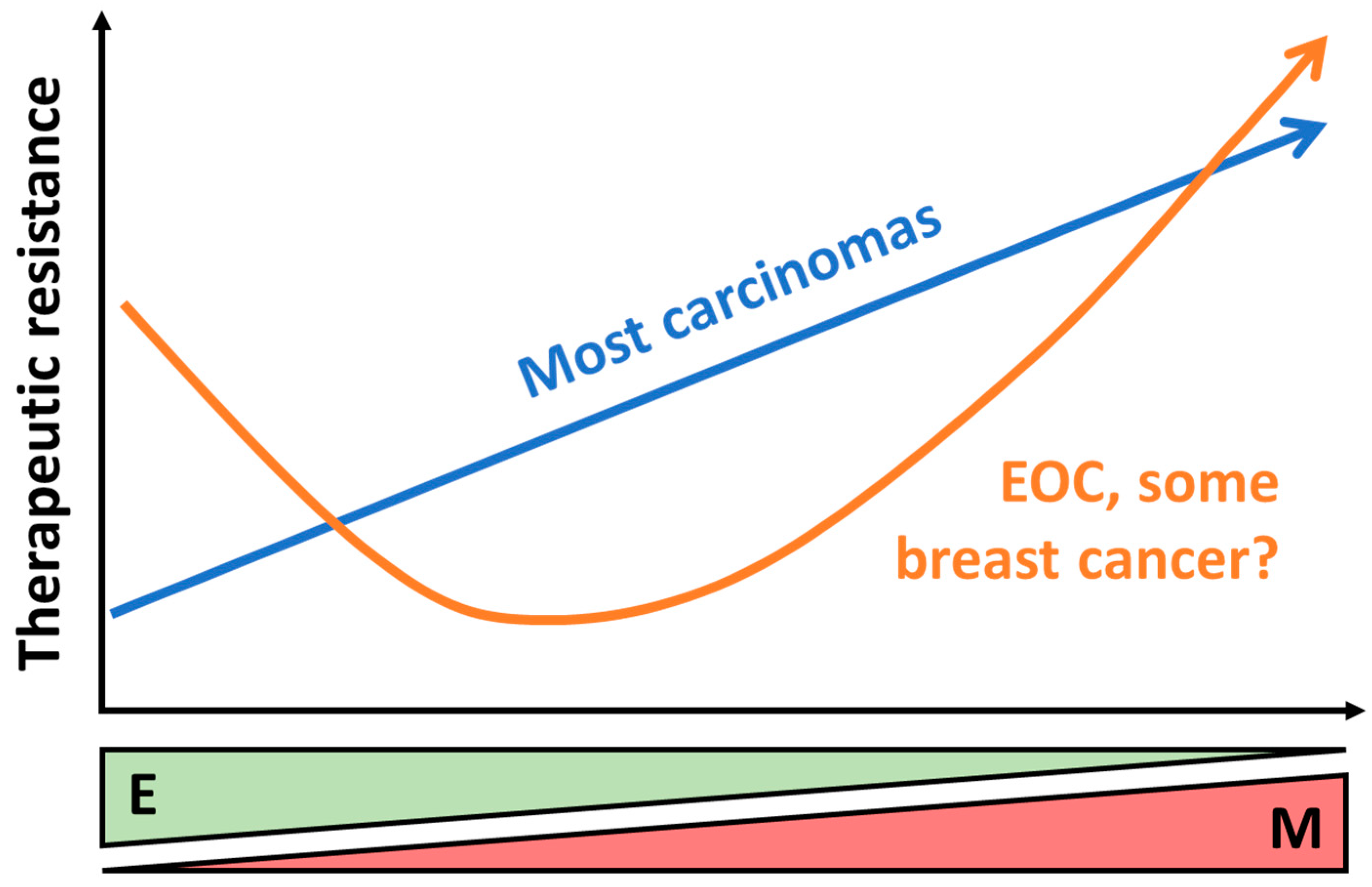
4.1. Overview
4.2. EMT and Cytotoxic Drugs
4.3. Regulation of EMT Protein Expression
4.4. EMT and Multi-Drug Resistance
4.5. EMT and Response to Anti-Angiogenic Therapy
4.6. EMT and Response to Growth Factor Inhibition
4.7. EMT and Ferroptosis
4.8. EMT and Drug Sensitivity
4.9. Intersection of CSC and Drug-Resistant Phenotypes

{kind=link}
{kind=link}
{kind=link}
| Drug | Cancers | Genes/Pathways Impacted | References |
|---|---|---|---|
| Cisplatin | Ovarian *, mesothelioma, gastric, NSCLC, breast | Akt, miRNA, Rab31, eIF5A2, CDK1 | [85,86,88,89,90,93,97,98,103] |
| Oxaliplatin | CRC | Akt, MFAP2, HK2, FHL3, Slug | [87,94,104,107] |
| 5-Fluorouracil | CRC | Akt | [87] |
| Paclitaxel | Breast, ovarian | Akt2, CDK1 | [86,91,103] |
| Resveratrol | Ovarian | Akt, hedgehog | [90] |
| Pterostilbene | Ovarian | STAT3 | [91] |
| Doxorubicin | HCC | eIF5A2 | [96] |
| Decitabine | AML | OGT | [105] |
| Pazopanib | Renal | Vascular mimicry, androgen receptor | [112] |
| Apatinib | Gastric | VEGFR2, COL1A2 | [114] |
| Erlotinib | NSCLC | EGFR, Akt, MAPK | [115] |
| Gefitinib | NSCLC | EGFR, Akt, non-coding RNA, microRNA | [116,117] |
| Erastin | Gastric | CPEB1, ferroptosis | [118] |
| Olaparib | Ovarian * | HR | [119] |
| Nu-7441 | Ovarian * | HR | [119] |
| ABT-888 | Breast * | HR | [121] |
| Ophiobolin A | Breast * | EMT | [122] |
| Milademetan | Intimal sarcoma * | MDM2/p53 | [123] |
| Irenotecan | CRC | CSC | [124] |
| Eribulin mesylate | Breast | Circulating tumor cells, CSCs | [125] |
| Multiple | HCC, CRC, gastric | MDR1 | [108,109,110] |
5. TWIST1 and EMT as Therapeutic Targets
5.1. Overview
5.2. Drugs Targeting EMT
5.3. Gene Knockdown and Nanoparticle Approaches
5.4. Existing Drugs with Effects against EMT
| Drug/Approach | Cancers | Pathways Impacted | References |
|---|---|---|---|
| COM33 | Ovarian | Migration, invasion, resistance | [130] |
| Nisin | Liver | Frizzled | [131] |
| α-Linolenic acid | Breast | TWIST1 turnover | [130] |
| Ophiobin A | Breast | CSCs | [122] |
| Harmine and derivatives | Breast, NSCLC (ulcerative colitis) | Invasion, TWIST1 turnover, DNA intercalation, CSCs | [53,133,134,135,136,137,138] |
| Gene knockdown | Ovarian, breast, melanoma | DCLK1, TWIST1 | [143,144,145,146,147,148] |
| BZ6 nanoparticles | Breast, pancreatic | Migration, TWIST1 | [149] |
| Pirfenidone | Breast | TGFβ, Smad2 | [150] |
| Metformin | Endometrial | TGFβ, ERK/MAPK | [151] |
| Antrodia salmonea | Head and neck | HIF1α, glycolysis | [152] |
| Formoterol | Lung | Reactive oxygen species (ROS) | [153] |
| Aprepitant | Cervical, glioblastoma, breast, prostate | ROS, NFκB | [154,155,156,157] |
| Paclitaxel nanoparticles | Breast | EMT and CSCs, mammospheres | [158] |
| Entinostat | Gastric | Histone deacetylase, SALL4 | [159] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AML | acute myeloid leukemia |
| AR | androgen receptor |
| AS | antrodia salmonea |
| CAFs | cancer-associated fibroblasts |
| CPEB1 | cytoplasmic polyadenylation element binding protein 1 |
| CRC | colorectal cancer |
| CSCs | cancer stem cells |
| DCLK1 | doublecortin-like kinase 1 |
| EndMT | endothelial to mesenchymal transition |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial-to-mesenchymal transition |
| HCC | hepatocellular carcinoma |
| HK2 | hexokinase-2 |
| LPA | lysophosphatidic acid |
| LSCs | leukemia stem cells |
| MFAP2 | microfibrillar-associated protein 2 |
| MMPs | matrix metallopeptidases |
| NSCLC | non-small cell lung carcinoma |
| OGT | O-GlcNAc transferase |
| PDAC | pancreatic ductal adenocarcinoma |
| PLK1 | polo-like kinase 1 |
| PROTACs | proteolysis-targeting chimeras |
| TGFβ | transforming growth factor beta |
| TNBC | triple-negative breast cancer |
References
- Shook, D.; Keller, R. Mechanisms, Mechanics and Function of Epithelial–Mesenchymal Transitions in Early Development. Mech. Dev. 2003, 120, 1351–1383. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Abe, H.; Mochizuki, S.; Shimoda, M.; Okada, Y. SOX4, an Epithelial–Mesenchymal Transition Inducer, Transactivates ADAM28 Gene Expression and Co-localizes with ADAM28 at the Invasive Front of Human Breast and Lung Carcinomas. Pathol. Int. 2018, 68, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Platt, J.B. Ontogenetische Differenzirung Des Ektoderms in Necturus. Arch. für Mikrosk. Anat. 1894, 43, 911–966. [Google Scholar] [CrossRef]
- Lim, J.; Thiery, J.P. Epithelial-Mesenchymal Transitions: Insights from Development. Development 2012, 139, 3471–3486. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.T.; Liu, Y.; Ayyanathan, K.; Benner, C.; Jiang, Y.; Prokop, J.W.; Paz, H.; Wang, D.; Li, H.R.; Fu, X.D.; et al. An Evolutionarily Conserved DNA Architecture Determines Target Specificity of the TWIST Family BHLH Transcription Factors. Genes. Dev. 2015, 29, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P. Maternal-Zygotic Gene Interactions during Formation of the Dorsoventral Pattern in Drosophila Embryos. Genetics 1983, 105, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.S.; Glackin, C.A.; Winters, K.A.; Gazit, D.; Kahn, A.J.; Murray, E.J. Expression of Helix-Loop-Helix Regulatory Genes during Differentiation of Mouse Osteoblastic Cells. J. Bone Miner. Res. 1992, 7, 1131–1138. [Google Scholar] [CrossRef]
- Lee, M.; Lowe, G.N.; Strong, D.D.; Wergedal, J.E.; Glackin, C.A. TWIST, a Basic Helix-loop-helix Transcription Factor, Can Regulate the Human Osteogenic Lineage. J. Cell. Biochem. 1999, 75, 566–577. [Google Scholar] [CrossRef]
- Chen, Z.F.; Behringer, R.R. Twist Is Required in Head Mesenchyme for Cranial Neural Tube Morphogenesis. Genes. Dev. 1995, 9, 686–699. [Google Scholar] [CrossRef]
- Bildsoe, H.; Loebel, D.A.; Jones, V.J.; Chen, Y.T.; Behringer, R.R.; Tam, P.P. Requirement for Twist1 in Frontonasal and Skull Vault Development in the Mouse Embryo. Dev. Biol. 2009, 331, 176–188. [Google Scholar] [CrossRef]
- Zhang, Z.; Sui, P.; Dong, A.; Hassell, J.; Cserjesi, P.; Chen, Y.T.; Behringer, R.R.; Sun, X. Preaxial Polydactyly: Interactions among ETV, TWIST1 and HAND2 Control Anterior-Posterior Patterning of the Limb. Development 2010, 137, 3417–3426. [Google Scholar] [CrossRef] [PubMed]
- Krawchuk, D.; Weiner, S.J.; Chen, Y.T.; Lu, B.C.; Costantini, F.; Behringer, R.R.; Laufer, E. Twist1 Activity Thresholds Define Multiple Functions in Limb Development. Dev. Biol. 2010, 347, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.D.; Paznekas, W.A.; Green, E.D.; Chiang, L.C.; Ma, N.; de Luna, R.I.O.; Delgado, C.G.; Gonzalez-Ramos, M.; Kline, A.D.; Jabs, E.W. Mutations in TWIST, a Basic Helix-Loop-Helix Transcription Factor, in Saethre-Chotzen Syndrome. Nat. Genet. 1997, 15, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Horsley, S.W.; Moloney, D.M.; Oldridge, M.; Twigg, S.R.; Walsh, S.; Barrow, M.; Njolstad, P.R.; Kunz, J.; Ashworth, G.J.; et al. A Comprehensive Screen for TWIST Mutations in Patients with Craniosynostosis Identifies a New Microdeletion Syndrome of Chromosome Band 7p21. 1. Am. J. Hum. Genet. 1998, 63, 1282–1293. [Google Scholar] [CrossRef]
- Cai, J.; Goodman, B.K.; Patel, A.S.; Mulliken, J.B.; Maldergem, L.V.; Hoganson, G.E.; Paznekas, W.A.; Ben-Neriah, Z.; Sheffer, R.; Cunningham, M.L.; et al. Increased Risk for Developmental Delay in Saethre-Chotzen Syndrome Is Associated with TWIST Deletions: An Improved Strategy for TWIST Mutation Screening. Hum. Genet. 2003, 114, 68–76. [Google Scholar] [CrossRef]
- Song, Y.; Tian, T.; Fu, X.; Wang, W.; Li, S.; Shi, T.; Suo, A.; Ruan, Z.; Guo, H.; Yao, Y. GATA6 Is Overexpressed in Breast Cancer and Promotes Breast Cancer Cell Epithelial–Mesenchymal Transition by Upregulating Slug Expression. Exp. Mol. Pathol. 2015, 99, 617–627. [Google Scholar] [CrossRef]
- Bates, R.C. Colorectal Cancer Progression: Integrin Alphavbeta6 and the Epithelial-Mesenchymal Transition (EMT). Cell Cycle 2005, 4, 1350–1352. [Google Scholar] [CrossRef]
- Domínguez, D.; Montserrat-Sentís, B.; Virgós-Soler, A.; Guaita, S.; Grueso, J.; Porta, M.; Puig, I.; Baulida, J.; Francí, C.; Herreros, A.G. de Phosphorylation Regulates the Subcellular Location and Activity of the Snail Transcriptional Repressor. Mol. Cell. Biol. 2003, 23, 5078–5089. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable β-Catenin Expression in Colorectal Cancers Indicates Tumor Progression Driven by the Tumor Environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef]
- Vesuna, F.; van Diest, P.; Chen, J.H.; Raman, V. Twist Is a Transcriptional Repressor of E-Cadherin Gene Expression in Breast Cancer. Biochem. Biophys. Res. Commun. 2008, 367, 235–241. [Google Scholar] [CrossRef]
- Meng, J.; Chen, S.; Han, J.-X.; Qian, B.; Wang, X.-R.; Zhong, W.-L.; Qin, Y.; Zhang, H.; Gao, W.-F.; Lei, Y.-Y.; et al. Twist1 Regulates Vimentin through Cul2 Circular RNA to Promote EMT in Hepatocellular Carcinoma. Cancer Res. 2018, 78, 4150–4162. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Hu, W.; Fu, W.; Dai, L.; Jiang, Z.; Zhong, S.; Deng, B.; Zhao, J. HGF/MET Regulated Epithelial-Mesenchymal Transitions And Metastasis By FOSL2 In Non-Small Cell Lung Cancer. OncoTargets Ther. 2019, 12, 9227–9237. [Google Scholar] [CrossRef] [PubMed]
- Lv, N.; Shan, Z.; Gao, Y.; Guan, H.; Fan, C.; Wang, H.; Teng, W. Twist1 Regulates the Epithelial–Mesenchymal Transition via the NF-ΚB Pathway in Papillary Thyroid Carcinoma. Endocrine 2016, 51, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.C.; Baxter, E.W.; Loadman, P.M.; Hull, M.A.; Knowles, M.A. FGFR1-Induced Epithelial to Mesenchymal Transition through MAPK/PLCγ/COX-2-Mediated Mechanisms. PLoS ONE 2012, 7, e38972. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.H.; Xu, G.L.; Jia, W.D.; Li, J.S.; Ma, J.L.; Ren, W.H.; Ge, Y.S.; Yu, J.H.; Liu, W.B.; Wang, W. Activation of STAT3 Signal Pathway Correlates with Twist and E-Cadherin Expression in Hepatocellular Carcinoma and Their Clinical Significance. J. Surg. Res. 2012, 174, 120–129. [Google Scholar] [CrossRef]
- Huang, C.; Yang, G.; Jiang, T.; Zhu, G.; Li, H.; Qiu, Z. The Effects and Mechanisms of Blockage of STAT3 Signaling Pathway on IL-6 Inducing EMT in Human Pancreatic Cancer Cells in Vitro. Neoplasma 2011, 58, 396–405. [Google Scholar] [CrossRef]
- Cheng, G.Z.; Zhang, W.Z.; Sun, M.; Wang, Q.; Coppola, D.; Mansour, M.; Xu, L.M.; Costanzo, C.; Cheng, J.Q.; Wang, L.H. Twist Is Transcriptionally Induced by Activation of STAT3 and Mediates STAT3 Oncogenic Function. J. Biol. Chem. 2008, 283, 14665–14673. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 Induces an Epithelial-Mesenchymal Transition Phenotype in Human Breast Cancer Cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef]
- Hsu, K.W.; Hsieh, R.H.; Huang, K.H.; Li, A.F.-Y.; Chi, C.W.; Wang, T.Y.; Tseng, M.J.; Wu, K.J.; Yeh, T.S. Activation of the Notch1/STAT3/Twist Signaling Axis Promotes Gastric Cancer Progression. Carcinogenesis 2012, 33, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct Regulation of TWIST by HIF-1alpha Promotes Metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kendall, S.E.; Raices, R.; Finlay, J.; Covarrubias, M.; Liu, Z.; Lowe, G.; Lin, Y.H.; Teh, Y.H.; Leigh, V.; et al. TWIST1 Associates with NF-KappaB Subunit RELA via Carboxyl-Terminal WR Domain to Promote Cell Autonomous Invasion through IL8 Production. BMC Biol. 2012, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Mironchik, Y.; Winnard, P.T., Jr.; Vesuna, F.; Kato, Y.; Wildes, F.; Pathak, A.P.; Kominsky, S.; Artemov, D.; Bhujwalla, Z.; Diest, P.V.; et al. Twist Overexpression Induces in Vivo Angiogenesis and Correlates with Chromosomal Instability in Breast Cancer. Cancer Res. 2005, 65, 10801–10809. [Google Scholar] [CrossRef]
- Niu, R.F.; Zhang, L.; Xi, G.M.; Wei, X.Y.; Yang, Y.; Shi, Y.R.; Hao, X.S. Up-Regulation of Twist Induces Angiogenesis and Correlates with Metastasis in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2007, 26, 385–394. [Google Scholar] [PubMed]
- Low-Marchelli, J.M.; Ardi, V.C.; Vizcarra, E.A.; van Rooijen, N.; Quigley, J.P.; Yang, J. Twist1 Induces CCL2 and Recruits Macrophages to Promote Angiogenesis. Cancer Res. 2013, 73, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Ramundo, V.; Zanirato, G.; Palazzo, M.L.; Riganti, C.; Aldieri, E. APE-1/Ref-1 Inhibition Blocks Malignant Pleural Mesothelioma Cell Proliferation and Migration: Crosstalk between Oxidative Stress and Epithelial Mesenchymal Transition (EMT) in Driving Carcinogenesis and Metastasis. Int. J. Mol. Sci. 2023, 24, 12570. [Google Scholar] [CrossRef]
- Fan, Q.; Qiu, M.-T.; Zhu, Z.; Zhou, J.-H.; Chen, L.; Zhou, Y.; Gu, W.; Wang, L.-H.; Li, Z.-N.; Xu, Y.; et al. Twist Induces Epithelial-Mesenchymal Transition in Cervical Carcinogenesis by Regulating the TGF-β/Smad3 Signaling Pathway. Oncol. Rep. 2015, 34, 1787–1794. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-Mesenchymal Transitions in Tumour Progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Nawshad, A.; LaGamba, D.; Polad, A.; Hay, E.D. Transforming Growth Factor-β Signaling during Epithelial-Mesenchymal Transformation: Implications for Embryogenesis and Tumor Metastasis. Cells Tissues Organs 2005, 179, 11–23. [Google Scholar] [CrossRef]
- Zhao, J.; Dong, D.; Sun, L.; Zhang, G.; Sun, L. Prognostic Significance of the Epithelial-to-Mesenchymal Transition Markers e-Cadherin, Vimentin and Twist in Bladder Cancer. Int. Braz. J. Urol. 2014, 40, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Francou, A.; Anderson, K.V. The Epithelial-to-Mesenchymal Transition in Development and Cancer. Annu. Rev. Cancer Biol. 2019, 4, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P. The Epithelial-Mesenchymal Transition (EMT) Phenomenon. Ann. Oncol. 2010, 21 (Suppl. 7), vii89–vii92. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Schaffhauser, B.; Christofori, G. Cadherins and the Tumour Progression: Is It All in a Switch? Cancer Lett. 2002, 176, 123–128. [Google Scholar] [CrossRef]
- Yan, X.; Chang, J.; Sun, R.; Meng, X.; Wang, W.; Zeng, L.; Liu, B.; Li, W.; Yan, X.; Huang, C.; et al. DHX9 Inhibits Epithelial-Mesenchymal Transition in Human Lung Adenocarcinoma Cells by Regulating STAT3. Am. J. Transl. Res. 2019, 11, 4881–4894. [Google Scholar]
- Watanabe, K.; Liu, Y.; Noguchi, S.; Murray, M.; Chang, J.-C.; Kishima, M.; Nishimura, H.; Hashimoto, K.; Minoda, A.; Suzuki, H. OVOL2 Induces Mesenchymal-to-Epithelial Transition in Fibroblasts and Enhances Cell-State Reprogramming towards Epithelial Lineages. Sci. Rep. 2019, 9, 6490. [Google Scholar] [CrossRef]
- Jahanbin, B.; Sarmadi, S.; Ghasemi, D.; Nili, F.; Moradi, J.-A.; Ghasemi, S. Pathogenic Role of Twist-1 Protein in Hydatidiform Molar Pregnancies and Investigation of Its Potential Diagnostic Utility in Complete Moles. Diagn. Pathol. 2023, 18, 40. [Google Scholar] [CrossRef]
- Moussa, R.A.; Eesa, A.N.; Abdallah, Z.F.; Abdelmeged, A.; Mahran, A.; Bahaa, H. Diagnostic Utility of Twist1, Ki-67, and E-Cadherin in Diagnosing Molar Gestations and Hydropic Abortions. Am. J. Clin. Pathol. 2018, 149, 442–455. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence That Fibroblasts Derive from Epithelium during Tissue Fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Xue, C.; Plieth, D.; Venkov, C.; Xu, C.; Neilson, E.G. The Gatekeeper Effect of Epithelial-Mesenchymal Transition Regulates the Frequency of Breast Cancer Metastasis. Cancer Res. 2003, 63, 3386–3394. [Google Scholar]
- Kida, Y.; Asahina, K.; Teraoka, H.; Gitelman, I.; Sato, T. Twist Relates to Tubular Epithelial-Mesenchymal Transition and Interstitial Fibrogenesis in the Obstructed Kidney. J. Histochem. Cytochem. 2007, 55, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Valenzi, E.; Bahudhanapati, H.; Tan, J.; Tabib, T.; Sullivan, D.I.; Nouraie, M.; Sembrat, J.; Fan, L.; Chen, K.; Liu, S.; et al. Single-Nucleus Chromatin Accessibility Identifies a Critical Role for TWIST1 in Idiopathic Pulmonary Fibrosis Myofibroblast Activity. Eur. Respir. J. 2023, 62, 2200474. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Mo, L.-H.; Feng, B.-S.; Jin, Q.-R.; Li, Y.; Lin, J.; Shu, Q.; Liu, Z.-G.; Liu, Z.; Sun, X.; et al. Twist1 Contributes to Developing and Sustaining Corticosteroid Resistance in Ulcerative Colitis. Theranostics 2021, 11, 7797–7812. [Google Scholar] [CrossRef] [PubMed]
- Górska, A.; Urbanowicz, M.; Grochowalski, Ł.; Seweryn, M.; Sobalska-Kwapis, M.; Wojdacz, T.; Lange, M.; Gruchała-Niedoszytko, M.; Jarczak, J.; Strapagiel, D.; et al. Genome-Wide DNA Methylation and Gene Expression in Patients with Indolent Systemic Mastocytosis. Int. J. Mol. Sci. 2023, 24, 13910. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Zhu, X.; Han, B.; Weng, J.; Wang, Y.; Liu, Y. The SIK1/CRTC2/CREB1 and TWIST1/PI3K/Akt/GSK3β Signaling Pathways Mediated by MicroRNA-25-3p Are Altered in the Schizophrenic Rat Brain. Front. Cell. Neurosci. 2023, 17, 1087335. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Xiao, X.; Li, R.; Lv, C.; Zhang, Y.; Wang, L.; Hong, T.; Zhang, H.; Wang, Y. Single-Cell Sequencing Reveals That Endothelial Cells, EndMT Cells and Mural Cells Contribute to the Pathogenesis of Cavernous Malformations. Exp. Mol. Med. 2023, 55, 628–642. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Gu, T.; Shen, Z.; Zhou, C.; Guo, Y.; Wang, J.; Li, B.; Xu, X.; Li, F.; Zhang, Q.; et al. Escherichia Coli Promotes Endothelial to Mesenchymal Transformation of Liver Sinusoidal Endothelial Cells and Exacerbates Nonalcoholic Fatty Liver Disease Via Its Flagellin. Cell. Mol. Gastroenterol. Hepatol. 2023, 16, 857–879. [Google Scholar] [CrossRef]
- Roberts, C.M.; Shahin, S.A.; Loeza, J.; Dellinger, T.H.; Williams, J.C.; Glackin, C.A. Disruption of TWIST1-RELA Binding by Mutation and Competitive Inhibition to Validate the TWIST1 WR Domain as a Therapeutic Target. BMC Cancer 2017, 17, 184. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Roberts, C.M.; Cardenas, C.; Tedja, R. The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers 2019, 11, 1083. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Wang, X.; Ling, M.T.; Guan, X.-Y.; Tsao, S.W.; Cheung, H.W.; Lee, D.T.; Wong, Y.C. Identification of a Novel Function of TWIST, a BHLH Protein, in the Development of Acquired Taxol Resistance in Human Cancer Cells. Oncogene 2004, 23, 474–482. [Google Scholar] [CrossRef]
- Fabregat, I.; Malfettone, A.; Soukupova, J. New Insights into the Crossroads between EMT and Stemness in the Context of Cancer. J. Clin. Med. 2016, 5, 37. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial–Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Yang, M.-H.; Hsu, D.S.-S.; Wang, H.-W.; Wang, H.-J.; Lan, H.-Y.; Yang, W.-H.; Huang, C.-H.; Kao, S.-Y.; Tzeng, C.-H.; Tai, S.-K.; et al. Bmi1 Is Essential in Twist1-Induced Epithelial–Mesenchymal Transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef]
- Dang, H.; Ding, W.; Emerson, D.; Rountree, C.B. Snail1 Induces Epithelial-to-Mesenchymal Transition and Tumor Initiating Stem Cell Characteristics. BMC Cancer 2011, 11, 396. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Hausen, A.Z.; et al. The EMT-Activator ZEB1 Promotes Tumorigenicity by Repressing Stemness-Inhibiting MicroRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Hojo, N.; Huisken, A.L.; Wang, H.; Chirshev, E.; Kim, N.S.; Nguyen, S.M.; Campos, H.; Glackin, C.A.; Ioffe, Y.J.; Unternaehrer, J.J. Snail Knockdown Reverses Stemness and Inhibits Tumour Growth in Ovarian Cancer. Sci. Rep. 2018, 8, 8704. [Google Scholar] [CrossRef]
- Garg, M. Targeting MicroRNAs in Epithelial-to-Mesenchymal Transition-Induced Cancer Stem Cells: Therapeutic Approaches in Cancer. Expert. Opin. Ther. Targets 2015, 19, 285–297. [Google Scholar] [CrossRef]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef]
- Gudjonsson, T.; Villadsen, R.; Nielsen, H.L.; Rønnov-Jessen, L.; Bissell, M.J.; Petersen, O.W. Isolation, Immortalization, and Characterization of a Human Breast Epithelial Cell Line with Stem Cell Properties. Genes Dev. 2002, 16, 693–706. [Google Scholar] [CrossRef]
- Choi, S.; Yu, J.; Park, A.; Dubon, M.J.; Do, J.; Kim, Y.; Nam, D.; Noh, J.; Park, K.-S. BMP-4 Enhances Epithelial Mesenchymal Transition and Cancer Stem Cell Properties of Breast Cancer Cells via Notch Signaling. Sci. Rep. 2019, 9, 11724. [Google Scholar] [CrossRef]
- Hollier, B.G.; Tinnirello, A.A.; Werden, S.J.; Evans, K.W.; Taube, J.H.; Sarkar, T.R.; Sphyris, N.; Shariati, M.; Kumar, S.V.; Battula, V.L.; et al. FOXC2 Expression Links Epithelial–Mesenchymal Transition and Stem Cell Properties in Breast Cancer. Cancer Res. 2013, 73, 1981–1992. [Google Scholar] [CrossRef]
- Zhou, P.; Li, B.; Liu, F.; Zhang, M.; Wang, Q.; Liu, Y.; Yao, Y.; Li, D. The Epithelial to Mesenchymal Transition (EMT) and Cancer Stem Cells: Implication for Treatment Resistance in Pancreatic Cancer. Mol. Cancer 2017, 16, 52. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Matsuda, Y.; Yoshimura, H.; Ueda, J.; Naito, Z.; Korc, M.; Ishiwata, T. Nestin Delineates Pancreatic Cancer Stem Cells in Metastatic Foci of NOD/Shi-Scid IL2Rγnull (NOG) Mice. Am. J. Pathol. 2014, 184, 674–685. [Google Scholar] [CrossRef]
- Wang, F.; Ma, L.; Zhang, Z.; Liu, X.; Gao, H.; Zhuang, Y.; Yang, P.; Kornmann, M.; Tian, X.; Yang, Y. Hedgehog Signaling Regulates Epithelial-Mesenchymal Transition in Pancreatic Cancer Stem-Like Cells. J. Cancer 2016, 7, 408–417. [Google Scholar] [CrossRef]
- Lee, Y.; Yoon, J.; Ko, D.; Yu, M.; Lee, S.; Kim, S. TMPRSS4 Promotes Cancer Stem–like Properties in Prostate Cancer Cells through Upregulation of SOX2 by SLUG and TWIST1. J. Exp. Clin. Cancer Res. 2021, 40, 372. [Google Scholar] [CrossRef]
- Wang, M.; Ren, D.; Guo, W.; Huang, S.; Wang, Z.; Li, Q.; Du, H.; Song, L.; Peng, X. N-Cadherin Promotes Epithelial-Mesenchymal Transition and Cancer Stem Cell-like Traits via ErbB Signaling in Prostate Cancer Cells. Int. J. Oncol. 2015, 48, 595–606. [Google Scholar] [CrossRef]
- Chen, J.; Wu, Z.; Deng, W.; Tang, M.; Wu, L.; Lin, N.; Chen, L.; Fu, Y.; Zhao, M.; Chen, C.; et al. USP51 Promotes Non-Small Cell Lung Carcinoma Cell Stemness by Deubiquitinating TWIST1. J. Transl. Med. 2023, 21, 453. [Google Scholar] [CrossRef]
- Wu, K.; Ding, J.; Chen, C.; Sun, W.; Ning, B.; Wen, W.; Huang, L.; Han, T.; Yang, W.; Wang, C.; et al. Hepatic Transforming Growth Factor Beta Gives Rise to Tumor-initiating Cells and Promotes Liver Cancer Development. Hepatology 2012, 56, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Bertran, E.; Crosas-Molist, E.; Sancho, P.; Caja, L.; Lopez-Luque, J.; Navarro, E.; Egea, G.; Lastra, R.; Serrano, T.; Ramos, E.; et al. Overactivation of the TGF-β Pathway Confers a Mesenchymal-like Phenotype and CXCR4-dependent Migratory Properties to Liver Tumor Cells. Hepatology 2013, 58, 2032–2044. [Google Scholar] [CrossRef] [PubMed]
- Khales, S.A.; Mozaffari-Jovin, S.; Geerts, D.; Abbaszadegan, M.R. TWIST1 Activates Cancer Stem Cell Marker Genes to Promote Epithelial-Mesenchymal Transition and Tumorigenesis in Esophageal Squamous Cell Carcinoma. BMC Cancer 2022, 22, 1272. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yin, J.; You, N.; Zhu, W.; Guo, N.; Liu, X.; Zhang, P.; Huang, W.; Xie, Y.; Ren, Q.; et al. Twist Family BHLH Transcription Factor 1 Is Required for the Maintenance of Leukemia Stem Cell in MLL-AF9+ Acute Myeloid Leukemia. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.M.; Tran, M.A.; Pitruzzello, M.C.; Wen, W.; Loeza, J.; Dellinger, T.H.; Mor, G.; Glackin, C.A. TWIST1 Drives Cisplatin Resistance and Cell Survival in an Ovarian Cancer Model, via Upregulation of GAS6, L1CAM, and Akt Signalling. Sci. Rep. 2016, 6, 37652. [Google Scholar] [CrossRef] [PubMed]
- Aptecar, L.; Puech, C.; Lopez-Crapez, E.; Peter, M.; Coopman, P.; D’Hondt, V.; Freiss, G. PTPN13 Participates in the Regulation of Epithelial–Mesenchymal Transition and Platinum Sensitivity in High-Grade Serous Ovarian Carcinoma Cells. Int. J. Mol. Sci. 2023, 24, 15413. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, H.; Liang, Y.; Zhao, L.; Li, Y.; Yan, Y.; Zhao, H.; Zhang, X.; Zou, F. MiR-15a-5p Enhances the Malignant Phenotypes of Colorectal Cancer Cells through the STAT3/TWIST1 and PTEN/AKT Signaling Pathways by Targeting SIRT4. Cell. Signal. 2023, 101, 110517. [Google Scholar] [CrossRef]
- Cioce, M.; Rutigliano, D.; Puglielli, A.; Fazio, V.M. Butein-Instigated MiR-186-5p-Dependent Modulation of TWIST1 Affects Resistance to Cisplatin and Bioenergetics of Malignant Pleural Mesothelioma Cells. Cancer Drug Resist. 2022, 5, 814–828. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, H.; Yin, X.; Long, L.; Xie, C.; Liu, Y.; Hui, L.; Lin, X.; Fang, Y.; Cao, Y.; et al. MiR-186 Regulation of Twist1 and Ovarian Cancer Sensitivity to Cisplatin. Oncogene 2016, 35, 323–332. [Google Scholar] [CrossRef]
- Parashar, D.; Geethadevi, A.; Mittal, S.; McAlarnen, L.A.; George, J.; Kadamberi, I.P.; Gupta, P.; Uyar, D.S.; Hopp, E.E.; Drendel, H.; et al. Patient-Derived Ovarian Cancer Spheroids Rely on PI3K-AKT Signaling Addiction for Cancer Stemness and Chemoresistance. Cancers 2022, 14, 958. [Google Scholar] [CrossRef]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist Transcriptionally Up-Regulates AKT2 in Breast Cancer Cells Leading to Increased Migration, Invasion, and Resistance to Paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Ferraresi, A.; Esposito, A.; Girone, C.; Vallino, L.; Salwa, A.; Ghezzi, I.; Thongchot, S.; Vidoni, C.; Dhanasekaran, D.N.; Isidoro, C. Resveratrol Contrasts LPA-Induced Ovarian Cancer Cell Migration and Platinum Resistance by Rescuing Hedgehog-Mediated Autophagy. Cells 2021, 10, 3213. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Lowe, G.; Roberts, C.M.; Finlay, J.; Han, E.S.; Glackin, C.A.; Dellinger, T.H. Pterostilbene Suppresses Ovarian Cancer Growth via Induction of Apoptosis and Blockade of Cell Cycle Progression Involving Inhibition of the STAT3 Pathway. Int. J. Mol. Sci. 2018, 19, 1983. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, M.; Wu, B. TWIST1 Promotes Colorectal Carcinoma Stemness and Oxaliplatin Resistance by Activating Microfibrillar-Associated Protein 2. ASSAY Drug Dev. Technol. 2023, 21, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Xu, J.; Tong, Y.; Yan, J.-F.; Pan, Y.; Wang, W.; Zheng, L.; Zheng, X.; Hu, C.; Hu, X.; et al. Rab31 Promotes Metastasis and Cisplatin Resistance in Stomach Adenocarcinoma through Twist1-Mediated EMT. Cell Death Dis. 2023, 14, 115. [Google Scholar] [CrossRef] [PubMed]
- Lou, B.; Fan, J.; Wang, K.; Chen, W.; Zhou, X.; Zhang, J.; Lin, S.; Lv, F.; Chen, Y. N1-Guanyl-1,7-Diaminoheptane (GC7) Enhances the Therapeutic Efficacy of Doxorubicin by Inhibiting Activation of Eukaryotic Translation Initiation Factor 5A2 (EIF5A2) and Preventing the Epithelial–Mesenchymal Transition in Hepatocellular Carcinoma Cells. Exp. Cell Res. 2013, 319, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xu, Z.; Lv, H.; Wang, Y.; Wang, L.; Ni, Y.; Wang, X.; Hu, C.; Chen, S.; Teng, F.; et al. EIF5A2 Regulates the Resistance of Gastric Cancer Cells to Cisplatin via Induction of EMT. Am. J. Transl. Res. 2018, 10, 4269–4279. [Google Scholar] [PubMed]
- Xu, G.; Chen, H.; Wu, S.; Chen, J.; Zhang, S.; Shao, G.; Sun, L.; Mu, Y.; Liu, K.; Pan, Q.; et al. Eukaryotic Initiation Factor 5A2 Mediates Hypoxia-Induced Autophagy and Cisplatin Resistance. Cell Death Dis. 2022, 13, 683. [Google Scholar] [CrossRef]
- Nushtaeva, A.; Ermakov, M.; Abdurakhmanova, M.; Troitskaya, O.; Belovezhets, T.; Varlamov, M.; Gayner, T.; Richter, V.; Koval, O. “Pulsed Hypoxia” Gradually Reprograms Breast Cancer Fibroblasts into Pro-Tumorigenic Cells via Mesenchymal–Epithelial Transition. Int. J. Mol. Sci. 2023, 24, 2494. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhu, Y. FAP, CD10, and GPR77-Labeled CAFs Cause Neoadjuvant Chemotherapy Resistance by Inducing EMT and CSC in Gastric Cancer. BMC Cancer 2023, 23, 507. [Google Scholar] [CrossRef]
- Xiang, X.; Hao, Y.; Cheng, C.; Hu, H.; Chen, H.; Tan, J.; Wang, Y.; Liu, X.; Peng, B.; Liao, J.; et al. A TGF-β-Dominant Chemo-Resistant Phenotype of Hepatoblastoma Associated with Aflatoxin Exposure in Children. Hepatology 2023. [Google Scholar] [CrossRef] [PubMed]
- Tedja, R.; Roberts, C.M.; Alvero, A.B.; Cardenas, C.; Yang-Hartwich, Y.; Spadinger, S.; Pitruzzello, M.; Yin, G.; Glackin, C.A.; Mor, G. Protein Kinase Calpha-Mediated Phosphorylation of Twist1 at Ser-144 Prevents Twist1 Ubiquitination and Stabilizes It. J. Biol. Chem. 2019, 294, 5082–5093. [Google Scholar] [CrossRef] [PubMed]
- Guan, T.; Li, M.; Song, Y.; Chen, J.; Tang, J.; Zhang, C.; Wen, Y.; Yang, X.; Huang, L.; Zhu, Y.; et al. Phosphorylation of USP29 by CDK1 Governs TWIST1 Stability and Oncogenic Functions. Adv. Sci. 2023, 10, 2205873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chan, S.; Liu, X.; Shi, Y.; Dong, Z.; Shao, X.; Zheng, L.; Mai, Z.; Fang, T.; Deng, L.; et al. Targeting Hexokinase 2 Increases the Sensitivity of Oxaliplatin by Twist1 in Colorectal Cancer. J. Cell. Mol. Med. 2021, 25, 8836–8849. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Feng, S.; Chang, K.; Yu, X.; Yang, F.; Huang, H.; Wang, Y.; Li, X.; Guan, F. Reciprocal Regulation of TWIST1 and OGT Determines the Decitabine Efficacy in MDS/AML. Cell Commun. Signal. 2023, 21, 255. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Ogura, K.; Wang, Z.; Inuzuka, H. Degradation of the Transcription Factor Twist, an Oncoprotein That Promotes Cancer Metastasis. Discov. Med. 2013, 15, 7–15. [Google Scholar] [PubMed]
- Cao, G.; Li, P.; He, X.; Jin, M.; Li, M.; Chen, S.; Xu, X.; Sun, Q.; Xiong, M.; Chen, B. FHL3 Contributes to EMT and Chemotherapy Resistance Through Up-Regulation of Slug and Activation of TGFβ/Smad-Independent Pathways in Gastric Cancer. Front. Oncol. 2021, 11, 649029. [Google Scholar] [CrossRef]
- Lai, H.; Hung, L.; Yen, C.; Hung, H.; Chen, R.; Ku, Y.; Lo, H.; Tsai, H.; Lee, Y.; Yang, T.; et al. NEIL3 Promotes Hepatoma Epithelial–Mesenchymal Transition by Activating the BRAF/MEK/ERK/TWIST Signaling Pathway. J. Pathol. 2022, 258, 339–352. [Google Scholar] [CrossRef]
- Sousa-Squiavinato, A.C.M.; Ramos, D.A.A.; Wagner, M.S.; Tessmann, J.W.; de-Freitas-Junior, J.C.M.; Morgado-Díaz, J.A. Long-Term Resistance to 5-Fluorouracil Promotes Epithelial–Mesenchymal Transition, Apoptosis Evasion, Autophagy, and Reduced Proliferation Rate in Colon Cancer Cells. Eur. J. Pharmacol. 2022, 933, 175253. [Google Scholar] [CrossRef]
- Solanes-Casado, S.; Cebrián, A.; Rodríguez-Remírez, M.; Mahíllo, I.; García-García, L.; Río-Vilariño, A.; Baños, N.; de Cárcer, G.; Monfort-Vengut, A.; Castellano, V.; et al. Overcoming PLK1 Inhibitor Resistance by Targeting Mevalonate Pathway to Impair AXL-TWIST Axis in Colorectal Cancer. Biomed. Pharmacother. 2021, 144, 112347. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.G.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J.C. Vascular Channel Formation by Human Melanoma Cells in Vivo and in Vitro: Vasculogenic Mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Xie, M.; Zhou, Y.; Li, T.; Liu, W.; Yu, W.; Jia, M.; Yu, S.; Chen, L.; Dai, R.; et al. Vascular Mimicry Induced by M6A Mediated IGFL2-AS1/AR Axis Contributes to Pazopanib Resistance in Clear Cell Renal Cell Carcinoma. Cell Death Discov. 2023, 9, 121. [Google Scholar] [CrossRef] [PubMed]
- You, B.; Sun, Y.; Luo, J.; Wang, K.; Liu, Q.; Fang, R.; Liu, B.; Chou, F.; Wang, R.; Meng, J.; et al. Androgen Receptor Promotes Renal Cell Carcinoma (RCC) Vasculogenic Mimicry (VM) via Altering TWIST1 Nonsense-Mediated Decay through LncRNA-TANAR. Oncogene 2021, 40, 1674–1689. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Chen, W.; Li, X.; Yu, L.; Xu, Y.; Ruan, Q.; He, Y.; Wang, Y. TWIST1-EP300 Expedites Gastric Cancer Cell Resistance to Apatinib by Activating the Expression of COL1A2. Anal. Cell. Pathol. 2022, 2022, 5374262. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhou, A.; Hu, X.; Xiang, Y.; Huang, M.; Huang, J.; Yang, D.; Tang, Y. LMNA Reduced Acquired Resistance to Erlotinib in NSCLC by Reversing the Epithelial–Mesenchymal Transition via the FGFR/MAPK/c-Fos Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 13237. [Google Scholar] [CrossRef]
- Peng, M.; Zheng, Z.; Chen, S.; Fang, L.; Feng, R.; Zhang, L.; Tang, Q.; Liu, X. Sensitization of Non-Small Cell Lung Cancer Cells to Gefitinib and Reversal of Epithelial–Mesenchymal Transition by Aloe-Emodin Via PI3K/Akt/TWIS1 Signal Blockage. Front. Oncol. 2022, 12, 908031. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Fu, X.; Zhang, X.; Bai, C.; Wang, Y. Circ_0001658 Regulates Gefitinib Resistance of Non-Small Cell Lung Cancer through MiR-409-3p/TWIST1 Axis. Anti-Cancer Drugs 2022, 33, 158–166. [Google Scholar] [CrossRef]
- Wang, J.; Wang, T.; Zhang, Y.; Liu, J.; Song, J.; Han, Y.; Wang, L.; Yang, S.; Zhu, L.; Geng, R.; et al. CPEB1 Enhances Erastin-induced Ferroptosis in Gastric Cancer Cells by Suppressing Twist1 Expression. IUBMB Life 2021, 73, 1180–1190. [Google Scholar] [CrossRef]
- Roberts, C.M.; Rojas-Alexandre, M.; Hanna, R.E.; Lin, Z.P.; Ratner, E.S. Transforming Growth Factor Beta and Epithelial to Mesenchymal Transition Alter Homologous Recombination Repair Gene Expression and Sensitize BRCA Wild-Type Ovarian Cancer Cells to Olaparib. Cancers 2023, 15, 3919. [Google Scholar] [CrossRef]
- Alvero, A.B.; Chen, R.; Fu, H.-H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.-A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular Phenotyping of Human Ovarian Cancer Stem Cells Unravels the Mechanisms for Repair and Chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, W.; Cheng, C.-T.; Ren, X.; Somlo, G.; Fong, M.Y.; Chin, A.R.; Li, H.; Yu, Y.; Xu, Y.; et al. TGFβ Induces “BRCAness” and Sensitivity to PARP Inhibition in Breast Cancer by Regulating DNA-Repair Genes. Mol. Cancer Res. 2014, 12, 1597–1609. [Google Scholar] [CrossRef] [PubMed]
- Reisenauer, K.N.; Tao, Y.; Das, P.; Song, S.; Svatek, H.; Patel, S.D.; Mikhail, S.; Ingros, A.; Sheesley, P.; Masi, M.; et al. Epithelial-Mesenchymal Transition Sensitizes Breast Cancer Cells to Cell Death via the Fungus-Derived Sesterterpenoid Ophiobolin A. Sci. Rep. 2021, 11, 10652. [Google Scholar] [CrossRef]
- Koyama, T.; Shimizu, T.; Kojima, Y.; Sudo, K.; Okuma, H.S.; Shimoi, T.; Ichikawa, H.; Kohsaka, S.; Sadachi, R.; Hirakawa, A.; et al. Clinical Activity and Exploratory Resistance Mechanism of Milademetan, an MDM2 Inhibitor, in Intimal Sarcoma with MDM2 Amplification: An Open-Label Phase Ib/II Study. Cancer Discov. 2023, 13, 1814–1825. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, G.; Zhu, D.; Huang, Y.; Luo, Y.; Su, P.; Chen, X.; Wang, Q. Epithelial-Mesenchymal Transition and Cancer Stem Cell-like Phenotype Induced by Twist1 Contribute to Acquired Resistance to Irinotecan in Colon Cancer. Int. J. Oncol. 2017, 51, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M.A.; Mala, A.; Merodoulaki, A.C.; Vassilakopoulou, M.; Mavroudis, D.; Agelaki, S. Investigating the Role of CTCs with Stem/EMT-like Features in Metastatic Breast Cancer Patients Treated with Eribulin Mesylate. Cancers 2022, 14, 3903. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, M.; Nimura, K.; Mori, M.; Nakagami, H.; Kaneda, Y. The Transcription Factors Tbx18 and Wt1 Control the Epicardial Epithelial-Mesenchymal Transition through Bi-Directional Regulation of Slug in Murine Primary Epicardial Cells. PLoS ONE 2013, 8, e57829. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Abadi, A.J.; Gholami, M.H.; Hashemi, F.; Zabolian, A.; Hushmandi, K.; Zarrabi, A.; Entezari, M.; Aref, A.R.; Khan, H.; et al. The Involvement of Epithelial-to-Mesenchymal Transition in Doxorubicin Resistance: Possible Molecular Targets. Eur. J. Pharmacol. 2021, 908, 174344. [Google Scholar] [CrossRef]
- Khan, M.A.; Chen, H.; Zhang, D.; Fu, J. Twist: A Molecular Target in Cancer Therapeutics. Tumor Biol. 2013, 34, 2497–2506. [Google Scholar] [CrossRef]
- Tabatabaee, A.; Nafari, B.; Farhang, A.; Hariri, A.; Khosravi, A.; Zarrabi, A.; Mirian, M. Targeting Vimentin: A Multifaceted Approach to Combatting Cancer Metastasis and Drug Resistance. Cancer Metastasis Rev. 2023. [Google Scholar] [CrossRef]
- Zhou, Z.; Jin, L.; Shen, J.; Shi, W.; Xu, Y.; Ye, L.; Liu, J.; Pan, J. COM33 Suppresses Carboplatin-Induced Epithelial-Mesenchymal Transition via Inhibition of Twist1 in Ovarian Cancer. Acta Biochim. Biophys. Sin. 2022, 55, 34–42. [Google Scholar] [CrossRef]
- Balcik-Ercin, P.; Sever, B. An Investigation of Bacteriocin Nisin Anti-Cancer Effects and FZD7 Protein Interactions in Liver Cancer Cells. Chem.-Biol. Interact. 2022, 366, 110152. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Sun, H.-L.; Hsu, Y.-H.; Liu, S.-H.; Lii, C.-K.; Tsai, C.-H.; Liu, K.-L.; Huang, C.-S.; Li, C.-C. α-Linolenic Acid Inhibits the Migration of Human Triple-Negative Breast Cancer Cells by Attenuating Twist1 Expression and Suppressing Twist1-Mediated Epithelial-Mesenchymal Transition. Biochem. Pharmacol. 2020, 180, 114152. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Chen, S.; Yu, T.; Chen, W.; Huang, J.; Peng, C.; Ding, Y. Harmine Suppresses Breast Cancer Cell Migration and Invasion by Regulating TAZ-Mediated Epithelial-Mesenchymal Transition. Am. J. cancer Res. 2021, 12, 2612–2626. [Google Scholar]
- Nafie, E.; Lolarga, J.; Lam, B.; Guo, J.; Abdollahzadeh, E.; Rodriguez, S.; Glackin, C.; Liu, J. Harmine Inhibits Breast Cancer Cell Migration and Invasion by Inducing the Degradation of Twist1. PLoS ONE 2021, 16, e0247652. [Google Scholar] [CrossRef] [PubMed]
- Cordani, N.; Mologni, L.; Piazza, R.; Tettamanti, P.; Cogliati, V.; Mauri, M.; Villa, M.; Malighetti, F.; Bella, C.D.; Jaconi, M.; et al. TWIST1 Upregulation Is a Potential Target for Reversing Resistance to the CDK4/6 Inhibitor in Metastatic Luminal Breast Cancer Cells. Int. J. Mol. Sci. 2023, 24, 16294. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Yang, Y.; Yang, J.; Cui, Y.; Cao, Z.; Zuo, D.; Zhai, X. Harmine-Inspired Design and Synthesis of Benzo[d]Imidazo [2,1-b]Thiazole Derivatives Bearing 1,3,4-Oxadiazole Moiety as Potential Tumor Suppressors. Bioorg. Med. Chem. 2021, 46, 116367. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Qu, L.; Wang, C.; Luo, H.; Li, S.; Yin, F.; Liu, X.; Chen, X.; Luo, Z.; Cui, N.; et al. Harmine-Based Dual Inhibitors Targeting Histone Deacetylase (HDAC) and DNA as a Promising Strategy for Cancer Therapy. Bioorg. Chem. 2022, 120, 105604. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Mi, D.; Ling, J.; Li, H.; He, P.; Liu, N.; Chen, Q.; Chen, Y.; Huang, L. Discovery of YH677 as a Cancer Stemness Inhibitor That Suppresses Triple-Negative Breast Cancer Growth and Metastasis by Regulating the TGFβ Signaling Pathway. Cancer Lett. 2023, 560, 216142. [Google Scholar] [CrossRef]
- Reisenauer, K.N.; Aroujo, J.; Tao, Y.; Ranganathan, S.; Romo, D.; Taube, J.H. Therapeutic Vulnerabilities of Cancer Stem Cells and Effects of Natural Products. Nat. Prod. Rep. 2023, 40, 1432–1456. [Google Scholar] [CrossRef]
- Bobbin, M.L.; Rossi, J.J. RNA Interference (RNAi)-Based Therapeutics: Delivering on the Promise? Annu. Rev. Pharmacol. Toxicol. 2016, 56, 103–122. [Google Scholar] [CrossRef]
- Simão, A.Y.; Gonçalves, J.; Duarte, A.P.; Barroso, M.; Cristóvão, A.C.; Gallardo, E. Toxicological Aspects and Determination of the Main Components of Ayahuasca: A Critical Review. Medicines 2019, 6, 106. [Google Scholar] [CrossRef] [PubMed]
- Marwat, S.K.; ur Rehman, F. Chapter 70—Medicinal and Pharmacological Potential of Harmala (Peganum harmala L.) Seeds. In Nuts and Seeds in Health and Disease Prevention; Part 2: Effects of Specific Nuts and Seeds; Elsevier: Amsterdam, The Netherlands, 2011; pp. 585–599. [Google Scholar] [CrossRef]
- Dogra, S.; Elayapillai, S.P.; Qu, D.; Pitts, K.; Filatenkov, A.; Houchen, C.W.; Berry, W.L.; Moxley, K.; Hannafon, B.N. Targeting Doublecortin-like Kinase 1 Reveals a Novel Strategy to Circumvent Chemoresistance and Metastasis in Ovarian Cancer. Cancer Lett. 2023, 578, 216437. [Google Scholar] [CrossRef] [PubMed]
- Bahar, E.; Kim, J.-Y.; Kim, D.-C.; Kim, H.-S.; Yoon, H. Combination of Niraparib, Cisplatin and Twist Knockdown in Cisplatin-Resistant Ovarian Cancer Cells Potentially Enhances Synthetic Lethality through ER-Stress Mediated Mitochondrial Apoptosis Pathway. Int. J. Mol. Sci. 2021, 22, 3916. [Google Scholar] [CrossRef] [PubMed]
- Finlay, J.; Roberts, C.M.; Lowe, G.; Loeza, J.; Rossi, J.J.; Glackin, C.A. RNA-Based TWIST1 Inhibition via Dendrimer Complex to Reduce Breast Cancer Cell Metastasis. Biomed. Res. Int. 2015, 2015, 382745. [Google Scholar] [CrossRef] [PubMed]
- Finlay, J.; Roberts, C.M.; Dong, J.; Zink, J.I.; Tamanoi, F.; Glackin, C.A. Mesoporous Silica Nanoparticle Delivery of Chemically Modified SiRNA against TWIST1 Leads to Reduced Tumor Burden. Nanomedicine 2015, 11, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.M.; Shahin, S.A.; Wen, W.; Finlay, J.B.; Dong, J.; Wang, R.; Dellinger, T.H.; Zink, J.I.; Tamanoi, F.; Glackin, C.A. Nanoparticle Delivery of SiRNA against TWIST to Reduce Drug Resistance and Tumor Growth in Ovarian Cancer Models. Nanomedicine 2017, 13, 965–976. [Google Scholar] [CrossRef]
- Shahin, S.A.; Wang, R.; Simargi, S.I.; Contreras, A.; Echavarria, L.P.; Qu, L.; Wen, W.; Dellinger, T.; Unternaehrer, J.; Tamanoi, F.; et al. Hyaluronic Acid Conjugated Nanoparticle Delivery of SiRNA against TWIST Reduces Tumor Burden and Enhances Sensitivity to Cisplatin in Ovarian Cancer. Nanomedicine 2018, 14, 1381–1394. [Google Scholar] [CrossRef]
- Dhanwal, V.; Katoch, A.; Nayak, D.; Chakraborty, S.; Gupta, R.; Kumar, A.; Gupta, P.N.; Singh, N.; Kaur, N.; Goswami, A. Benzimidazole-Based Organic–Inorganic Gold Nanohybrids Suppress Invasiveness of Cancer Cells by Modulating EMT Signaling Cascade. ACS Appl. Bio Mater. 2021, 4, 470–482. [Google Scholar] [CrossRef]
- Luo, D.; Zeng, X.; Zhang, S.; Li, D.; Cheng, Z.; Wang, Y.; Long, J.; Hu, Z.; Long, S.; Zhou, J.; et al. Pirfenidone Suppressed Triple-negative Breast Cancer Metastasis by Inhibiting the Activity of the TGF-β/SMAD Pathway. J. Cell. Mol. Med. 2023, 27, 456–469. [Google Scholar] [CrossRef]
- Ruiz-Mitjana, A.; Vidal-Sabanés, M.; Navaridas, R.; Perramon-Güell, A.; Yeramian, A.; Nicholson-Sabaté, N.; Egea, J.; Encinas, M.; Matias-Guiu, X.; Dolcet, X. Metformin Exhibits Antineoplastic Effects on Pten-Deficient Endometrial Cancer by Interfering with TGF-β and P38/ERK MAPK Signalling. Biomed. Pharmacother. 2023, 168, 115817. [Google Scholar] [CrossRef]
- Hseu, J.-H.; Lin, Y.-A.; Pandey, S.; Vadivalagan, C.; Ali, A.; Chen, S.-J.; Way, T.-D.; Yang, H.-L.; Hseu, Y.-C. Antrodia Salmonea Suppresses Epithelial-Mesenchymal Transition/Metastasis and Warburg Effects by Inhibiting Twist and HIF-1α Expression in Twist-Overexpressing Head and Neck Squamous Cell Carcinoma Cells. J. Ethnopharmacol. 2024, 318, 117030. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, M.; Vincenzo, S.D.; Lazzara, V.; Pinto, P.; Patella, B.; Inguanta, R.; Bruno, A.; Pace, E. Formoterol Exerts Anti-Cancer Effects Modulating Oxidative Stress and Epithelial-Mesenchymal Transition Processes in Cigarette Smoke Extract Exposed Lung Adenocarcinoma Cells. Int. J. Mol. Sci. 2023, 24, 16088. [Google Scholar] [CrossRef] [PubMed]
- Mozafari, M.; Ebrahimi, S.; Darban, R.A.; Hashemy, S.I. Potential in Vitro Therapeutic Effects of Targeting SP/NK1R System in Cervical Cancer. Mol. Biol. Rep. 2022, 49, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Jafarinezhad, S.; Darban, R.A.; Javid, H.; Hashemy, S.I. The SP/NK1R System Promotes the Proliferation of Breast Cancer Cells through NF-ΚB-Mediated Inflammatory Responses. Cell Biochem. Biophys. 2023, 81, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, S.; Darban, R.A.; Javid, H.; Hashemy, S.I. The Therapeutic Potential of Aprepitant in Glioblastoma Cancer Cells through Redox Modification. BioMed Res. Int. 2022, 2022, 8540403. [Google Scholar] [CrossRef] [PubMed]
- Akbari, S.; Darban, R.A.; Javid, H.; Esparham, A.; Hashemy, S.I. The Anti-Tumoral Role of Hesperidin and Aprepitant on Prostate Cancer Cells through Redox Modifications. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2023, 396, 3559–3567. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, J.; Xie, L.; Zhou, S.; Huang, Y.; Chen, Y.; Gao, X.; Xiao, W.; Chen, J. Biguanide-Anchored Albumin-Based Nanoplatform Inhibits Epithelial-Mesenchymal Transition and Reduces the Stemness Phenotype for Metastatic Cancer Therapy. Acta Biomater. 2023, 171, 565–579. [Google Scholar] [CrossRef]
- Du, L.; Xie, F.; Han, H.; Zhang, L. Targeting SALL4 by Entinostat Inhibits the Malignant Phenotype of Gastric Cancer Cells by Reducing EMT Signaling. Anticancer Res. 2023, 43, 4389–4401. [Google Scholar] [CrossRef]
- Trimboli, A.J.; Fukino, K.; de Bruin, A.; Wei, G.; Shen, L.; Tanner, S.M.; Creasap, N.; Rosol, T.J.; Robinson, M.L.; Eng, C.; et al. Direct Evidence for Epithelial-Mesenchymal Transitions in Breast Cancer. Cancer Res. 2008, 68, 937–945. [Google Scholar] [CrossRef]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The Epithelial–Mesenchymal Transition: New Insights in Signaling, Development, and Disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Reed, I.G. Mechanism and Regulation of Epithelial—Mesenchymal Transition in Cancer. Cell Health Cytoskelet. 2015, 7, 155–166. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Migrating Cancer Stem Cells—An Integrated Concept of Malignant Tumour Progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhu, H.; Wan, H.; Zou, X.; Ma, X.; Gao, G. Harmine Suppresses the Proliferation and Migration of Human Ovarian Cancer Cells through Inhibiting ERK/CREB Pathway. Oncol. Rep. 2017, 38, 2927–2934. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Cades, J.; Mazzacurati, L.; Neumann, N.M.; Khetarpal, S.K.; Chatterjee, S.; Wang, H.; Attar, M.A.; Huang, E.H.-B.; Chatley, S.N.; et al. A First-in-Class TWIST1 Inhibitor with Activity in Oncogene-Driven Lung Cancer. Mol. Cancer Res. 2017, 15, 1764–1776. [Google Scholar] [CrossRef]
- Lu, X.; Jin, J.; Wu, Y.; Liu, X.; Liang, X.; Lin, J.; Sun, Q.; Qin, J.; Zhang, W.; Luan, X. Progress in RAS-targeted Therapeutic Strategies: From Small Molecule Inhibitors to Proteolysis Targeting Chimeras. Med. Res. Rev. 2023, 1–21. [Google Scholar] [CrossRef]
- Condeelis, J.; Segall, J.E. Intravital Imaging of Cell Movement in Tumours. Nat. Rev. Cancer 2003, 3, 921–930. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirjat, D.; Kashif, M.; Roberts, C.M. Shake It Up Baby Now: The Changing Focus on TWIST1 and Epithelial to Mesenchymal Transition in Cancer and Other Diseases. Int. J. Mol. Sci. 2023, 24, 17539. https://doi.org/10.3390/ijms242417539
Mirjat D, Kashif M, Roberts CM. Shake It Up Baby Now: The Changing Focus on TWIST1 and Epithelial to Mesenchymal Transition in Cancer and Other Diseases. International Journal of Molecular Sciences. 2023; 24(24):17539. https://doi.org/10.3390/ijms242417539
Chicago/Turabian StyleMirjat, Dureali, Muhammad Kashif, and Cai M. Roberts. 2023. "Shake It Up Baby Now: The Changing Focus on TWIST1 and Epithelial to Mesenchymal Transition in Cancer and Other Diseases" International Journal of Molecular Sciences 24, no. 24: 17539. https://doi.org/10.3390/ijms242417539